
Abstract
In the first part of this review, the basic principles of chemical glycosylation reactions are discussed; this way the advantages of O‑glycosyl trichloroacetimidates and related systems as glycosyl donors become obvious. Many new methods for the generation of O‑glycosyl trichloroacetimidates and for their use as glycosyl donors have been introduced which are compiled as well as their use in solid-phase oligosaccharide synthesis. The power of these glycosyl donors is demonstrated by their application in complex oligosaccharide and glycoconjugate synthesis, as outlined in the second part of this review. Recent applications in glycolipid, glycosyl amino acid and glycopeptide, nucleoside and nucleotide glycosidation, glycosamino glycan, cell wall constituent, and GPI anchor synthesis, glycosylation of various natural products and their metabolites, and finally cyclooligosaccharide generation are compiled. In the last section, related glycosyl donors are briefly discussed.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others
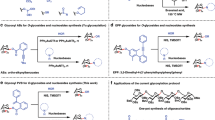
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The biological significance of oligosaccharides and glycoconjugates has stimulated many activities in carbohydrate chemistry. Most of these activities have been devoted to the development of methods for glycoside bond formation as the assembly of monosaccharide building blocks to complex oligosaccharides and glycoconjugates is the most difficult task in this endeavor.
Glycosyl Donor Generation Through Anomeric-Oxygen Exchange Reactions
For a long time the methods developed for glycosylation have essentially favored approaches which require for glycosyl donor generation an anomeric oxygen exchange reaction on the half-acetal moiety of pyranoses and furanoses [Fig. 1, methods (A) and (B)] [1,2,3,4]. The Fischer–Helferich method (A), as a direct acid‐catalyzed anomeric‐oxygen replacement reaction has been very successfully applied to the synthesis of simple alkyl glycosides. However, because of its reversibility, it is of limited usefulness in the synthesis of complex oligosaccharides and glycoconjugates. For this endeavor, irreversible methods are required, which can be attained by preactivation of the anomeric center by introducing good leaving groups which can be released by leaving group specific promoters or—even better—by catalytic amounts of an activator.

Synthesis of glycosides and saccharides
The best‐known of these methods is the Koenigs–Knorr method (B) in which the anomeric hydroxy group is replaced by chlorine or bromine. Thus, an α‑halogen‐ether is generated which in the glycosylation step can be readily activated by halophilic promoters frequently incorrectly termed “catalyst.” Generally, from one up to four equivalents of heavy metal salts are employed resulting in an irreversible transfer of the glycosyl donor moiety to the acceptor. On the basis of this general method many valuable techniques for the synthesis of complex oligosaccharides and glycoconjugates have been introduced which have been extensively reviewed. However, the obvious limitations of this method were the reason for the search for alternative methods [1,2,3,4,5,6,7].
Therefore, other anomeric oxygen exchange reactions closely related to the Koenigs–Knorr method have been extensively investigated. Particularly, the introduction of fluorine and alkyl- and arylthio groups as leaving groups gained great interest because these groups also tolerate manipulations of orthogonal protecting groups. Differences in thio leaving group tendencies (in combination with the “armed”/“disarmed” principle ) could be even employed for one‑pot consecutive glycosylations leading to more or less pure oligosaccharide products. However, the basic drawbacks of the Koenigs–Knorr method are also associated with these promoter systems. For instance, the large amounts of promoter required (and often additional reagents) limit their usefulness particularly in large‐scale oligosaccharide and glycoconjugate synthesis.
Direct Anomeric Oxygen Alkylation
For more than a century glycosylations were essentially based on methods where the anomeric carbon of the sugar residue to be coupled served as the electrophile (the glycosyl donor ) and the alcohol (the glycosyl acceptor ) as the nucleophile [Fig. 1, (A), (B)]. Alternatively, base‐mediated deprotonation of the anomeric hydroxy group generating at first an anomeric oxide structure from a pyranose or a furanose and then anomeric O‑alkylation leading directly and irreversibly to glycosides should be also available [Fig. 1, (C)]. Surprisingly, no studies employing this simple ‘anomeric O‑alkylation method’ as termed by us [1,2,3,4], for the synthesis of complex glycosides and glycoconjugates had been reported before our work. Only a few scattered examples with simple alkylating agents, for instance, excess methyl iodide or dimethyl sulfate, have been found in the literature [1]. However, in our hands, direct anomeric O‑alkylation of variously protected and totally unprotected sugars in the presence of a base and triflates or Michael acceptors, respectively, as alkylating agents has become a very convenient method for glycoside bond formation [8,9,10,11]. Base‐promoted decomposition reactions, particularly of the acyclic form, were practically not observed. Often even high anomeric diastereocontrol was available. The high diastereocontrol in pyranoses is based on the enhanced nucleophilicity of equatorial oxygen atoms (due to steric effects and the stereoelectronic kinetic anomeric effect ) [1,2,3,4] and on the higher stability of axial oxygen atom‐derived products (due to the thermodynamic anomeric effect ). Chelation effects can be also employed to design anomeric stereocontrol. The availability and to some extent the stability of the carbohydrate‐derived alkylating agents precluded the general applicability of this simple method to the synthesis of complex oligosaccharides and glycoconjugates.
Glycosyl Donor Generation Through Retention of the Anomeric Oxygen
The requirements for an efficient glycosylation method are the following:
-
high chemical and stereochemical yield,
-
applicability to large‐scale preparations, and
-
avoidance of large amounts of waste materials, i. e. activation of the donor by catalytic amounts of reagent.
These demands were not met by any of the above‐described methods for the synthesis of complex oligosaccharides and glycoconjugates. However, the general strategy for glycoside bond formation seems to be correct:
-
The first step ( activation step ) should consist of an activation of the anomeric center under formation of a stable glycosyl donor—best by a catalyzed attachment of a leaving group to the anomeric hydroxy group.
-
The second step ( glycosylation step ) should consist of a sterically uniform high‐yielding glycosyl transfer to the glycosyl acceptor based on glycosyl donor activation with catalytic amounts of promoter, i. e. a catalyst. Obviously, this catalytic procedure has to be orthogonal to the glycosyl donor preparation procedure. Diastereocontrol in the glycosylation step may be derived from the anomeric configuration of the glycosyl donor (by inversion or retention), by anchimeric assistance, by the solvent influence, by thermodynamic and/or stereoelectronic effects, or by any other effects.
The experience with the direct anomeric O‑alkylation exhibited that these demands can be fulfilled by a simple base‐catalyzed anomeric O‑transformation into a leaving group and its acid‐catalyzed activation in the glycosylation step. This approach should also satisfy the demand for simplicity in combination with efficiency which is decisive for general acceptance.
Obviously, for achieving stereocontrolled activation of the anomeric oxygen atom, the anomerization of the anomeric hydroxy group or the anomeric oxide ion, respectively, has to be considered. Thus, in a reversible activation process and with the help of kinetic and thermodynamic reaction control, possibly both activated anomers should be accessible. From these considerations it was concluded that suitable triple‐bond systems A≡B (or compounds containing cumulative double bond systems A=B=C) might be found that add pyranoses and furanoses under base catalysis directly and reversibly in a stereocontrolled manner [Fig. 1, (D)].

Trichloroacetimidate formation and its acid‐catalyzed transformations
Electron‐deficient nitriles , such as for instance trichloroacetonitrile (and trifluoroacetonitrile [11]), are known to undergo direct and reversible, base‐catalyzed addition of alcohols providing O‑alkyl trichloroacetimidates (or O‑alkyl trifluoroacetimidate, respectively) [1,12,13] [Fig. 2, (1)]. This imidate synthesis has the advantage that the imidates can be isolated as stable adducts which are less sensitive to hydrolysis than the corresponding salts. On acid addition leading to imidate activation [Fig. 2, (2)], hydrolysis with water (R2–OH = H2O) is a fast reaction furnishing amide CCl3–CONH2 and alcohol R1–OH [Fig. 2, (3A)]; mechanistically this is an acylative attack of the imidate at the nucleophile. With other nucleophiles, after acylation by the imidate, other transformations are possible, as for instance ortho‐ester formation. However, the basic question is: are these activated imidates also good alkylating agents [Fig. 2, (3B)] as required for being effective in glycosidation reactions? On consideration of the influence of the substituents R and R1 on these two competing reactions an attack of R2–OH leads to the following expectations:
-
Acylation [reaction (3A)] is supported by R being a small electron‐withdrawing group, R1 destabilizing carbenium ion formation.
-
Alkylation [reaction (3B)] is supported by R being a sterically demanding electron‐withdrawing group, R1supporting carbenium ion formation.
From these considerations it can be deduced that the bulky and strongly electron‐withdrawing trichloromethyl group as R and the glycosyl group as R1, which through the α‑oxygen atom supports oxocarbenium ion formation at the anomeric center, should provide excellent alkylating agents; hence, O‑glycosyl trichloroacetimidates should on acid activation exhibit excellent glycosyl donor properties.
As expected, on base‐catalyzed addition of the anomeric hydroxy group to trichloroacetonitrile the O‑glycosyl trichloroacetimidate is formed for which, due to the reversibility of the addition, the different nucleophilicities of the anomeric oxides, and the different thermodynamic stabilities of the O‑glycosyl trichloroacetimidates, anomeric stereocontrol is possible. Thus, the weak base potassium carbonate could be employed for preferential or exclusive formation of the β‑anomer and the strong base sodium hydride could be employed for the thermodynamically more stable α‑anomer (Fig. 3). This stereocontrol could be successfully extended to SN2‑type glycosidation reactions in solvents of low donicity and under low temperature conditions and particularly to glycosylations of O=X–OH nucleophiles with X being RC or R2P (see below). However, for many cases anomeric stereocontrol is based on other effects such as for instance neighboring group participation and/or steric effects, stereoelectronic effects, solvent participation effects, etc.

O‑Glycosyl trichloroacetimidate formation and acid‐catalyzed glycosylation of acceptors HOR′
The experience with the trichloroacetimidate method exhibited that the demands on a new glycosylation methodology are fulfilled:
-
(i)
The O‑glycosyl trichloroacetimidates are readily formed and generally stable under room temperature conditions. However, on acid catalysis they exhibit extraordinary high glycosyl donor properties.
-
(ii)
The release of nonbasic trichloroacetamide fulfills the criteria for acid catalysis: The acid is not consumed by the leaving group, therefore generally only catalytic amounts of (Lewis) acid are required (∼0.001 to 0.1 equivalents).
-
(iii)
The released trichloroacetamide is also not acidic, therefore the acidity provided by the catalyst amount is maintained throughout the reaction course. Hence, negative effects of increasing acidity in the reaction mixture (found for instance for phosphate, sulfate, sulfonate leaving groups, Fig. 1, D) are avoided.
-
(iv)
Glycosidation is basically a condensation reaction. In this procedure, water is bound to trichloroacetonitrile under trichloroacetamide formation. Hence, drying agents are not required. Often molecular sieves are used in glycosidation reactions. Because they may affect the acidity of the reaction in an unpredictable fashion, their use is not even recommended in this method.
-
(v)
Trichloroacetamide can be removed from the reaction mixture and transformed back to trichloroacetonitrile, thus exhibiting the cost‐effectiveness and ecofriendliness of this method in large‐scale preparations.
-
(vi)
Neither in the formation of the O‑glycosyl trichloroacetimidates nor in the glycosidation reactions are equivalent or even higher amounts of salts produced—a disadvantage of most of the above‐mentioned methods, particularly in large‐scale preparations. Also, highly expensive sterically hindered bases are not required.

Reaction of O‑glycosyl trichloroacetimidates with O-, C-, N-, S-, and P‑nucleophiles—activation by (Lewis)acid catalysis
This discussion exhibits that O‑glycosyl N‑methyl‐acetimidates , introduced by the Sinaÿ group [14,15], are poor glycosyl donors: they lack the strongly electron‐withdrawing sterically demanding trichloromethyl group. Hence, they are rather better acylating agents than alkylating agents. In addition, their formation via O‑glycosylation of N‑methyl‐acetamide with excess amounts of silver oxide is quite cumbersome.
These favorable aspects of O‑glycosyl trichloroacetimidates led to their frequent use as glycosyl donors for various types of glycosyl acceptors such as nucleophiles (Fig. 4). Besides hydroxy groups (of standard alcohols, phenols, sugars, etc.), also carboxylic acids, phosphorous acids, and sulfonic and sulfuric acids, respectively, were successfully employed as acceptors; due to the acidity of these acidic acceptors, generally catalysts are not required for the activation of the glycosyl donors and—presumably via an eight‐membered transition state—often the inversion product at the anomeric center is generated. However, also various C-, N-, S-, and P‑nucleophiles have been successfully glycosylated by O‑glycosyl trichloroacetimidates [1,2].
Methodological Aspects
As outlined above, the anomeric hydroxy groups add under base catalysis readily to the electron‐deficient nitrile group of trichloroacetonitrile to furnish generally via equilibration at the anomeric center O‑glycosyl trichloroacetimidates. As bases mainly potassium carbonate, sodium hydride, DBU, and cesium carbonate are employed. Weak bases, such as for instance potassium carbonate, permit kinetic product control (formation of the equatorial product) whereas strong bases, as for instance sodium hydride, lead to the thermodynamically more stable product (generally the axially oriented trichloroacetimidate group). Because of convenience, DBU has become the most popular base, which also favors axial product formation. Therefore, polystyrene‐supported DBU (PS‑DBU) [16,17] and various other solid‐supported nitrogen bases were successfully employed in this reaction as shown for the particularly difficult trichloroacetimidate formation of 4‑dimethylamino‐2,4,6‑trideoxy sugar kedarosamine (1a → 1b) and its use in the glycosylation of the ansamacrolide substructure (1b + 1c → 1d) of the kedarcidin chromophore (Scheme1) [17]. Also Dowex 1‑X8 OH− is a good catalyst for the trichloroacetimidate formation of a glucosamine derivative [18,19]. Polymer‐supported 1,5,7‑triazabicyclo[4.4.0]dec‐5‑ene (PTBD) turned out to be a powerful catalyst for trichloroacetimidate formation as well [17,19]; this base catalyst in combination with Nafion®‐SAC resin (a nanocomposite of Nafion® resin with porous silica) could be successfully employed in one‐pot trichloroacetimidate formation and following glycosylation reactions [19].

Scheme1
O‑Glycosyl trichloroacetimidates are quite stable under neutral but also under basic conditions. Hence, trichloroacetimidate formation is tolerated by standard O - and N‑protecting groups, such as O‑acyl, O‑benzyl, O,O‑alkylidene, O‑silyl, N‑acyl and N,N‑diacyl, N‑phthaloyl (Phth), N‑dimethylmaleoyl (DMM), and the latent amino functionality azido. It is particularly worth mentioning that trichloroacetimidate formation is also tolerated by Fmoc‐protected hydroxy groups; this group has become an important temporary protecting group which is orthogonal to other temporary protecting groups, thus permitting in oligosaccharide synthesis not only regioselective chain extension but also branching [20,21]. The useful 3‑O‑Fmoc‐protected galactosyl donor 2d can be readily prepared from compound 2a via intermediates 2b and 2c (Scheme2) [20]. Additional protecting groups have been recently probed particularly at 2‑O in order to influence the stereochemical outcome, such as for instance the diphenylmethyl (DPM), the 9‑fluorenyl (Fl) group [22], and the 4‑acetoxy‐2,2‑dimethylbutanoyl (ADMB) group [23]. The ADMB group is a useful alternative to the pivaloyl group; it combines the strong 1,2‑trans‐selection in glycosylation reactions of the pivaloyl group with the ease of removal of more reactive acyl groups. Other important new groups are the O‑trifluoroethylsulfonate group which could be successfully employed to release the sulfate group and the replacement of the 6‑hydroxy group by a phenylthio group which is employed for uronate formation [25].

Scheme2

Scheme3
O‑Glycosyl trichloroacetimidates are under acidic conditions very powerful glycosyl donors. Reaction with nonacidic nucleophiles is generally performed with catalytic amounts of Brønsted or Lewis acids . TMSOTf and BF3OEt2 are the most frequently employed catalysts. TMSOTf is the catalyst of first choice and it is generally used in 0.001 to 0.1 equivalents based on the glycosyl donor. Commonly 1.0 to 1.5 equivalents of glycosyl acceptor are added. Dichloromethane is the solvent of first choice. Because of the high reactivity of O‑glycosyl trichloroacetimidates often reaction temperatures between 0 and −40 ℃ are selected. For highly sensitive glycosyl donors and for the anomeric stereocontrol many variations have been probed which are shortly summarized.
Numerous acid catalysts have been investigated. Brønsted acids, such as for instance p‑TsOH and TfOH, can lead—as previously discussed [1,2,3,4,26]—to O‑glycosyl sulfonates as reactive intermediates which are in most cases not the glycosyl donors but rather some type of glycosyl oxocarbenium ion intermediates (SN1‑type reactions). Similarly, pyridinium p‑toluenesulfonate (PPTS) [27,28] and perchloric acid [29,30] are working as catalysts. Metal triflates such as AgOTf [31,32], Cu(OTf)2 [33], Sn(OTf)2 [34,35], Sm(OTf)3 [36], and Yb(OTf)3 [37], are particularly valuable in glycosylations with acid sensitive glycosyl donors and/or acceptors. On Cu(OTf)2‑activation of various donors the trichloroacetimidates have proven to be the most potent glycosyl donors [33]. For instance, AgOTf has proven to be useful, as a catalyst for highly reactive deoxysugars such as glycosyl donors, as shown in Scheme3 for the reaction of fucosyl donor 3a with fucosyl acceptor 3b giving the α‑oligosaccharide 3c [32]. Sn(OTf)2 permitted successful glycosylations of acid sensitive glycal derivatives as acceptors as shown for the synthesis of 4c from 4a and 4b (Scheme4) [35]. Various other catalyst systems have proven to be useful such as for instance HB(C6F5)4 [29,30], N‑acyl‐sulfonamides, phenols [38], I2 [39], I2/Et3SiH [40], and electrophilic carbonyl compounds [41], such as for instance chloral. Silica‐supported perchloric acid [42,43], acid‐washed molecular sieves (MS AW 300) [44], and Nafion®‑SAC resin [19] (see above) are highly valuable solid‐supported acid catalysts. This is nicely demonstrated in an avermectin B1a analog synthesis [43]: Rhamnosylation of intermediate 5b with donor 5a gave target molecule 5c in quantitative yield (Scheme5).

Scheme4

Scheme5
The solvent of choice in the glycosylation reactions is dichloromethane. In combination with a solvent of low polarity and/or of low donicity (such as for instance cyclohexane, petroleum ether, etc.), BF3OEt2 as a mild catalyst and at low temperature, SN2‑type reactions could be carried out [1,2,3,4]. Solvents of high donicity (such as for instance ethers and nitriles) permit—based on the exo‐anomeric effect—a different anomeric stereocontrol: As found and previously explained, in ethers at room temperature generally the axial product (for most cases the α‑product) is favored whereas in nitriles at low temperature, based on the nitrile effect , the equatorial product (for most cases the β‑product) is preferentially or exclusively obtained [3,4,45,46]. Hence, solvent and temperature selection play also an important role in anomeric stereocontrol.
Another interesting solvent effect is associated with the application of the ‘inverse procedure ’ (IP) which consists of the addition of the glycosyl donor to a mixture of acceptor and acid catalyst [47,48]. In this way, particularly with highly reactive glycosyl donors often dramatic glycosylation yield improvements could be obtained [49]. This result seems to be based on a cluster effect between the catalyst and acceptor molecules; hence, on penetration of the donor into this cluster, activation of the donor and following glycoside bond formation takes place within the cluster in a practically intramolecular fashion. Obviously, this effect is critically dependent on the acceptor type, the solvent, and the temperature; therefore it requires often some experimentation for its successful application.
A dramatic polarity increase of the solvent can be reached by adding LiClO4 to organic solvents (for instance, ether, dichloromethane, etc.) which enabled O‑glycosyl trichloroacetimidate activation under essentially neutral conditions [50,51]. High polarity solvents are also ionic liquids (ILs) which have gained a lot of interest as solvents for organic reactions [52]. ILs have been probed in glycosylation reactions with O‑glycosyl trichloroacetimidates and excellent glycosylation yields have been obtained (Scheme6; 6a + 6b → 6c) [53]; for reactive systems again no acid activation is required [53]. Microwave heating has been employed for O‑glycosyl trichloroacetimidate activation as well, furnishing glycosides in high yields [54]; however, presumably due to the temperature effects only modest anomeric stereocontrol was available.

Scheme6
Besides solvent, temperature, and (kinetic and thermodynamic) stereoelectronic effects, anchimeric assistance by neighboring acyl groups and/or by steric shielding is decisive for the anomeric stereocontrol [1,2,3,4,5,6,7,22]. This way 1,2‑trans- (β‑gluco, α‑manno) and 1,2‑cis-type (α‑gluco) glycosides have been generally obtained at wish in good to excellent stereochemical yields. However, a 2‑O‑acyl group does not guarantee β‑glucopyranoside or α‑mannopyranoside formation. Besides undesired ortho‐ester formation, which can often be overcome by using more catalyst or by varying the acyl group, once in a while still the α‑glucopyranoside or even some β‑mannopyranoside, respectively, is obtained. Interesting cases of α‑linkage on attempted 2‑O‑acyl supported β(1–3)‑glucan synthesis were recently reported which were explained by ‘remote control ’ [55,56,57]. 7aβ + 7bβ gave 7cα whereas 7aα + 7bα gave 7cβ (Scheme7). However, this problem could be readily overcome by employing the ADMB group [23]. The influence of the structure of the glycosyl acceptors on the anomeric stereocontrol has often been discussed and since the work of van Boeckel et al. [58,59] the potential importance of matched and mismatched donor‐acceptor pairs on the stereochemical outcome of glycosylation reactions has become evident. Recent work on 2‑azido‐2‑deoxy‐glucopyranosyl trichloroacetimidates exhibited that the structure of the glycosyl donor has generally the major influence [60].

Scheme7
Other anchimerically assisting groups at C‑2, such as–SPh, -SePh, -Br, and -I, have been investigated [61,62]. Particularly worth mentioning is the work on equatorial iodo substitution of glucopyranosyl trichloroacetimidates which strongly favored 1,2‑trans- i. e. β‑glycosidation. This result could be due to generation of an iodonium intermediate or due to the steric demand of the iodo group, thus favoring a twist‐boat type oxocarbenium ion intermediate which—as previously discussed in β‑mannopyranoside synthesis (see below) [26]—for steric and stereoelectronic reasons favors nucleophilic attack from the β‑side [63,64].
An interesting addition to the repertoire of anchimerically assisting groups is the chiral 1‑phenyl‐2‑(phenylsulfanyl)ethyl group [65]. Coupled as (S)‑isomer 8a to the 2‑hydroxy group of the glucopyranosyl trichloroacetimidates with acceptor 8b 1,2‑cis- i. e. α‑glucopyranosides 8c have been obtained in excellent yields and stereoselectivities (Scheme8). The formation of a cyclic β‑linked sulfonium ion intermediate 8a*, having a trans‐decalin type structure has been confirmed by NMR experiments. This intermediate seems to be preferentially or exclusively attacked by the nucleophile from the α‑side.

Scheme8
A particularly difficult problem is 1,2‑cis- i. e. β‑linkage formation in β‑mannopyranoside synthesis . The presence of β‑linked mannopyranosides in various natural products [66], particularly in the N‑glycan core structure of glycoproteins [66,67], led to the search for efficient methodologies for generating this target structure. Investigation of mannopyranosyl donors with nonparticipating protecting groups and different leaving groups led in general mainly or exclusively to α‑products [1,66,67,68]. Several specific methods led to some success in this endeavor [69,70,71,72,73,74], however finally epimerization of β‑glucopyranosides at 2‑O [74,75,76,77,78,79] and intramolecular aglycone delivery [80,81,82,83,84] have led to very successful results. Mannopyranosyl donors with diol O‑protecting groups leading to ring annelation had already been investigated but again with limited success [85]. Therefore, it was a big surprise that 2,3‑di‐O‑alkyl‐4,6‑O‑benzylidene‐protected mannopyranosyl sulfoxides as glycosyl donors gave preferentially β‑products with various acceptors at low temperatures [86,87]. The same result is more conveniently obtained with the corresponding trichloroacetimidates as shown in Scheme9 with glycosyl donor 9a furnishing with acceptor 9b preferentially the β‑disaccharide 9c [26]. The results obtained on varying the reaction parameters were not compatible with the reaction mechanism proposed for sulfoxide activation, in which an α‑triflate intermediate is thought to play the decisive role [87]. Rather the anomeric stereocontrol is caused by a conformational effect enforced by the 4,6‑O‑benzylidene group on the pyranosyl ring, which favors the generation of a flattened twist‐boat conformation as the intermediate. For stereoelectronic and steric reasons, this twist‐boat intermediate will be preferentially attacked from the β‑side, thus forming after equilibration the 4C1‑conformer of the β‑mannopyranoside [26]. This mechanistic proposal is also confirmed by l‑rhamnosylation reactions with bulky 3‑O- and 4‑O‑protecting groups which enforce the 4C1‑conformation and this way favor β‑l‑rhamnoside formation [88]. On the basis of these mechanistic considerations, β‑mannopyranoside formation should be facilitated by nonparticipating, strongly electron‐withdrawing groups at the 2‑O atom because generation of the twist‐boat intermediate would gain from a strong dipole effect. This expectation could be confirmed [89].

Scheme9
Regioselectivity in glycosylation reactions is generally based on a sequence consisting first of regioselective functional group protection, making use of various principles, second glycosylation of the O‑unprotected hydroxy group, and finally of O‑deprotection. Direct regioselective glycosylation using the difference in reactivity of sugar hydroxy groups is of great interest as it often avoids cumbersome protection and deprotection steps. Many regioselective glycosylations of partly unprotected carbohydrate acceptors have been reported in the literature [1,2,3,4,5,6,7]. Generally, they make use of the higher reactivity of primary hydroxy groups over secondary hydroxy groups (particularly those in axial orientation) and the higher reactivity of equatorial 3‑hydroxy groups, for instance in galacto- and glucopyranosides. This was again confirmed in the glycosylation of 4,6‑O‑benzylidene‐glucopyranosides which gave with various glycosyl trichloroacetimidates as donors the (1–3)‐linked disaccharides in very good yields [90]. Another interesting alternative is one‐pot regioselective protection and following O‑glycosylation with O‑glycosyl trichloroacetimidates which, as shown in Scheme10 in the glycosylation of 10a with donors 10b and 10c affording disaccharides 10d and 10e, respectively, makes use of this reactivity difference [91].

Scheme10
Separation of the target glycosides from the reaction mixture generally requires column chromatography purification. Alternatively, highly fluorinated compounds are readily separated from nonfluorinated compounds by a simple phase separation . Therefore, organic synthesis with substrates having fluorinated tags has become a valuable alternative to solid‐phase synthesis (see below). This concept has been already applied to oligosaccharide synthesis [92]. On the basis of the novel fluorous acyl protecting group Bfp (bis fluorous chain type propanoyl) highly successful oligosaccharide syntheses could be performed [93], which were extended to the synthesis of globo‐triaosyl ceramide 11e from lactoside 11a as shown in Scheme11 [94]. All fluorous intermediates (11b and 11d) were extracted with FC‑72 (perfluorohexane isomers) and an organic solvent (toluene or methanol) and were purified without silica gel chromatography. After removal of the Bfp groups by simple base‐catalyzed methanolysis the pure allyl trisaccharide 11e was obtained by only one silica gel column chromatography in 34% total yield after five steps (∼81% per step).

Scheme11
Glycoside Synthesis on Polymer Supports

Scheme12
Successful solid‐phase oligosaccharide syntheses (SPOS) have been developed by several research groups [95,96,97,98,99,100,101,102,103,104,105], which exhibit the inherent advantages over solution phase synthesis, such as (i) higher reaction yields due to the use of excess building blocks, (ii) shorter reaction times for the completion of total syntheses, and (iii) convenient purification procedures by just washing the resin. In addition, methods to avoid undesired byproduct formation in the synthesis of the target molecules have been introduced [106,107,108,109], such as for instance capping procedures of unreacted intermediates (Scheme12) [108]. Low reactive acceptor 12a after glycosylation with donor 12b is capped with benzoyl isoyanate yielding 12c and 12d. Chain extension with 12e (→ 12c + 12f) and cleavage led to readily separable 12g and 12h. So far, no generally accepted strategy has yet appeared for the efficient construction of various complex oligosaccharides on polymer supports, thus limiting the commercialization of automated synthesizers. However, it seems that O‑glycosyl trichloroacetimidates and the less reactive O‑glycosyl phosphates have the potential to become the glycosyl donors of choice because the requirement of catalytic amounts of just one activator is a major advantage over the other glycosylation methods.
After various attempts of the Schmidt group with different linkers between solid support and the carbohydrate groups, such as for instance
-
thioglycosides and their cleavage by thiophilic reagents,
-
silyl glycosides and their cleavage by fluorides,
-
pentenyl‐type glycosides and their cleavage by electrophilic reagents, and
-
allyl‐type glycosides and their cleavage by cross metathesis or by ring‐closing metathesis, respectively [21,110,111],

Solid phase synthesis of high mannose, complex, and hybrid type N‑glycans: increased donor and acceptor reactivity


Solid phase synthesis of a small library of N‑glycans: typical constituents

Scheme15

Scheme16

(continued)
finally the ester‐based SPOS design was introduced which gave excellent results in chain extension and product cleavage from the resin [110,111]. However, to cope with complex oligosaccharide synthesis, besides the ester‐linker and -spacer system, three types of glycosyl donor building blocks for controlled chain extension (suffix e), branching (suffix b), and chain termination (suffix t) are required. An efficient solution to these requirements for SPOS of a small library of N‑glycans is exhibited in Scheme13 which shows the retrosynthesis to the required building blocks and the required reaction steps. A particularly important role played the selection of the N‑protecting group for the glucosamine residue.
The ester‐based SPOS methodology (Scheme13) comprises (i) different types of esters, that is, the benzoate group as a linker and for chain termination and the Fmoc and PA (phenoxyacetyl) group as temporary protecting groups for chain extension and branching which can be chemoselectively cleaved (in the sequence Fmoc and then PA), (ii) the benzyl group for permanent O‑protection and for the spacer between the anomeric center at the reducing end sugar, thus providing after final product cleavage from the resin a structurally defined target molecule, (iii) O‑glycosyl trichloroacetimidates of type e, b, or t (for chain extension, branching, or termination) as powerful glycosyl donors, which can be readily activated by catalytic amounts of (Lewis) acid, and (iv) benzoic acid residues on the Merrifield resin for the linkage of the hydroxymethylbenzyl spacer. Hence, retrosynthesis of a typical N‑glycan molecule containing the core pentasaccharide and some antennae leads to spacer‐linker connected Merrifield resin SLP and to glycosyl donors G e, G t, Gal t, Gal b, M b, M t, and MG b which can be selectively converted into acceptors on resin (e and b‑type donor building blocks). Thus, as indicated in Scheme13, only four simple procedures are required for successful SPOS: (a) glycosidation under TMSOTf catalysis; (b) product cleavage under transesterification conditions; (c) selective Fmoc cleavage under basic conditions; and (d) selective PA cleavage under milder transesterification conditions.
On the basis of this concept, as shown in Scheme14a–c, a small library of N‑glycans (compounds 14a to 14q) was successfully synthesized: (i) The Merrifield resin as solid support exhibited excellent results during all stages of the assembly; (ii) all glycosylations, including those with N‑DMM protected glycosyl donors, gave high yields; (iii) the methodology presented herein shows the desired versatility in terms of efficient chain extension and branching requiring only two standard (in one direction) orthogonal protecting groups; (iv) cleavage of the product from the resin was feasible leading to stable 1‑O‑benzyl type products with only benzyl, DMM and, after acetylation, acetyl protection; (v) the crude products were already of high purity; therefore, standard silica gel chromatography and MPLC were sufficient for purification; (vi) yields of isolated products were high, ranging from 97% per step (after three steps) to 89% per step (after 13 steps) on solid‐phase; (vii) the methodology is technically simple, thus lending itself available to automation.

Scheme17

(continued)

Scheme18
Very good SPOS results have also been obtained by the Seeberger group with O‑glycosyl phosphates and O‑glycosyl trichloroacetimidates, respectively, as glycosyl donors and (Z)‑4‑octene‐1,8‑diol as the linker between the Merrifield resin and the sugar residue. The linker was cleaved by cross‐metathesis with ethylene, thus providing pentenyl glycosides as cleavage products (Schemes 15 and 16) [112]. Scheme15 shows the synthesis of pentenyl cellotrioside 15g with 15a as the donor over seven steps with 15b–f as intermediates in 53% overall yield. As a temporary protecting group in the glycosyl donor the TBS group in the 4‑position was employed. The β‑selectivity was supported by a 2‑O‑pivaloyl group. This method was also employed in an automated SPOS which was based on a modified peptide synthesizer and O‑glycosyl trichloroacetimidates as glycosyl donors (Scheme16) [96,113,114]. This way a glycosylphosphatidyl inositol (GPI) (16n) was synthesized which functions as a malarial toxin. Starting from three different O‑glycosyl trichloroacetimidates (16a, d, f) having O‑acetyl groups as temporary protection for chain extension and one per‑O‑benzylated O‑mannopyranosyl trichloroacetimidate (16j) for chain termination a tetrasaccharide 16k was obtained which was cleaved from the resin (→ 16l) by cross metathesis. Transformation into a trichloroacetimidate 16m and glycosylation of a pseudodisaccharide led after deprotection to the target molecule 16n.
For the real‐time monitoring of the glycosylation result by the Ito group sensitive color tests were introduced with p‑nitrobenzylpyridine (PNBP), reacting with chloroacetyl (CAc) as the temporary protecting group under red color formation, and with cyanuric chloride‐Disperse Red conjugate, which readily reacts with hydroxy or amino groups to give a red color on the resin [115]. This way, as shown in Scheme17, the repeating unit 17e of the immuno‐active oligosaccharide schizophyllan was synthesized based on a partly CAc‐protected O‑glucosyl trichloroacetimidate donor 17f with 17a as starting material and 17b to 17d as intermediates; the presence of the CAc groups could be readily monitored with PNBP and the presence or absence of hydroxy groups was monitored by the Disperse‐Red method. As a linker to piperazine‐modified TentaGel, a 2‑nitrophenyloxyacetyl group [99] was employed that can be readily cleaved under transesterification conditions. This led also to loss of other ester protecting groups furnishing the target molecule 17f in very good overall yield.
The synthesis of oligosaccharides on soluble supports has been further investigated as well. Particularly worth mentioning is the synthesis of heparin‐like oligosaccharides on polyethylene glycol ω‑monomethyl ether (MPEG) resin with a succinoyl ester linker and O‑glycosyl trichloroacetimidates as glycosyl donors [116,117]. A typical example is shown in Scheme18 [117]. After the glycosylation steps of 18a with polymer bound 18b and 18c with 18d, respectively, catalyzed by TMSOTf, a capping step was included in which unreacted MPEG‐bound acceptor was captured onto the solid‐phase of succinate‐functionalized Merrifield resin (PS–Suc‐CO2H) by a chemoselective ester formation (→ 18c and 18f). Alkaline hydrolysis released the octasaccharide from the resin and also all other ester groups were cleaved. By further known transformations [118] the octasaccharide fragment 18g, containing the structural motif of the regular region of heparin, was obtained.
An interesting comparative study between solution phase synthesis and synthesis of the same oligosaccharides on MPEG as soluble support was carried out by the Furneaux group [119]. This study clearly reflected the incompatibility of MPEG as a soluble support with some common reagents, reaction conditions, and choice of standard protecting groups. Hence, the shortcomings associated with this support seem to outbalance the merits [119,120].
To avoid some limitations of PEGs, as for instance low loading, hyperbranched polyester such as Boltorn H40 and H50 have been successfully investigated in glycosylation reactions with O‑glycosyl trichloroacetimidates [121]. For simple mono- and disaccharide synthesis optimization of the reaction conditions led to practically quantitative transformations.
Recent Applications of O-Glycosyl Trichloroacetimidates in Complex Oligosaccharide and Glycoconjugate Synthesis
Several comprehensive reviews have been devoted to the use of O‑glycosyl trichloroacetimidates in glycosylation reactions. In this overview, so far the basic principles of the glycosylation methods, the strengths of the trichloroacetimidate method, differences to other methods, and methodological variations have been discussed. In this chapter recent applications of this method in complex glycoconjugate synthesis will be highlighted.
Glycolipids
Glycosphingolipid synthesis has remained a target of great interest because of the biological importance of these compounds. Assembly of the sugar residues with O‑glycosyl trichloroacetimidates as donors and attachment of the ceramide residue via the azidosphingosine glycosylation procedure [122,123] has become the method of choice. On the basis of this approach, aminodeoxy analogs of globotriosyl ceramide [124], globo- and isoglobotriosides bearing cinnamoylphenyl tags [125], and a quite practical globotriose synthesis [126], which is essentially based on O‑acyl protection, have been carried out. An efficient glycosylation protocol for the attachment of an α‑linked galactosamine residue for asialo GM2 synthesis has been reported [127]. A novel ether‐bridged GM3 lactone analog has been successfully prepared and used in antibody‐based cancer therapy studies [128]. Also ganglioside GD3 was synthesized [129]; comparison with bovine‐brain derived GD3 showed that the effects in GD3‐triggered uncoupling of mitochondrial respiration and induction of apoptosis in oligodendrocytes are very similar. Also ganglioside mimics for binding studies with myelin‐associated glycoprotein were prepared [130].
The lacto- and neolacto‐series of glycosphingolipids also attracted further attention. The synthesis of the lacto‐N‑neotetraose and lacto‐N‑tetraose building blocks has been improved by using the N‑dimethylmaleoyl protecting group [131]. A total synthesis of the natural antigen 19k involved in a hyperacute rejection response to xenotransplants has been carried out as shown in Scheme19 [132]. Glycosyl donor 19a, also obtained via the trichloroacetimidate method, gave with acceptor 19b pentasaccharide 19c. Standard transformation via 19d, 19e, and 19f led to pentasaccharide donor 19g. Application of the azidosphingosine glycosylation procedure with 19h as acceptor furnished 19i which was transformed via 19j into the target molecule 19k.

Scheme19

Scheme20

Scheme21

Reagents and conditions: (a) SAT(N), CMP‐NANA, MOPS (pH 7.4), Triton S4, BSA, H2O, CIP, 55%; (b) FucT(V), GDP‐Fuc, MnCl2, DTT, Tris‐HCl (pH 7.4), qu

Scheme23
Further, for microdomain formation studies fluorescence labeled sialyl Lewis X [133], for cluster effect studies dimeric sialyl Lewis X [134], and for carbohydrate–carbohydrate recognition studies a dimer of Lewis X [135] were synthesized (Scheme20, 20k). To this aim, from building blocks 20a, 20b, and 20f via intermediates 20c, d, e protected Lewis X intermediate 20g was obtained which gave monomer 20h. For the dimerization, via 20i, j, the target molecule 20k was obtained. The synthesis of sialyl Lewis X containing glycolipids with different core structures [136] exhibited the importance of the spacer in selectin‐binding studies [137]. Sulfated sialyl Lewis X variants, containing a lactamized sialyl residue, were obtained by the Kiso‐Ishida group (Scheme21) [138]; these compounds were found to be potent antigenic determinants. Following the standard procedure 21a and 21b gave 21c, which was transformed via 21d–h into lactamized target molecules 21i and 21j. Also chemoenzymatic synthesis was successfully engaged in the synthesis of sulfated sialyl Lewis X connected to a core 1 mucin (T antigen) structure (Scheme22) [139]. From 22a, b disaccharide 22c was obtained, which was transformed into donor 22d giving with acceptor 22e tetrasaccharide 22f. Partial deprotection (→ 22g, h), regioselective sulfation and final deprotection gave 22i; enzymatic sialylation and fucosylation led to target molecules 22j and 22k.
Some α‑galactosyl ceramides with a phytosphingosine residue, isolated from the marine sponge Agelas mauritianus, were found to possess strong in vivo activities against several murine tumor cells [140,141]. The synthesis of analogs exhibited that compound 23e (Scheme23) is a potent candidate for clinical trials [142]; 23e was also found to have immunostimulatory activity [143]. These findings promoted a great demand for this compound, therefore, several syntheses of 23e and analogs have been reported [144]. The Schmidt group reported an efficient synthesis which is based on galactosyl trichloroacetimidate 23a as donor and phytosphingosine derivative 23b as acceptor which led to the α‑linked intermediate 23c. Azide introduction (→ 23d), hydrogenolysis of protecting groups and N‑acylation furnished target molecule 23e very efficiently.
Lipid‐linked T and Tn antigens [145] and glycosidated phosphoglycerolipids [146] were also synthesized based on O‑glycosyl trichloroacetimidates donors. In a comparative study glucopyranosylation of methyl ω‑hydroxy‐hexadecanoate with different glucosyl donors was carried out which demonstrated the advantageous properties of the trichloroacetimidate donor; this way the desired β‑linked target molecule for biological studies was readily obtained [147]. 3,4,5‐Tris(alkyloxy)benzyl glycosides were prepared with standard O‑glycosyl trichloroacetimidates as donors [148]. The three lipid chains permitted the immobilization of these compounds on a hydrophobic surface and lectin affinity studies by surface plasmon resonance.
Glycosyl Amino Acids and Glycopeptides

Scheme24

Scheme25

Scheme26

Scheme27
The availability of O - or N‑glycosyl amino acids as building blocks for glycopeptide or eventually glycoprotein synthesis is of great importance. Therefore, various synthetic methods have been reported [149,150]. Direct O‑glycosylations of serine and threonine derivatives with O‑glycosyl trichloroacetimidates as donors have been reported [151,152]. The straightforward hexafluoroacetone O,N‑protection of Ser, Thr, Pyp, and Tyr gave generally the best results with trichloroacetimidate donors as shown in Scheme24 [153] on glucosylation of 24a, d, g with per‑O‑acetylated glucopyranosyl donors affording 24b, e, h. As this protection also leads to an activated carboxylate group, protecting group cleavage and peptide chain extension can be combined furnishing dipeptides 24c, f, i.
The presence of the Glcα(1–2)Galβ(1‑O)Hyl moiety in collagen was reason to synthesize this building block [154]. Replacement of the ω‑amino group of Hyl by an azido group and protection of the α‑amino group by the Z group and the carboxylate group by a t‐butyl group led cleanly to the target molecule after two glycosylation steps. Glycosylation of Fmoc‐protected Ser, Hse, and Thr with TN, T, and ST‐derived glycosyl donors were also successfully carried out, partly by following known procedures [155,156]. Also an important Galβ(1–4)GlcNAcβ(1–3)‑l‑Fuc moiety was prepared, which is part of O‑linked chains of human clotting factor IX [157].
More demanding is direct glycosylation of peptides, which was so far not very successful because of solubility problems and side reactions with the functional groups [158,159,160]. The Meldal group expected that a solid‐phase approach would suffer less from these drawbacks, therefore, they undertook a study with Ser, Thr, and Tyr containing hexapeptides, having no other functional side chains (!), leading to direct reaction in the order Tyr > Ser > Thr, as shown in Scheme25 [161]. The peptide was linked via a photolabile linker (PLL) to the resin (→ 25a). Galactosylation with 25b (→ 25c), then O‑deprotection (→ 25d), and galactosylation with 25b gave 25e; threonine did not undergo reaction in this case.
A particularly interesting case is the glycosylation of the vancomycin aglycone to achieve the vancomycin total synthesis as successfully investigated by the Nicolaou group [162,163]. They obtained very good results on glycosylation of aglycone 26a with glycosyl donors 26b and 26g affording intermediates 26c and 26g (Scheme26), which were further transformed, thus furnishing via 26d target molecule 26e. Similarly, vancomycin analogs were prepared by the Wong group [164]; biological studies exhibited growth inhibition of vancomycin sensitive bacteria by several of these compounds.
N‑Glycopeptides are generally obtained by treatment of reducing sugars with ammonium bicarbonate [165] or reduction of glycosyl azides [166] with activated aspartic acid. This way a heptasaccharide synthesized with O‑glycosyl trichloroacetimidates as donors was linked to Asp [167,168,169,170]; further work along these lines was carried out [171]. Direct formation of the N‑glycosidic linkage with Asn by chemical glycosylation is still an important task. An interesting method was investigated by Ito et al. [172] based on hydroxyamination of the Asp side chain and then glycosylation with a glycosyl donor. The results show, for standard glycosyl donors direct N- glycosylation is possible, however so far yields and anomeric stereoselection are not yet satisfactory.
A number of C - and N‑linked tryptophan glycoconjugates were discovered as constituents of natural products [173,174,175,176]. Also tryptophan N‑glucoside 27d has been detected for which Unverzagt et al. [177] reported a successful synthesis (Scheme27). With the help of 2‑O‑pivaloyl protection in glucosyl donor 27a the undesired acetal formation could be overcome; thus with acceptor 27b N‑glucoside 27c could be obtained and then transformed into target molecule 27d.
Nucleoside and Nucleotide Glycosidation
The presence of carbohydrate moieties in biomolecules influences many biological functions, thus also modulation of the functions of the aglycone. Therefore, studies have been undertaken to glycosylate nucleosides [178] and nucleotides [179]. Particularly interesting is the direct glycosylation of CPG‐bound oligo‐deoxynucleotides containing pyrimidine residues leading to reaction preferentially at the 5′‑end.
The occurrence at 5‑(β‑d‑glucopyranosyloxymethyl‐2′‑deoxyuridine (βdJ) in DNA, for instance of Trypanosoma brucei [180,181,182], led to the development of an efficient route for the synthesis of βdJ (Scheme28) and its phosphoramidite as building blocks for DNA synthesis [183]. The synthesis was based on glucosyl donor 28a (R = R′ = Bz) which gave with acceptor 28b intermediate 28cβ and after deprotection βdJ. Similarly, from 28a (R = Bn, R′ = Ac) and 28b α‑linked 28cα was obtained which led to αdJ.

Scheme28
Synthesis of Glycosaminoglycans

Scheme29
Glycosaminoglycans (GAGs) are bioactive oligosaccharides that are highly functionalized, linear, and anionically charged. Because of their complexity their chemical synthesis is a demanding task. The chondroitin sulfates (CSs) are a member of the GAG family. They are found in the extracellular matrix of connective tissues at the surface of many cells and in intracellular secretory granules [184,185]. They are linear heteroglycans built mainly from d‑GlcA and GalNAc dimers which are β(1–3)‐linked; chain extension employs a β(1–4)‐linkage. In addition, the sugar residues are sulfated at various positions (→ A- E- and K‑type CSs). Stereocontrolled shark cartilage CS (d‑type) synthesis has been successfully carried out by Jacquinet et al. (Scheme29) [186,187]. The GlcA building block 29a possesses as temporary protecting group for chain extension at 4‑O the chloroacetyl group, and for sulfation at 2‑O the benzoyl group. Similarly, the galactosamine residue 29b carries for chain extension at 1‑O an MP group and for sulfation at O‑6 the TBDMS group; the amino group is protected by the trichloroacetyl group. Glycosylation and further transformations furnish donor 29c which on chain extension with 29d led to the tetrasaccharide 29e (n = 1) and higher oligomers which could be deprotected to yield, for instance, target molecule 29f. On the basis of a similar strategy E‑type CS was obtained which was found to stimulate neuronal outgrowth [188]. A hexameric E‑type CS was also synthesized by Tamura et al. [189]; in this synthetic approach the amino group of the galactosamine residue was replaced by an azido group.
Hyaluronic acids possess a repeating unit consisting of GlcA β(1–3)‐linked to GlcNAc; chain extension of this dimer employs a β(1–4)‐linkage. The dimeric unit has been successfully synthesized based on GlcA trichloroacetimidate as the donor [190]. A trimer consisting of two GlcNAc residues and one GlcA residue was also successfully obtained [191]. In this synthesis design the carboxylate function was introduced at a late stage, after construction of the trisaccharide backbone.

Scheme30
Fibroblast growth factors (FGFs) display high binding affinities for GAGs [185,192,193,194,195] such as heparan sulfate (HS) and heparin . Heparin is a linear, heterogeneously sulfated, anionic polysaccharide composed of alternating l‑iduronic acid and d‑glucosamine residues which is found in almost all animal tissues. The biological roles of several members of the FGFs have been extensively investigated [196,197], which also called for synthetic endeavors; they are still ongoing. The synthesis of a highly sulfated tetrasaccharide has been reported by Lay et al. (Scheme30) [198]. It is based on a versatile α(1–4)‐linked GalN3‑IdoA disaccharide building block from which four differently protected disaccharide acceptors and donors (30a–d) are obtained, thus leading to 30e, f, g. The deprotection scheme is shown for 30g, leading to target molecules 30h, i, j. An interesting study was also reported by Bonnaffé et al. [199]; for successful glycosylation a remote N‑acetyl group had to be replaced by an azido group. Tri-, penta-, hepta-, and nonasaccharides of heparin have been synthesized by Hung et al. (Scheme31) [200]. In this synthetic approach, the carboxylate function is again introduced at a late stage by oxidation of the hydroxymethyl group of the idose moiety. Glycosylation of Ido derivative 31b with donor 31a led via further transformations to disaccharide donor 31c which was chain extended with glucosamine acceptor 31d to yield 31e with n = 1. Further chain extension is based on 31c having naphthylmethyl (NAP) temporary protection; chemoselective NAP cleavage is readily performed with DDQ. Thus via the higher oligomers of 31e after deprotection target molecules 31f (n = 1, 2, 3, 4) are obtained. Heparin‐like oligosaccharide syntheses were also reported by Martin‐Lomas et al. [201], by Seeberger et al. [202], and by Yu et al. [203]. The low reactivity of the axial 4‑hydroxy group in the iduronate residue in the Seeberger approach led to trichloroacetimidate rearrangement because the donor and acceptor reactivity did not match.

Scheme31
Proteoglycans are other important glycoconjugates with various roles [204], such as lubrication and blood anticoagulation. Several proteoglycans possess a highly conserved tetrasaccharide linkage region joining a GAG to a core protein. This GlcAβ(1–3)Galβ(1–3)Galβ(1–4)Xylβ(1‑O)Me tetrasaccharide was also successfully synthesized based on the trichloroacetimidate method [205].
Cell Wall Constituents
Bacterial cell wall peptidoglycan is known as a strong immunopotentiator which induces various mediators such as cytokines, prostaglandins, platelet activation factor, and NO, thus stimulating the immune system [206]. The receptor for peptidoglycans was shown to be TLR2 [207], the same as for lipoteichoic acids (LTAs) and lipoproteins. Therefore, the synthesis of peptidoglycan fragments is of continuing interest. Recently up to an octasaccharide fragment was obtained as shown in Scheme32 [208,209]. Starting material 32a was transformed into donor 32b and acceptor 32c based on the temporary protection at O‑1 with allyl and at O‑4′ with benzylidene. Glycosylation afforded tetrasaccharide 32d which by similar transformation to 32e and 32f led to octasaccharide 32g which could be deprotected to yield target molecule 32h. This procedure of chain extension could be even extended to the hexadecasaccharide, however the Troc deprotection of this compound failed so far. To the octasaccharide 32h also small peptides were attached via the lactate residue and the biological activity of the derived compounds was tested.

Scheme32

Scheme33

(continued)
Many bacterial and fungal cell walls contain homo - and heteroglycans . The synthesis of fragments of these cell wall constituents has gained increasing interest in the last years. The Manα(1–3)Manα(1–2)Manα(1–6)Manα(1–2)Man heptasaccharide obtained by mild acetolysis from C. glabrata IFO 0622 has been successfully synthesized by Kong et al. [210]; they synthesized also a related sulfated pentasaccharide [211]. Also mannan repeating units of Trichophytan mentagrophytes, T. rubrum [212], Saccharomyces cerevisiae X 2180–1A [213], and Candida kefyr IFO 0586 [214] have been prepared. Another important class of glycans are β(1–3)- and β(1–6)‐linked glucans which are frequently branched. Glucans consisting of β(1–3)‐linked glucose residues having β(1–6)‐linked branches have been found in many plants and fungi [215,216]. Since these glucans show antitumor activity, the synthesis of at least minimal structural units for biological activity studies has gained great interest [217]. Recently, several successful syntheses of such molecules have been reported mainly by Kong et al. [218,219,220,221,222,223]. Also a 2‑branched β(1–3)‐glucan was found and dimers of the trisaccharide repeating unit were prepared [224]. 3‑Branched β(1–6)‐linked glucans were found to be phytoalexin elicitors; they possess antitumor activities as well. A highly efficient synthesis of tetradecasaccharide 33t was reported again by the Kong group [225] (Scheme33). The synthesis is based on readily available glucose‐derived building blocks 33a, 33b, and 33e and leads via disaccharides 33c and 33d, trisaccharides 33f–33i, and 33n, hexasaccharides 33j, k, o, p, heptasaccharides 33l, m, q, r to tetradecasaccharide 33s which was deprotected to furnish target molecule 33t in good overall yield. Such compounds were also prepared in large scale and with different aglycones [226,227,228].
Mannoglucan from Microellabosperia grisea possesses also high antitumor activity, therefore the repeating unit {[Glcα(1–3)][Glcα(1–6)]Glcβ(1–4)Glcβ(1–4)}n has been synthesized via an efficient route [229]. β(1–6)‐Linked galactofuranosyl oligosaccharides are constituents of the cell wall of bacteria and fungi including some clinically significant pathogens [230,231,232]. The highly immunogenic arabinogalactans contain arabinofuranosyl and galactofuranosyl residues. β(1–6)‐Linked galactofuranose oligomers, also found in the cell wall of the fungus Fusarium, exhibited in plants elicitor activity. Therefore, a β(1–6)‐linked O‑galactofuranosyl hexasaccharide was prepared based on the trichloroacetimidate method in high yield [233,234]. The Kong group also synthesized α(1–5)‑linked l‑arabinofuranosyl oligosaccharides up to the octamer based on related methodologies [235]. 2‑O‑Arabinofuranosylated β(1–6)‐linked galactopyranosyl oligosaccharides, found in arabiogalactans, could be obtained also by Kong et al. [236] who also reported the successful synthesis of other arabinogalactan linkage types based on the trichloroacetimidate method [237,238,239,240]. Lipoarabinomannans have attracted great interest as well, because they are part of the cell surface oligosaccharide of mycobacterial species that cause many diseases, including tuberculosis and leprosy. The largest heteroglycan synthesis for this type of compound was recently reported in which the trichloroacetimidate method plays an important role [241].

Scheme34
In algal cell wall polysaccharides β(1–3)‐linked xylans were found. Fragments up to the hexamer were successfully synthesized [242]. The specific O‑chain of the lipopolysaccharide of many bacteria contains repeating units composed of various sugar residues. Several of these repeating units or fragments thereof have been synthesized based on the trichloroacetimidate method. Rhamnosylated rhamnan [243], the d‑Rhapα(1–3)[l‑Xylpβ(1–2)]d‑Rhapα(1–3)[l‑Xylpβ(1–2)]d‑Rha unit [244], (Scheme34, 34i), a more complex xylorhamnan [245], a xylosylated GlcNAc rhamnan [246], a rhamnan with GlcNAc in the branch [247,248], the pentasaccharide d‑Glcβ(1–2)‐[d‑Ribfβ(1–3)]l‑Rhapα(1–3)l‑Rhapα(1–3)l‑Rhapα(1–2)l‑Rhap [249], the glucurono xylomannan hexasaccharide repeating unit of C. neoformans serotype A (Scheme35) [250], a heptasaccharide fragment of C. neoformans serotype C [251], a repeating unit of lactosillan [252], the Shigella flexneri serotype 2a [253,254,255,256,257], and serotype 5a [258] have been efficiently obtained (Scheme36). The straight forward synthesis of heptasaccharide 34i is outlined in Scheme34 [244]. Rhamnosyl donor 34a readily reacts with rhamnose acceptor 34b to give mainly disaccharide 34c which is transformed into donor 34d. Chain extension with 34b leads to 34e and then to acceptor 34f which on reaction with xylosyl donor 34g furnishes pentasaccharide 34h; deprotection leads to target molecule 34i. The hexasaccharide repeating unit synthesis of C. neoformans type A is outlined in Scheme35 [29]. Mannosyl donor 35a reacts with acceptor 35b to afford trisaccharide 35c leading to donor 35d which on reaction with 35b gives pentasaccharide 35e and acceptor 35f. Glucuronidation with donor 35g furnished hexasaccharide 35h which gave on deprotection target molecule 35i. The Shigella flexneri serotype 2a pentasaccharide fragment is outlined in Scheme36 [36]. Rhamnosyl donor 36b and acceptor 36a yield disaccharide 36c which is transformed via 36d into donor 36e. On reaction with acceptor 36f tetrasaccharide 36g is obtained which is again transformed via 36h into donor 36i. Reaction with acceptor 36j pentasaccharide 36k is obtained which led to target molecule 36m having an aminoethyl group at O‑1 for glycoconjugate synthesis. Linkage to the PADRE sequence, acting as a universal T‑cell epitope, was successfully performed and immunogenicity studies were carried out. Also successful investigations towards the synthesis of a tetrasaccharide rhamnogalacturonan related to an antiulcer pectic polysaccharide have been reported [259]. The Neisseria lipooligosaccharide contains two heptose residues within the conserved core structure; one heptose residue is 3,4‑branched. On the basis of the trichloroacetimidate method a successful synthesis of a branched tetrasaccharide unit was successfully performed by Yamasaki et al. [260,261].

Scheme35

Scheme36
Synthesis of Glycosylphosphatidyl Inositol Anchors
Glycosylphosphatidyl inositol anchors constitute a class of glycolipids that link proteins and glycoproteins via their C‑terminus to eukaryotic cell membranes. The first structure of a GPI anchor, that of Trypanosoma brucei, was published by Ferguson et al. [262]. Since then quite a few examples of GPI anchors were described, allowing the definition of the core structure depicted in Scheme37 [263].
The diversity within GPI anchors is mainly reflected in the location and nature of the branching groups of the glycan residue (R2, R3, R4). Additional ethanolamine phosphates (R1) seem to be specific for higher eukaryotes [264]. Concerning the lipid residue, many of the structures of GPI anchors contain a diacylglycerol moiety but alkylacylglycerol residues are not uncommon and ceramide structures have also been identified [263]. These modifications of the evolutionary conserved structure give rise to species-, stage-, and tissue‐specific GPI structures.
The function of GPI anchors has been extensively discussed. A controversial aspect of GPI anchors is their ability to mediate signaling mechanisms or to function as second messengers, e. g. in insulin‐mediated signal transduction processes [265]. Therefore, to perform biological studies elucidating the functions of GPI anchors, it seems to be an important objective to have access to structurally homogeneous GPI anchors and their derivatives. For the total synthesis of GPI anchors, a combination of lipid, phosphate, and oligosaccharide chemistry is required. A highly versatile strategy has been successfully followed for a ceramide‐containing GPI anchor of yeast [266,267]. Similarly obtained were the acylglycerol‐containing GPI anchors of Trypanosoma brucei [268,269] and rat brain Thy‑1 [270].

General structure of GPI anchors (EA, ethanolamine; P, phosphate; DAG, diacylglycerol)

Scheme38

Scheme39

(continued)

Scheme40
On the basis of earlier work [266,267], the development of a highly variable synthetic strategy, which is also applicable to the preparation of branched GPI anchors, was reported by Schmidt et al. [271,272]. This strategy allows also for the attachment of peptide or protein residues to the GPI anchor. The focus was on 4,6‑branched mannose residues as there are several prominent examples in nature (Scheme37). Therefore, the fully phosphorylated GPI anchor of Toxoplasma gondii was prepared [271]. Also a new route to rat‐brain Thy‑1 [272] was developed which is outlined in Scheme38, showing important disconnections and building blocks; it is totally based on trichloroacetimidate donors. Two efficient routes starting from mannosyl acceptor 39a and glucosamine‐derived donor 39e leading to disaccharide 39g were developed (Scheme39). With variously protected mannosyl donors 39j and 39l, chain extension to pentasaccharide 39o could be efficiently performed. Transformation into donor 39n and reaction with disaccharide acceptor 39v led to pseudoheptasaccharide 39w. Introduction of two orthogonally protected aminoethyl phosphate residues led to 39aa to which the phospholipid was attached (→ 39ab). Debenzylation afforded N‑Boc‐protected 39ac which permits regioselective attachment of peptide residues to one aminoethylphosphate moiety and complete deprotection led to target molecule 39ad. This way a highly variable concept for the synthesis of branched GPI anchors could be established. It is based on versatile building blocks which are readily accessible and provide the desired regio- and stereocontrol. The concept further allows for the regioselective attachment of peptides or proteins.
The synthesis of a partial structure of a branched pseudohexasaccharide of an inositolphosphoglycan (IPG) has been reported by Martin‐Lomas et al. [273]. Linear oligosaccharides of GPIs were also synthesized based on the trichloroacetimidate method [274,275]. The GPI anchor of Trypanosoma cruzi containing an unsaturated fatty acid was obtained without any O‑benzyl protection in the decisive intermediate (Scheme40) [276]. Acyl‐protected donor 40a and acyl‐protected acceptor 40b gave disaccharide 40c which was transformed into acceptor 40d which on reaction with donor 40e gave trisaccharide 40f and then acceptor 40g. Glycosylation with 40h led to tetrasaccharide 40i which was transformed via 40j into donor 40k. From this compound the desired target molecule was prepared.
Glycosylation of Various Natural Products and Their Metabolites
Many antibiotics are glycosylated, however the structural complexity of many antibiotics and their glycosylation is a demanding task. On the basis of the trichloroacetimidate method a successful synthesis of landamycin A hexasaccharide was reported [277]. Novobiocin was glycosylated with three different O‑glycosyl trichloroacetimidates as donors at the 7‑hydroxy group [278]. Also the quite sensitive (+)‑Neo‐carzinostatin aglycone 41a (Scheme41) could be successfully glycosylated [279]. Reaction of 41a with glycosyl donor 41b furnished the desired glycoside 41c in good yield which on deprotection afforded target molecule 41d. Hydroquinone glycosylation leading to α‑arbutin was also performed [280]; this compound is an inhibitor of human tyrosinase and therefore of interest in the cosmetic industry.

Scheme41
Aminoglycoside antibiotics are of special interest as glycosylation targets. Interesting neomycin trisaccharide mimetics have been prepared by Boons et al. [281]. Glycosidase inhibitors have also been combined with additional carbohydrate residues in order to fine tune their biological properties. Thus, deoxynojirimycin has been combined with β(1–3)- and β(1–6)‐linked glucan residues [282].
Many metabolites are glucuronidated , therefore the introduction of the glucuronic acid residue is a major task because of the low reactivity of the derived donors and their tendency to 4,5‑elimination. Therefore, often compounds are first glycosidated and at a later stage of the synthesis the hydroxymethyl group is selectively oxidized to the carboxylate group. Excellent results in direct glucuronidation based on O‑isobutyryl‐protected trichloroacetimidate have been obtained by Scheinmann and Stachulski et al., who glucuronidated for instance an anti‑estrogenic steroid [283,284] and morphine [285]. Also MS 209, a quinoline derivative exhibiting multidrug resistance in cancer therapy was directly glucuronidated based on an O‑pivaloyl‐protected donor [286]. 5‑Hydroxypyridine derivative 42b (Scheme42) which is part of ABT‐724, a potent D4 dopamine receptor agonist, could be directly glucuronidated [287] leading to 42c. Further transformations via intermediates 42e and 42f gave the desired glycosylated metabolite 42g.

Scheme42
Dihydroxyphenyl glycosides are a family of plant components . Some representatives have also been synthesized based on the trichloroacetimidate method, such as for instance the conandroside [288]. Other plant metabolites, such as for instance the ellagitannins, contain benzoyl groups at the anomeric oxygen. The derived coriariin A (43d, Scheme43) was obtained as discussed above just by heating 43b with glycosyl donor 43a, leading to 43c, which was transformed into target molecule 43d [289]. Macrophylloside D [290] and buprestin A [291] were similarly prepared.

Scheme43

Scheme44
N‑Glycosylation of nitrogen‐containing heterocycles has also been investigated. An interesting synthesis of N‑indigoglycosides was undertaken which, as analogs of akashines, exhibit activity against various human tumor cell lines [292].
Unique glycolipids produced by plants are the resin glycosides for which the first synthesis was based on the trichloroacetimidate method [293,294]. Other members of this family have been prepared with the help of closely related trichloroacetimidate building blocks and a similar strategy, such as for instance for woodrosin I [295] and tricolorin F [296]. Structurally related, highly bioactive antiviral compounds, such as for instance cycloviracin B1 or glucolipsin A were isolated and successfully synthesized [297,298,299]. The approach to the synthesis of glucolipsin A [299] is exhibited in Scheme44. Glucosylation of fatty acid derivative 44a with O‑glycosyl trichloroacetimidate 44b gave the desired glycoside 44c in good yield. Ester hydrolysis (→ 44d) and dimerization to 44e and then hydrogenolysis of the O‑benzyl groups led to target molecule 44f which had physical data in accordance with the natural product (see [299]).
Saponins are steroid- or triterpenoid‐based glycolipids which are found in terrestrial and marine plants. They possess various biological activities [300]. Several papers reported on successful glycosylations at the 3‑hydroxy group [301,302,303,304,305,306]. When this hydroxy group is sterically hindered due to disubstitution at C‑4, this glycosylation is a difficult task. However, with O‑glycosyl trichloroacetimidate donors good yields and anomeric selectivities have also been obtained for this glycosylation step [307,308,309,310,311,312]. Particularly worth mentioning is the total synthesis of QS 21 A published by Gin et al. [25] where the decisive step is carried out with a trisaccharide donor.
Most carbohydrates contain ethane‐1,2‑diol fragments, therefore they were also successfully included into crown ether macrocycles [313]. Polyvalency of carbohydrates was studied based on dendrimeric structures which were generated on successful glycosylations of highly branched polyethylene glycol units [314].
Cyclooligosaccharides
Cyclodextrins (CDs) have gained a lot of interest because they provide useful cavities for the generation of inclusion complexes. Therefore, branched CDs and CDs with various carbohydrate residues have been synthetic targets. The trichloroacetimidate method was successfully employed to reach such goals. Mannosyl, galactosyl, lactosyl, and the Galβ(1–4′)‑lactosyl residues were selectively introduced at O‑6 of β- and γ‑CD, respectively [315,316]. Also building blocks for oligomannoside attachment to CDs were prepared [317]. Most remarkable is the synthesis of a O‑6 branched cyclo(1–3)‐glucohexaose and octaose [318,319]. The ring closure of the prepared nona- and dodecasaccharide worked extremely well based on O‑glycosyl trichloroacetimidate donors (Scheme45) [5]. Dodecasaccharide 45a was transformed via 45b into trichloroacetimidate 45c having a 3‑hydroxy group at the nonreducing end. Acid‐catalyzed activation led to CD derivative 45d which gave after deacylation target molecule 45e.

Scheme45
C-Glycoside Synthesis
The synthesis of “ C‑glycosides ” has been of great interest [320]. C‑Glycosylation of electron‐rich compounds with O‑glycosyl trichloroacetimidates continues to be a method of choice. This work is not discussed here in detail. A few references give an entry to the work in this field [321,322,323,324].
O-Glycosyl Trichloroacetimidates of N,O- and S,O-Halfacetals
Not only O,O‑halfacetals but also N,O -, and S,O‑halfacetals have successfully been transformed into trichloroacetimidate derivatives [325,326]. Thus carbohydrates with sulfur in the ring have been successfully transformed into O‑glycosyl trichloroacetimidates and employed in glycosylation reactions by M. Hashimoto et al. [327,328] and by H. Hashimoto et al. [329,330]. Thy glycosyl donors could be readily obtained by base‐catalyzed trichloroacetonitrile addition to the anomeric hydroxy group and glycosylation yields under standard conditions were generally very good.
Related Activation Systems
O‑Glycosyl trichloroacetimidates have become popular because they are easily available, powerful glycosyl donors. Therefore, investigations were undertaken to provide glycosyl donors which follow the principle of O‑glycosyl trichloroacetimidate formation and activation. A short outline of this work is attached here.
Already previously trifluoroacetonitrile has been investigated for the generation of glycosyl donors [12,331]. However, because trifluoroacetonitrile is a gas, its use is not as convenient as the use of trichloroacetonitrile. In addition, preliminary glycosylation results were inferior to those obtained with O‑glycosyl trichloroacetimidates. Much more promising were studies with dichloromalonitrile which is a suitable reagent for the base‐catalyzed generation of O‑glycosyl dichloro‐cyanoacetimidates [332,333]. These compounds exhibited glycosyl donor properties closely related to those of O‑glycosyl trichloroacetimidates (Scheme46). A further important class of compounds is ketene imines , which should readily lead to O‑glycosyl imidates. However, so far only a few examples were investigated [12,13,330], therefore, the potential of these compounds has not been elucidated yet.

Scheme46
Another interesting class of compounds are imide halides having electron‐withdrawing carbon substituents and their heterocyclic equivalents. After some earlier work [12,334,335,336,337,338], recently imide halides have gained increased interest and excellent glycosylation results have been reported [45,339,340]. However, these systems have the disadvantage of furnishing equimolar amounts of salt on glycosyl donor generation. Additionally, glycosyl donor generation is not reversible, therefore α/β‑stereocontrol is more difficult or even impossible. Hence, matching or even surpassing the properties of O‑glycosyl trichloroacetimidates remains a demanding task.
Conclusions
The requirement for efficient glycosylation methods, as outlined at the beginning of this chapter, namely convenient diastereocontrolled anomeric O‑activation (first step) and subsequent efficient diastereocontrolled glycosylation promoted by catalytic amounts of a promoter (second step) are essentially completely fulfilled by the trichloroacetimidate method. In terms of reactivity and applicability toward different acceptors, the O‑glycosyl trichloroacetimidates have generally proven to be outstanding glycosyl donors, which resemble in various respects the nucleoside diphosphate sugar derivatives used by nature as glycosyl donors. Thus, base‐catalyzed generation of O‑glycosyl trichloroacetimidates followed by acid‐catalyzed glycosylation have become a very competitive alternative to other methods mainly requiring anomeric oxygen exchange reactions for glycosyl donor generation and at least equimolar amounts of a promoter system for the glycosylation step. Hence, the trichloroacetimidate method can readily be adapted for large‐scale preparations. Recently, related activation systems have attracted increasing interest, thus widening the scope of this glycosylation method.
Abbreviations
- ADMB:
-
4‑acetoxy-2,2‑dimethylbutanoyl
- CAc:
-
chloroacetyl
- CDs:
-
cyclodextrins
- CSs:
-
chondroitin sulfates
- DBU:
-
1,8‑diazabicyclo[5.4.0]undec-7-ene
- DDQ:
-
2,3-dichloro-5,6-dicyno-benzoquinone
- DMM:
-
N‑dimethylmaleoyl
- DPM:
-
diphenylmethyl
- FGFs:
-
fibroblast growth factors
- Fmoc:
-
9‑fluorenylmethyl carbamate
- GAGs:
-
glycosaminoglycans
- GPI:
-
glycosylphosphatidyl inositol
- HS:
-
heparan sulfate
- ILs:
-
ionic liquids
- IP:
-
inverse procedure
- IPG:
-
inositolphosphoglycan
- LTAs:
-
lipoteichoic acids
- MPEG:
-
mono-methyl polyethyleneglycol
- MPLC:
-
medium pressure liquid chromatography
- NAP:
-
naphthylmethyl
- PLL:
-
photolabile linker
- PNBP:
-
p-nitrobenzylpyridine
- PPTS:
-
pyridinium p-toluenesulfonate
- PTBD:
-
polymer‐supported 1,5,7‑triazabicyclo[4.4.0]dec-5-ene
- PS-DBU:
-
polystyrene‐supported DBU
- SPOS:
-
solid-phase oligosaccharide syntheses
- TBS:
-
tert-butyl-dimethylsilyl
- TMSOTf:
-
trimethylsilyl trifluoromethanesulfonate
References
Schmidt RR (1986) Angew Chem Int Ed Engl 25:89
Schmidt RR, Kinzy W (1994) Adv Carbohydr Chem Biochem 50:21
Schmidt RR, Jung KH (1997) Oligosaccharide Synthesis via Trichloroacetimidates. In: Hannessian S (ed) Preparative Carbohydrate Chemistry. Marcel Dekker, p 283
Schmidt RR, Jung KH (2000) Trichloroacetimidates. In: Ernst B, Hart GW, Sinaÿ P (eds) Carbohydrates in Chemistry and Biology, Part I: Chemistry of Saccharides, vol 1. Wiley‑VCH, Weinheim, p 5
Paulsen H (1982) Angew Chem Int Ed Engl 21:155
Kunz H (1987) Angew Chem Int Ed Engl 26:294; (1993) Pure Appl Chem 65:1223
Toshima K, Tatsuta K (1993) Chem Rev 93:1503
Schmidt RR, Reichrath M (1979) Angew Chem Int Ed Engl 18:466
Klotz W, Schmidt RR (1994) J Carbohydr Chem 13:1093
Schmidt RR, Klotz W (1991) Synlett 168
Klotz W, Schmidt RR (1993) Liebigs Ann Chem 683
Michel J (1983) PhD thesis, Universität Konstanz
Schmidt RR, Michel J (1980) Angew Chem Int Ed Engl 19:731
Pougny JR, Sinaÿ P (1976) Tetrahedron Lett 4073
Sinaÿ P (1978) Pure Appl Chem 50:1437
Ohashi I, Lear MJ, Yoshimura F, Dirama M (2004) Org Lett 6:719
Chiara JL, Encinas L, Díaz B (2005) Tetrahedron Lett 46:2445
Yoshizaki H, Fukuda N, Sato K, Oikawa M, Fukase K, Suda Y, Kusumoto S (2001) Angew Chem Int Ed 40:1475
Oikawa M, Tanaka T, Fukuda N, Kusumoto S (2004) Tetrahedron Lett 45:4039
Roussel F, Knerr L, Grathwohl M, Schmidt RR (2000) Org Lett 2:3043
Knerr L, Schmidt RR (2001) The Use of O‑Glycosyl Trichloroacetimidates for the Polymer Supported Synthesis of Oligosaccharides. In: Seeberger PH (ed) Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries. Wiley, New York, pp 67
Ali IAI, El‐Ashry ESH, Schmidt RR (2003) Eur J Org Chem 4121
Yu H, Williams DL, Ensley HE (2005) Tetrahedron Lett 46:3417
Karst NA, Islam TF, Avci FY, Linhardt RJ (2004) Tetrahedron Lett 45:6433
Yu B, Zhu X, Hui Y (2001) Tetrahedron 57:9403
Weingart R, Schmidt RR (2000) Tetrahedron Lett 41:8753
Nicolaou KC, Daines RA, Ogawa Y, Chakraborty TK (1987) J Am Chem Soc 109:2821
Nicolaou KC, Daines RA, Ogawa Y, Chakraborty TK (1988) J Am Chem Soc 110:4696
Jona H, Mandai H, Chavasiri W, Takeuchi K, Mukaiyama T (2002) Bull Chem Soc Jpn 75:291
Jona H, Mandai H, Mukaiyama T (2001) Chem Lett 426
Douglas SP, Whitfield DM, Krepinsky JJ (1993) J Carbohydr Chem 12:131
Wei G, Gu G, Du Y (2003) J Carbohydr Chem 22:385
Yamada H, Hayashi T (2002) Carbohydr Res 337:581
Castro‐Palomino JC, Schmidt RR (1995) Tetrahedron Lett 36:5343
Geiger J, Barroca N, Schmidt RR (2004) Synlett 836
Adinolfi M, Barone G, Guariniello L, Iadonisi A (2000) Tetrahedron Lett 41:9005
Adinolfi M, Barone G, Iadonisi A, Mangoni L, Schiattarella M (2001) Tetrahedron Lett 42:5967
Griswold KS, Horstmann TE, Miller SJ (2003) Synlett 1923
Kartha KPR, Karkkainen TS, March SJ, Field RA (2001) Synlett 260
Adinolfi M, Barone G, Iadonisi A, Mangoni L, Schiattarella M (2002) Synlett 269
Schmidt RR, Gaden H, Jatzke H (1990) Tetrahedron Lett 31:327
Mukhopadyay B, Maurer SV, Rudolph N, van Well RM, Russell DA, Field RA (2005) J Org Chem 70:9059
Du Y, Wei G, Cheng S, Hua Y, Linhardt RJ (2006) Tetrahedron Lett 47:307
Adinolfi M, Barone G, Iadonisi A, Mangoni L, Schiattarella M (2003) Org Lett 5:987
Schmidt RR, Rücker E (1980) Tetrahedron Lett 21:1421
Schmidt RR, Behrendt M, Toepfer A (1990) Synlett 694
Schmidt RR (1992) New Aspects of Glycosylation Reactions. In: Ogura H, Hasegawa A, Suami T (eds) Carbohydrates—Synthetic Methods and Applications in Medicinal Chemistry. Kodanasha Ltd., Tokyo, 68
Toepfer A, Schmidt RR (1991) Tetrahedron Lett 32:3353
Bommer R, Kinzy W, Schmidt RR (1991) Liebigs Ann Chem 425
Böhm G, Waldmann H (1995) Tetrahedron Lett 36:3843
Schmid U, Waldmann H (1997) Liebigs Ann Chem 2573
Jain N, Kumar A, Chauhan S, Chauhan SMS (2005) Tetrahedron 61:1015
Rencurosi A, Lay L, Russo G, Caneva E, Poletti L (2005) J Org Chem 70:7765
Larsen K, Worm‐Leonhard k, Olsen P, Hoel A, Jensen KJ (2005) Org Biomol Chem 3:3966
Zeng Y, Ning J, Kong F (2002) Tetrahedron Lett 43:3729
Yang F, He H, Du Y, Lü M (2002) Carbohydr Res 337:1165
Zeng Y, Ning J, Kong F (2003) Carbohydr Res 338:307
Spijker NM, van Boeckel CAA (1991) Angew Chem Int Ed Engl 30:180
Masamuni S, Choy W, Petersen JC, Sita LR (1985) Angew Chem Int Ed Engl 24:1
Cid MB, Alfonso F, Martín‐Lomas M (2005) Chem Eur J 11:928
Castro‐Palomino JC, Schmidt RR (1998) Synlett 501
Marzabadi CH, Franck RW (2000) Tetrahedron 56:8385
Roush WR, Gung BW, Bennett CE (1999) Org Lett 1:891
Chong PY, Roush WR (2002) Org Lett 4:4523
Kim JH, Yang H, Park J, Boons GJ (2005) J Am Chem Soc 127:12090
Gridley JJ, Osborn HMI (200) J Chem Soc Perkin Trans 1:1471
Dwek RA (1996) Chem Rev 96:683
Barresi F, Hindsgaul O (1996) In: Khan SH, O'Neill RA (eds) Modern Methods in Carbohydrate Synthesis. Harwood Academic Publishers, Amsterdam, p 251
Paulsen H, Lockhoff O (1981) Chem Ber 114:3102
Garegg PJ, Ossowski P (1983) Acta Chem Scand 337:249
Danishefsky SJ, Hu S, Cirilo PF, Eckhardt M, Seeberger PH (1997) Chem Eur J 3:1617
Lichtenthaler FW, Schneider‐Adams T (1994) J Org Chem 59:6728
Schmidt RR, Moering U, Reichrath M (1982) Chem Ber 115:39
Hodosi G, Kovác P (1997) J Am Chem Soc 119:2335
Ekborg G, Lindberg B, Lonngren J (1972) Acta Chem Scand B 26:3287
Matsuo I, Isomura M, Walton R, Ajisaka K (1996) Tetrahedron Lett 37:8795
Kunz H, Günther W (1988) Angew Chem Int Ed Engl 27:1086
Weiler S, Schmidt RR (1998) Tetrahedron Lett 39:2299
Twaddle GWJ, Yashunsky DV, Nikolaev AV (2003) Org Biomol Chem 1:623
Barresi F, Hindsgaul O (1991) J Am Chem Soc 113:9376
Stork G, LaChair JJ (1996) J Am Chem Soc 118:247
Ito Y, Ogawa T (1994) Angew Chem Int Ed Engl 33:1765
Jung KH, Müller M, Schmidt RR (2000) Chem Rev 100:4423
Ziegler T, Lemanski G, Rakoczy A (1995) Tetrahedron Lett 36:8973
Caregg PJ, Iversen T, Johansson R (1980) Acta Chem Scand B 34:505
Crich D, Sun S (1996) J Org Chem 61:4506
Crich D, Smith M (2001) J Am Chem Soc 123:9015
Ikeda T, Yamada H (2000) Carbohydr Res 329:889
Abdel‐Rahman AAH, Jonke S, El Ashry ESH, Schmidt RR (2002) Angew Chem Int Ed 41:2972
Zhou FY, Huang JY, Yuan Q, Wang YG (2005) Chem Lett 34:878
Wang CC, Lee JC, Luo SY, Fan HF, Pai CL, Yang WC Lu LD Hung SC (2002) Angew Chem Int Ed 41:2360
Curran DP, Ferritto R, Hua Y (1998) Tetrahedron Lett 39:4937
Miura T, Hirose Y, Ohmae M, Inazu T (2001) Org Lett 3:3947
Miura T, Inazu T (2003) Tetrahedron Lett 44:1819
Haase WC, Seeberger PH (2000) Chem Rev 100:4349
Plante OJ, Palmacci ER, Seeberger PH (2001) Science 291:1523
Roussel F, Takhi M, Schmidt RR (2001) J Org Chem 66:8540
Roussel F, Knerr L, Schmidt RR (2001) Eur J Org Chem 2066
Wu X, Grathwohl M, Schmidt RR (2001) Org Lett 3:747
Rademann J, Geyer A, Schmidt RR (1998) Angew Chem Int Ed 37:1241
Rademann J, Schmidt RR (1997) J Org Chem 62:3650
Zhu T, Boons GJ (2001) Chem Eur J 7:2382
Zhu T, Boons GJ (2000) J Am Chem Soc 122:10222
Nicolaou KC, Watanabe N, Li J, Pastor J, Winssinger N (1998) Angew Chem Int Ed 37:1559
Mogemark M, Elofsson M, Kihlberg J (2003) J Org Chem 68:7281
Ando H, Manabe S, Nakahara Y, Ito Y (2001) Angew Chem Int Ed 40:4725
Pamacci ER, Hewitt MC, Seeberger PH (2001) Angew Chem Int Ed 40:4433
Wu X, Schmidt RR (2004) J Org Chem 69:1853
Bauer J, Rademann (2005) J Am Chem Soc 127:7296
Wu X, Grathwohl M, Schmidt RR (2002) Angew Chem Int Ed 41:4489
Jonke S, Liu K, Schmidt RR (2006) Chem Eur J 12:1274
Andrade RB, Plante OJ, Melean LG, Seeberger PH (1999) Org Lett 1:1811
Ratner DM, Swanson ER, Seeberger PH (2003) Org Lett 5:4717
Hewitt MC, Snyder DA, Seeberger PH (2002) J Am Chem Soc 124:13434
Manabe S, Ito Y (2002) J Am Chem Soc 124:12638
Ojeda R, de Paz JL, Martín‐Lomas M (2003) Chem Commun 2486
Ojeda R, Terentí O, de Paz JL, Martín‐Lomas M (2004) Glycoconjugate J 21:179
de Paz JL, Angulo J, Lassaletta JM, Nieto PM, Ridondo‐Horcajo M, Lozano RM, Gimenez‐Gallego G, Martín‐Lomas M (2001) ChemBioChem 2:673
Blattner R, Furneaux RH, Ludewig M (2006) Carbohydr Res 341:299
Knust B (1996) Diploma thesis, Universität Konstanz
Kantchev EAB, Parquette JR (2005) Synlett 1567
Schmidt RR, Zimmermann P (1986) Angew Chem Int Ed Engl 25:725
Zimmermann P, Schmidt RR (1988) Liebigs Ann Chem 663
Hansen HC, Magnusson G (1999) Carbohydr Res 322:190
Aly MRE, Rochaix P, Amessou M, Johannes L, Florent JC (2006) Carbohydr Res 341:2026
Chen L, Zhao XE, Lai D, Song Z, Kong F (2006) Carbohydr Res 341:1174
Sun B, Pukin AV, Visser GM, Zuilhof H (2006) Tetrahedron Lett 14:7371
Tietze LF, Keim K, Janßen CO, Tappertzhofen C, Olschimke J (2000) Chem Eur J 6:2801
Castro‐Palomino JC, Simon B, Speer O, Leist M, Schmidt RR (2001) Chem Eur J 7:2178
Janssen S, Schmidt RR (2005) J Carbohydr Chem 24:611
Aly MRE, Ibrahim ESI, El Ashry ESH, Schmidt RR (1999) Carbohydr Res 316:121
Gege C, Kinzy W, Schmidt RR (2000) Carbohydr Res 328:459
Gege C, Oscarson S, Schmidt RR (2001) Tetrahedron Lett 42:377
Gege C, Schmidt RR (2002) Carbohydr Res 337:1089
Gege C, Geyer A, Schmidt RR (2002) Eur J Org Chem 2475
Gege C, Vogel J, Bendas G, Rothe U, Schmidt RR (2000) Chem Eur J 6:111
Gege C, Geyer A, Schmidt RR (2002) Chem Eur J 8:2454
Yamaguchi M, Ishida H, Kanamori A, Kannagi R, Kiso M (2003) Carbohydr Res 338:2793
Bélot F, Rabuka D, Fukuda M, Hindsgaul O (2002) Tetrahedron Lett 43:7743
Natori T, Morita M, Akimoto K, Koezuka Y (1994) Tetrahedron 50:2771
Natori T, Koezuka Y, Higa T (1993) Tetrahedron Lett 34:5591
Morita M, Motoki K, Akimoto K, Natori T, Sakai T, Sawa E, Yamaji K, Koezuka Y Kobayashi E, Fukushima H (1995) J Med Chem 38:2176
Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M (1997) Science 278:1626
Figueroa‐Pérez S, Schmidt RR (2000) Carbohydr Res 328:95
Laurent N, lafont D, Buillange P (2006) Carbohydr Res 341:823
Bartolmäs T, Heyn T, Mickeleit M, Fischer A, Reutter W, Danker K (2005) J Med Chem 48:6750
Gouin SG, Pilgrim W, Porter RK, Murphy PV (2005) Carbohydr Res 340:1547
Sato R, Toma K, Nomura K, Takagi M, Yoshida T, Azefu Y, Tamiaki H (2004) J Carbohydr Chem 23:375
Herzner H, Reipen T, Schultz M, Kunz H (2000) Chem Rev 100:4495
Röhrig CH, Retz OA, Hareng L, Hartung T, Schmidt RR (2005) ChemBioChem 6:1805
Saha UK, Schmidt RR (1997) J Chem Soc Perkin Trans 1:1855
Saha UK, Griffith LS, Rademann J, Geyer A, Schmidt RR (1997) Carbohydr Res 304:21
Burger K, Kluge M, Fehn S, Koksch B, Hennig L, Müller G (1999) Angew Chem Int Ed 38:1414
Allevi P, Anastasia M, Paroni R, Ragusa A (2004) Bioorg Med Chem Lett 14:3319
Komba S, Meldal M, Werdelin O, Jensen T, Bock K (1999) J Chem Soc Perkin Trans 1:415
St. Hilaire PM, Cipolla L, Franco A, Tedebark U, Tilly DA, Meldal M (1999) J Chem Soc Perkin Trans 1:3559
Xue J, Khaja SK, Locke RD, Matta KL (2004) Synlett 861
Paulsen H, Paal M, Schultz M (1983) Tetrahedron Lett 24:1759
Paulsen H, Paal M (1984) Carbohydr Res 135:53
Kessler H, Kottenhahn M, Kling A, Kolar C (1987) Angew Chem Int Ed Engl 99:919
Halkes KM, Gotfredsen CH, Grøtly M, Miranda LP, Duus JØ, Meldal M (2001) Chem Eur J 7:3584
Nicolaou KC, Mitchell HJ, Jain NF, Bando T, Hughes R, Winssinger N, Natarajan S, Koumbis AE (1999) Chem Eur J 5:2648
Nicolaou KC, Cho SY, Hughes R, Winssinger N, Smethurst C, Labischinski H, Endermann R (2001) Chem Eur J 7:3798
Ritter TK, Mong KKT, Liu H, Nakatani T, Wong CH (2003) Angew Chem Int Ed 42:4657
Meinjohanns E, Meldal M, Paulsen H, Dwek RA, Bock K (1998) J Chem Soc Perkin Trans 1: 549
Matsuo I, Nakahara Y, Ito Y, Nukada T, Nakahara Y, Ogawa T (1995) Bioorg Med Chem 3:1455
Chiesa MV, Schmidt RR (2000) Eur J Org Chem 3541
Aly MRE, Ibrahim ESI, El Ashry ESH, Schmidt RR (2001) Carbohydr Res 331:129
Zhu Y, Chen L, Kong F (2002) Carbohydr Res 337:207
Ratner DM, Plante OJ, Seeberger PH (2002) Eur J Org Chem 826
Mendoza VM, Agusti R, Gallo‐Rodriguez C, de Lederkremer RM (2006) Carbohydr Res 341:1488
Nakano J, Ichiyanagi T, Ohta H, Ito Y (2003) Tetrahedron Lett 44:1742
Hofsteenge J, Müller DR, de Beer T, Lüffler A, Richter WJ, Vliegenthart JFG (1994) Biochemistry 33:13524
Gäde G, Kellner R, Rinehart KL, Proefke ML (1992) Biochem Biophys Res Commun 189:1303
Hofsteenge J, Blommers M, Hess D, Furmanek A, Miroshnichenko O (1999) J Biol Chem 274:32786
Diem S, Albert J, Herderich M (2001) Eur Food Res Technol 213:439
Schnabel M, Römpp B, Ruckdeschel D, Unverzagt C (2004) Tetrahedron Lett 45:295
Adinolfi M, Barone G, De Napoli L, Guariniello L, Iadonisi A, Piccialli G (1999) Tetrahedron Lett 40:2607
Adinolfi M, De Napoli L, di Fabio G, Guariniello L, Iadonisi A, Messere A, Montesachio D, Piccialli G (2001) Synlett 745
Bernards A, van Harten‐Loosbroek N, Borst P (1984) Nucleic Acids Res 12:4153
Gommers‐Ampt JH, Lugterink J, Borst P (1991) Nucleic Acids Res 19:1745
Gommers‐Ampt JH, van Leeuwen F, de Beer ALJ, Vliegenthart JFG, Dizdaroglu M, Kowalak JA, Crain PF, Borst P (1993) Cell 75:1129
de Kort M, Ebrahimi E, Wijsman ER, van der Marel GA, van Boom JH (1999) Eur J Org Chem 2337
Fransson LÅ (1987) Trends Biochem Sci 12:406
Kjellen L, Lindahl U (1991) Annu Rev Biochem 60:443
Karst N, Jacquinet JC (2000) J Chem Soc Perkin Trans 1:2709
Karst N, Jacquinet JC (2002) Eur J Org Chem 815
Tully SE, Mabon R, Gama CI, Tsai SM, Liu X, Hsieh‐Wilson LC (2004) J Am Chem Soc 126:7736
Tamura J, Tokuyoshi M (2004) Biosci Biotechnol Biochem 68:2436
Soliman SE, Bassily RW, El‐Sokkary RI, Nashed MA (2003) Carbohydr Res 338:2337
Yeung BKS, Hill DC, Janicka M, Petillo PA (2000) Org Lett 2:1279
Casu B (1985) Adv Carbohydr Chem 43:51 and references cited therein
Faham S, Hileman RE, Fromm JR, Linhardt RJ, Rees DC (1996) Science 271:1116
Hileman RE, Fromm JR, Wiler JM, Linhardt RJ (1998) BioEssays 20:156
Pellegrine L, Burke DF, von Delft F, Mulloy B, Blundell TL (2000) Nature 107:1029
Mach H, Volkin DB, Burke CJ, Middaugh CR, Linhardt RJ, Fromm JR, Loganathan D, Mattsson L (1993) Biochemistry 32:5480
Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC (1993) J Biol Chem 268:23906
Poletti L, Fleischer M, Vogel C, Guerrini M, Torri G, Lay L (2001) Eur J Org Chem 2727
Lucas R, Hamza D, Lubineau A, Bonnaffé D (2004) Eur J Org Chem 2107
Lee JC, Lu XA, Kulkarni SS, Wen YS, Hung SC (2004) J Am Chem Soc 126:476
de Paz JL, Ojeda R, Reichardt N, Martín‐Lomas M (2003) Eur J Org Chem 3308
Lohman GJS, Seeberger PH (2004) J Org Chem 69:4081
Zhou Y, Lin F, Chen J, Yu B (2006) Carbohydr Res 341:1619
Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M (1999) Annu Rev Biochem 68:729
Chen L, Kong F (2002) Carbohydr Res 337:1373
Rietschel ET, Schletter J, Weidemann B, El‐Samalouti V, Mattern T, Zähringer U, Seydel U, Brade H, Flad HD, Kusumoto S, Gupta D, Dziarski R, Ulmer AJ (1998) Microb Drug Resist 4:37
Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S (1999) Immunity 11:443
Inamura S, Fukase K, Kusumoto S (2001) Tetrahedron Lett 42:7613
Inamura S, Fujimoto Y, Kawasake A, Shiokawa Z, Woelk V, Heine H, Lindner B, Inohara N, Kusumoto S, Fukase K (2006) Org Biomol Chem 4:232
Zeng Y, Zhang J, Kong F (2002) Carbohydr Res 337:1367
Gu G, Wie G, Du Y (2004) Carbohydr Res 339:1155
Ning J, Heng L, Kong F (2002) Carbohydr Res 337:1159
Zeng Y, Zhang J, Ning J, Kong F (2003) Carbohydr Res 338:5
Xing Y, Ning J (2003) Tetrahedron Asymmetry 14:1275
Bruneteau M, Fabre I, Perret J, Michel G, Ricci P, Joseleau J, Kraus J, Schneider M, Blaschek W, Franz G (1988) Carbohydr Res 175:137
Johnson J, Kirkwood S, Misaki A, Nelson TE, Scalleti JD, Smith F (1963) Chem Ind (London) 820
He H, Yang F, Du Y (2002) Carbohydr Res 337:1673
Zhu Y, Kong F (2000) Synlett 663
Zhang G, Fu M, Ning J (2005) Carbohydr Res 340:597
Zeng Y, Kong F (2003) Carbohydr Res 338:2359
Ning J, Zhang W, Yi Y, Yang G, Wu Z, Yi J, Kong F (2003) Bioorg Med Chem 11:2193
Wu Z, Kong F (2004) Carbohydr Res 339:2761
Wu Z, Kong F (2004) Carbohydr Res 339:377
Li A, Kong F (2004) Carbohydr Res 339:2499
Ning J, Yi Y, Kong F (2002) Tetrahedron Lett 43:5545
Ning J, Kong F, Lin B, Lei H (2003) J Agric Food Chem 51:987
Yi Y, Zhou Z, Ning J, Kong F, Li J (2003) Synthesis 491
Huang GL, Mei XY, Liu MX (2005) Carbohydr Res 340:603
Zhu Y, Kong F (2000) Carbohydr Res 329:199
McNeil M, Wallner SJ, Hunter SW, Brennan PJ (1987) Carbohydr Res 166:299
Nita‐Lazar M, Chevolot L, Iwahara S, Takegawa K, Furmanek A, Lienart Y (2002) Acta Biochim Pol 49:1019
Ramli N, Shinohara H, Takegawa K, Iwahara SJ (1994) Ferment Bioeng 78:341
Zhang G, Fu M, Ning J (2005) Carbohydr Res 340:155
Gandolfi‐Donadio L, Gallo‐Rodriguez C, de Lederkremer RM (2003) J Org Chem 68:6928
Du Y, Pan Q, Kong F (2000) Carbohydr Res 329:17
Ning J, Yi Y, Yao Z (2003) Synlett 2208
Li A, Zeng Y, Kong F (2004) Carbohydr Res 339:673
Ma Z, Zhang J, Kong F (2004) Carbohydr Res 339:1761
Li A, Kong F (2004) Carbohydr Res 339:1847
Li A, Kong F (2005) Carbohydr Res 340:1949
Borman S (2006) C&EN 84:80
Chen L, Kong F (2002) Carbohydr Res 337:2335
Bedini E, Carabellese A, Corsaro MM, De Castro C, Parrilli M (2004) Carbohydr Res 339:1907
Zhang J, Kong F (2002) Carbohydr Res 337:391
Zhang J, Ning J, Kong F (2003) Carbohydr Res 338:1023
Zhang J, Kong F (2003) Carbohydr Res 338:19
Zhang J, Kong F (2003) Tetrahedron 59:1429
Ma Z, Zhang J, Kong F (2004) Carbohydr Res 339:43
Zhang J, Kong F (2002) J Carbohydr Chem 21:579
Zhang J, Kong F (2003) Carbohydr Res 338:1719
Zhao W, Kong F (2005) Carbohydr Res 340:1673
Hua Y, Xiao J, Huang Y, Du Y (2006) Carbohydr Res 341:191
Bélot F, Costachel C, Wright K, Phalipon A, Mulard LA (2002) Tetrahedron Lett 43:8215
Segat‐Dioury F, Mulard LA (2002) Tetrahedron Asymmetry 13:2211
Mulard LA, Guerreiro C (2004) Tetrahedron 60:2475
Bélot F, Wright K, Costachel C, Phalipon A, Mulard LA (2004) J Org Chem 69:1060
Wright K, Guerreiro C, Laurent I, Baleux F, Mulard LA (2004) Org Biomol Chem 2:1518
Mulard LA, Clément MJ, Imberty A, Delepierre M (2002) Eur J Org Chem 2486
Maruyama M, Takeda T, Shimizu N, Hada N, Yamada H (2000) Carbohydr Res 325:83
Ishii K, Kubo H, Yamasaki R (2002) Carbohydr Res 337:11
Kubo H, Ishii K, Koshino H, Toubetto K, Naruchi K, Yamasaki R (2004) Eur J Org Chem 1202
Ferguson MAJ, Homans SW, Dwek RA, Rademacher TW (1988) Science 239:753
Frankhauser C, Homas SW, Oates JE, McConville MJ, Desponds C, Conzelmann A, Ferguson MAJ (1993) J Biol Chem 268:26365
Homans SW, Ferguson MAJ, Dwek RA, Rademacher TW, Anad R, Williams AF (1988) Nature 333:269
Caro HH, Martín‐Lomas M, Bernabé M (1993) Carbohydr Res 240:119
Mayer TG, Kratzer B, Schmidt RR (1994) Angew Chem Int Ed Engl 33:2177
Mayer TG, Schmidt RR (1999) Eur J Org Chem 1153
Murakata C, Ogawa T (1992) Carbohydr Res 234:75 and 235:95
Baeschlin DK, Chaperon AR, Green LG, Hahn ME, Ince SJ, Ley SV (2000) Chem Eur J 6:172
Campbell AS, Fraser‐Reid B (1995) J Am Chem Soc 117:10387
Pekari K, Tailler D, Weingart R, Schmidt RR (2001) J Org Chem 66:7432
Pekari K, Schmidt RR (2003) J Org Chem 68:1295
Martín‐Lomas M, Flores‐Mosquera M, Chiara JL (2000) Eur J Org Chem 1547
Ma Z, Zhang J, Kong F (2004) Carbohydr Res 339:29
Gandolfi‐Donadio L, Gallo‐Rodriguez C, de Lederkremer RM (2002) J Org Chem 67:4430
Yashunsky DV, Borodkin VS, Ferguson MAJ, Nikolaev AV (2006) Angew Chem 118:482
Roush WR, Bennett CE (2000) J Am Chem Soc 122:6124
Ješelnik M, Plavec J, Polanc S, Kočevar M (2000) Carbohydr Res 328:591
Myers AG, Liang J, Hammond M, Harrington PM, Wu Y, Kuo EY (1998) J Am Chem Soc 120:5319
Wang ZX, Shi XX, Chen GR, Ren ZH, Luo L, Yan J (2006) Carbohydr Res 341:1945
Rao Y, Venot A, Swayze EE, Griffey RH, Boons GJ (2006) Org Biomol Chem 4:1328
Blattner R, Furneaux RH, Pakulski Z (2006) Carbohydr Res 341:2115
Ferguson JR, Harding JR, Lumbard KW, Scheinmann F, Stachulski AV (2000) Tetrahedron Lett 41:389
Ferguson JR, Harding JR, Kilick DA, Lumbard KW, Scheinmann F, Stachulski AV (2001) J Chem: Soc Perkin Trans 1:3037
Brown RT, Carter NE, Mayalarp SP, Scheinmann F (2000) Tetrahedron 56:7591
Suzuki T, Mabuchi K, Fukazawa N (1999) Bioorg Med Chem Lett 9:659
Engstrom KM, Daanen JF, Wagaw S, Stewart AO (2006) J Org Chem 71:8378
Kawada T, Asano R, Makino K, Sakuno T (2000) Eur J Org Chem 2723
Feldman KS, Lawlor MD (2000) J Am Chem Soc 122:7396
Nicolaou KC, Pfefferkorn JA, Cao GQ (2000) Angew Chem Int Ed 39:734
Schramm S, Dettner K, Unverzagt C (2006) Tetrahedron Lett 47:7741
Hein M, Michalik D, Langer P (2005) Synthesis 3531
Jiang ZH, Geyer A, Schmidt RR (1995) Angew Chem Int Ed Engl 34:2520
Jiang ZH, Schmidt RR (1994) Liebigs Ann Chem 645
Fürstner A, Jeanjean F, Razon P (2002) Angew Chem Int Ed 41:2097
Brito‐Arias M, Pereda‐Miranda R, Heathcock CH (2004) J Org Chem 69:4567
Fürstner A, Albert M, Mlynarski J, Matheu M (2002) J Am Chem Soc 124:1168
Fürstner A, Mlynarski J, Albert M (2002) J Am Chem Soc 124:10274
Fürstner A, Ruiz‐Caro J, Prinz H, Waldmann H (2004) J Org Chem 69:459
Hostettmann K, Marston A (1995) Saponins, Chemistry and Pharmacology of Natural Products. University Press, Cambridge
Chwalek M, Plé K, Voutquenne‐Nazabadioko L (2004) Chem Pharm Bull 52:965
Deng S, Yu B, Xie J, Hui Y (1999) J Org Chem 36:7265
Plé K, Chwalek M, Voutquenne‐Nazabadioki L (2004) Eur J Org Chem 1588
Thompson MJ, Hutchinson EJ, Stratford TH, Bowler WB, Blackburn GM (2004) Tetrahedron Lett 45:1207
Ikeda T, Yamauchi K, Nakano D, Naknishi K, Miyashita H, Ito S, Nohara T (2006) Tetrahedron Lett 47:4355
Ikeda T, Miyashita H, Kajimoto T, Nohara T (2001) Tetrahedron Lett 42:2353
Zhu X, Yu B, Hui Y, Schmidt RR (2004) Eur J Org Chem 965
Schimmel J, Passos Eleutério MI, Ritter G, Schmidt RR (2006) Eur J Org Chem 1701
Passos Eleutério MI, Schimmel J, Ritter G, do Céu Costa M, Schmidt RR (2006) Eur J Org Chem 5293
Wang P, Kim YJ, Navarro‐Villalobos M, Rohde BD, Gin DY (2005) J Am Chem Soc 127:3256
Kim YJ, Wang P, Navarro‐Villalobos M, Rohde BD, Derryberry JM, Gin DY (2006) J Am Chem Soc 128:11906
Sawada N (2004) Synthesis of Deoxy‐QS‑21A, Universität Konstanz, to be published
Dumont‐Hornebeck B, Joly JP, Coulon J, Chapleur Y (1999) Carbohydr Res 320:147
Rele SM, Cui W, Wang L, Hou S, Barr‐Zarse G, Tatton D, Gnanou Y, Esko JD, Chaikof EL (2005) J Am Chem Soc 127:10132
Ikuta A, Tanimoto T, Koizumi K (2003) J Carbohydr Chem 22:297
Ikuta A, Mizuta N, Kitahata S, Murata T, Usui T, Koizumi K, Tanimoto T (2004) Chem Pharm Bull 52:51
Smiljanic N, Halila S, Moreau V, Djedaïni‐Pilard F (2003) Tetrahedron Lett 44:8999
Damager I, Olsen CE, Møller BL, Motawia MS (1999) Carbohydr Res 320:19
Wu Z, Kong F (2004) Synlett 2594
Levy DE, Tang C (1995) In: The Chemistry of C‑Glycosides, Pergamon, Elmsford NY
Dondoni A, Marra A, Massi A (1999) J Org Chem 64:933
Armitt DJ, Banwell MG, Freeman C, Parish CR (2002) J Chem Soc Perkin Trans 1:1743
Herzner H, Palmacci ER, Seeberger PH (2002) Org Lett 4:2965
Furuta T, Kumura T, Kondo S, Mihara H, Wakimoto T, Nukaya H, Tsuji K, Tanaka K (2004) Tetrahedron 60:9375
Ali IAI, El‑Ashry ESH, Schmidt RR (2004) Tetrahedron 40:4773; and references therein
Fuchss T, Schmidt RR (2000) J Carbohydr Chem 19:677; and references therein
Tsuruta O, Yuasa H, Hashimoto H, Kuromo S, Yazawa S (1999) Bioorg Med Chem Lett 9:1019
Izumi M, Tsuruta O, Kajihara Y, Yazawa S, Yuasa H, Hashimoto H (2005) Chem Eur J 11:3032
Ohara K, Matsuda H, Hashimoto M, Miyairi K, Okuno T (2002) Chem Lett 626
Morii Y, Matsuda H, Ohara K, Hashimoto M, Miyairi K, Okuno T (2005) Bioorg Med Chem 13:5113
Schmidt RR, Michel J, Roos M (1984) Liebigs Ann Chem 1343
Schmelzer U (1997) PhD thesis, Universität Konstanz
Schmelzer U, Zhang Z, Schmidt RR (2007) J Carbohydr Chem (submitted)
Huchel U (1998) PhD thesis, Universität Konstanz
Huchel U, Schmidt C, Schmidt RR (1995) Tetrahedron Lett 36:9457
Huchel U, Schmidt C, Schmidt RR (1998) Eur J Org Chem 1353
Hanessian S, Condé JJ, Lion B (1995) Tetrahedron Lett 36:5865
Hanessian S (1997) In: Hanessian S (ed) Preparative Carbohydrate Chemistry. Marcel Dekker, New York, p 381–388; and references therein
Yu B, Tao H (2001) Tetrahedron Lett 42:2405
Yu B, Tao H (2002) J Org Chem 67:9099
Acknowledgement
Our work reported in this chapter was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Springer-Verlag Berlin Heidelberg New York
About this entry
Cite this entry
Schmidt, R.R., Zhu, X. (2008). Glycosyl Trichloroacetimidates. In: Fraser-Reid, B.O., Tatsuta, K., Thiem, J. (eds) Glycoscience. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-30429-6_11
Download citation
DOI: https://doi.org/10.1007/978-3-540-30429-6_11
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-36154-1
Online ISBN: 978-3-540-30429-6
eBook Packages: Chemistry and Materials ScienceReference Module Physical and Materials ScienceReference Module Chemistry, Materials and Physics