
Abstract
Levulinate is an important hydroxyl protecting group in carbohydrate chemistry but has not previously been so employed in cellulose chemistry, perhaps because of challenges involved in synthesis of cellulose levulinates. Herein we describe homogeneous acylation of cellulose in N,N-dimethylacetamide/LiCl using differently activated levulinic acid derivatives, including in situ activation with dicyclohexylcarbodiimide, p-toluenesulfonyl chloride, 1,1′-carbonyldiimidazole, or trifluoroacetic anhydride, providing and comparing several methods to access cellulose levulinates. Degree of substitution (DS) has been determined by 1H NMR spectroscopy using perpropionylated cellulose levulinates, showing that DS values as high as 2.42 are attainable. Cellulose levulinate esters were deacylated selectively by hydrazine without detectable loss of other alkanoate ester groups (acetate or propionate), indicating strong promise for levulinate as a useful protecting group for the synthesis of regioselectively substituted cellulose and other polysaccharide derivatives.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Advances in the past two decades against the difficult problem of regioselective synthesis of polysaccharide derivatives, especially derivatives of cellulose (Fox et al. 2011; Iwata et al. 1992), have taught us that regioselectively substituted cellulose derivatives have properties that are often quite distinct from their randomly substituted equivalents. For example, key properties like crystallinity (Iwata et al. 1996a, b), thermal properties (Iwata et al. 1997), solubility (Kondo 1994), and optical properties (Buchanan et al. 2012) depend strongly on position of substitution. Initial regioselective syntheses employed protecting group chemistry, in particular exploiting the generally higher reactivity of the primary OH group at C-6 of cellulose to protect that position (Heinze et al. 1994; Klemm and Stein 1995); this methodology provided access to 2,3-disubstituted derivatives (Philipp et al. 1995) and through them to 2,3-A-6-B trisubstituted derivatives. Recently, we have developed routes to the same families of derivatives with similar regioselectivity but avoiding the necessity of protection/deprotection steps by regioselective 2,3-deacylation of cellulose (and amylose) esters using tetrabutyl ammonium fluoride (TBAF) or hydroxide (TBAOH) bases (Zheng et al. 2013a, b). While this provides an efficient route to these derivatives, full exploration of structure–property relationships will require synthetic access to other cellulose derivative homopolymers, such as cellulose 3,6-diesters and -diethers. This will require differentiation of the very similarly reactive 2-OH and 3-OH groups, and almost certainly will require the development of new protecting group chemistry. Sterically demanding silyl ethers like thexyldimethylsilyl have shown some promise for selective reaction at O-2 versus O-3, as pioneered by the Klemm and Heinze groups (Koschella et al. 2001, 2006; Koschella and Klemm 1997). Of equal importance, removal of these silyl ethers requires treatment with fluoride salts like TBAF (Petzold et al. 2004); as we note above TBAF is not compatible with ester substituents as it catalyzes their deacylation (Xu and Edgar 2012). As an illustrative example, in an attempt to deprotect the silyl ether group of 3-allyl-2-thexyldimethylsilylcellulose-6-O-acetate with TBAF in tetrahydrofuran (THF) (Xu and Edgar 2012), the reaction did not provide the expected product 3-O-allyl-6-O-acetyl-cellulose, but rather 3-O-allyl-cellulose (Scheme S1); TBAF removed not only the silyl group but also the acetate group.
It is of importance to develop better polysaccharide protective groups, which can be readily, ideally regioselectively, reacted with polysaccharide hydroxyl groups, and can then be removed chemoselectively without affecting other groups, especially rather sensitive ester groups, to permit regioselective synthesis of polysaccharide esters. For this purpose, our attention was drawn to levulinate esters, which are commonly used as protective groups in small molecule carbohydrate chemistry to protect hydroxyls. Derived from the acid hydrolysis of cellulose (Hegner et al. 2010), levulinic acid contains two reactive functional groups, a carboxylic acid and a ketone. Esters of levulinic acid are widely used in drug delivery systems, and as solvents, additives, plasticizers (Hegner et al. 2010), and as protecting groups applied in synthetic organic chemistry (Greene and Wuts 1999). More importantly, levulinate esters are acid stable and can, in small molecule chemistry, be removed by hydrazine selectively with respect to other esters (Jeker and Tamm 1988), due to the potential for reaction of a single hydrazine molecule with both the ketone and ester carbonyls of the levulinate group. These characteristics raised our interest in levulinate as a potential protecting group for the synthesis of regioselectively substituted cellulose esters. Alkyl and carbohydrate levulinates (Bart et al. 1994; Dharne and Bokade 2011; Fernandes et al. 2012; Ho and Wong 1975; Melero et al. 2013), and oligosaccharide levulinates (Koeners et al. 1980) have been well-explored. However, synthesis of polysaccharide levulinates has been little investigated. The Yamada lab (Tsukatani et al. 2003) reported in a Japanese patent the reaction of cellulose with a purported mixed anhydride of levulinic acid with p-toluenesulfonic acid in either pyridine or N,N-dimethylformamide (DMF), affording cellulose levulinates with average degree of substitution (DS) approaching 1. A Russian group (Vladimirova et al. 1965) described reaction of cellulose with a reagent they describe as “levulinoyl chloride”, which is not the dominant species from reaction of levulinic acid with acid chloride forming reagents such as the thionyl chloride used by the authors (vide infra). They obtained cellulose levulinate of DS up to 0.3 by this method. They reported synthesis of mixed acetate/levulinate esters by acid-catalyzed esterification using levulinic acid in the presence of acetic anhydride (maximum DSLev ca. 1), and also reported synthesis of cellulose levulinates by nucleophilic displacement of cellulose tosylates by levulinate ion (maximum DSLev ca. 1). Neither group systematically explored methods of cellulose levulinate synthesis, nor did they report the ability to synthesize high DS cellulose levulinates. Most importantly, they did not discuss potential methods for selective removal of the levulinate moiety.
While those experienced with synthesis of polysaccharide derivatives will hardly be surprised that some reactions of small molecule carbohydrates don’t work well on the less reactive, less soluble polysaccharides, especially the insoluble, crystalline, heavily hydrogen-bonded cellulose, still we were surprised at the lack of prior literature on the subject. Besides the poor reactivity of cellulose and to a lesser extent other polysaccharides, there is also the issue of the interesting cyclization chemistry available to levulinic acid. Exposure of levulinic acid to reagents like thionyl chloride does not result in the expected simple conversion to the acid chloride, but rather in chlorination of a cyclic intermediate, affording 5-chloro-5-methyl-dihydrofuran-2(3H)-one (ii, Scheme 1) (Newman et al. 1966). These chlorolactones are much less reactive towards hydroxyl acylation than are most acid chlorides. There is surprisingly little discussion of these issues with regard to acylation of carbohydrates with levulinate in that body of literature, however as we will see, the issues impact reaction with polysaccharides significantly and may help to explain the paucity of previous studies of polysaccharide levulinates.

Reaction of levulinic acid with thionyl chloride
We report herein initial studies of methods for synthesis and characterization of levulinic acid esters of cellulose. We attempt to synthesize cellulose levulinates of different degrees of substitution and avoid issues arising from levulinate cyclization to lactone species by using various methods for mild activation of levulinic acid, including dicyclohexylcarbodiimide (DCC), p-toluenesulfonyl chloride (TosCl), 1,1′-carbonyldiimidazole (CDI), and trifluoroacetic anhydride (TFAA) (Scheme 2). In addition to the essential issue of whether levulinates can be practically attached as polysaccharide esters, we also address the other critical issue of whether levulinate esters can be selectively removed in the presence of other alkanoate ester groups.

Reaction of cellulose with levulinic acid and DCC, TosCl, CDI or TFAA in DMAc/LiCl
Experimental
Materials
Microcrystalline cellulose (MCC, Avicel PH-101, DP 260) was vacuum-dried before use. 4-Dimethylaminopyridine (DMAP), levulinic acid, DCC, TosCl (99+ %), CDI (97 %), and TFAA were purchased from Acros Organics. N,N-Dimethylacetamide (DMAc), lithium chloride (LiCl), acetic anhydride, propionic anhydride, and pyridine (anhydrous) were purchased from Fisher; all reagents were used without further purification.
Measurements
1H, 13C NMR, HMBC and COSY spectra of the cellulose esters after peracetylation or perpropionylation were acquired in CDCl3 on a Bruker Avance II 500 MHz spectrometer at room temperature or 50 °C, with number of scans of 32, 10,000, 19,200 and 9,400 respectively. DS values were determined by means of 1H NMR spectroscopy.
Product yields were calculated using the following equation:
A is the mass of cellulose, and B is the mass of the product cellulose levulinate.
Molecular weight of the perpropionylated cellulose levulinate was determined by size exclusion chromatography (SEC) in chloroform on a Waters Alliance model 2690 chromatograph with Waters 2414 differential refractive index (RI) detector and Viscotek 270 dual detector, versus polystyrene standards.
General procedure for dissolution of cellulose in DMAc/LiCl
Following a known procedure (Edgar et al. 1995), a mixture of MCC (2.00 g, 12.32 mmol) and DMAc (74.8 mL) was heated to 156 °C over 26 min under nitrogen. Anhydrous LiCl (3.4 g) was added and the mixture was stirred at 165 °C for 8 min. Distillate (14.4 mL) was collected at 165 °C. The mixture was cooled down to room temperature and stirred overnight.
Synthesis of cellulose levulinate via in situ activation of levulinic acid with DCC
A solution of levulinic acid and DCC with stated molar ratio (Table 1) in DMAc (20 mL) was stirred for 30 min at 80 °C under N2. The mixture was slowly added to the pre-dissolved cellulose. DMAP (100 mg) was added as a catalyst, then the reaction mixture was stirred for 24 h at 80 °C. The reaction solution was cooled to room temperature and then precipitated into 200 mL ethanol. The product was isolated by filtration, washed several times with ethanol, collected, and dried under vacuum at 40 °C overnight. Example yield 82 % (DS 1.53).
Synthesis of cellulose levulinate via in situ activation of levulinic acid with TosCl
Levulinic acid and TosCl with stated molar ratio (Table 1) were dissolved in DMAc (20 mL). The reaction solution was stirred for 30 min at 80 °C under N2. The reaction mixture was then added dropwise into the pre-dissolved cellulose solution over 30 min and allowed to react at 80 °C for 24 h. The product was isolated by adding the reaction mixture slowly to ethanol (200 mL). The precipitate was isolated by filtration, washed with ethanol (3 × 100 mL), then dried under vacuum at 40 °C to yield the product. Example yield 80 % (DS 1.51).
Synthesis of cellulose levulinate via in situ activation of levulinic acid with CDI
A solution of levulinic acid and CDI with stated molar ratio (Table 1) in DMAc (20 mL) was stirred for 30 min at 80 °C under N2. The mixture was added dropwise from an addition funnel to the pre-dissolved cellulose solution and allowed to stir for 24 h at 80 °C. The homogeneous mixture was slowly added to ethanol (200 mL) to precipitate the product. The product was isolated by filtration and washed several times with ethanol. It was dried under vacuum at 40 °C overnight. Example yield 84 % (DS 1.64).
Synthesis of cellulose levulinate via in situ activation of levulinic acid with TFAA
A solution of TFAA and levulinic acid with stated molar ratio (Table 1) was stirred at 50 °C for 30 min under N2 and was immediately added dropwise into the pre-dissolved cellulose solution. The solution was stirred at 50 °C for 24 h. After cooling to room temperature, the reaction solution was slowly added to ethanol (500 mL) under vigorous stirring. The product was isolated by filtration, washed with ethanol several times, and dried under vacuum at 40 °C overnight. Example yield 78 % (DS 2.42).
General procedures for peracetylation or perpropionylation
Cellulose levulinates were peracylated for easier NMR analysis, according to literature procedures (Liebert et al. 2005; Xu and Edgar 2012; Xu et al. 2011). Cellulose levulinate (0.3 g), DMAP (15 mg) and acetic anhydride or propionic anhydride (4 mL) were added to pyridine (4 mL). After stirring at 80 °C for 24 h, the crude product was obtained by precipitation into ethanol (200 mL) and washed with several times by ethanol. The product was then re-dissolved in chloroform (5 mL), re-precipitated into ethanol (200 mL), and dried under vacuum to give the perpropionylated product for NMR analysis. DS values for the peracylated product were calculated based on the following equations by calculating the ratio of acetyl or propionyl proton integrals to the backbone hydrogens integral (Liebert et al. 2005; Xu et al. 2011).
The protons of the methyl group of the propionate moiety are located at δ = 0.95–1.25 ppm and the propionate methylene protons are at δ = 2.19 ppm. The levulinate methyl protons resonate at δ = 2.12 ppm and four levulinate methylene protons are visible at δ = 2.29–3.07 ppm. The signals between 3.35 and 5.30 ppm are the backbone protons.
General procedure for the reaction of peracylated cellulose levulinate with hydrazine
Peracylated cellulose levulinate (0.3 g) was dissolved in pyridine (5 mL) and propionic acid (1 mL). Hydrazine monohydrate [0.3 mL, 7.11 mol/mol anhydroglucose unit (AGU)] was added and the solution was allowed to stir at room temperature for 24 h. The product was dialyzed against DI water for 4 days and then freeze-dried.
Solubility test
Samples (10 mg) were mixed with a solvent (1 mL) under vigorous stirring at room temperature. THF, chloroform, dimethyl sulfoxide (DMSO), pyridine and ethanol were used for this test. Solubilities were determined by visual inspection.
Results and discussion
Approaches
We chose initially to explore the straightforward approach of reaction of levulinic acid with thionyl chloride, to see whether acylation via the chlorolactone intermediate ii (Scheme 1) might be feasible. Levulinic acid was reacted with thionyl chloride, and the product solution was then added dropwise to a cellulose solution in DMAc/LiCl, with pyridine as base. However, no levulinate signals were detected in the 1NMR spectra of the perpropionylated product. The chlorolactone is predominant and stable with respect to levulinoyl chloride formation; our observation indicates that direct acylation of cellulose with this reagent is not practical, consistent with experience in small molecule carbohydrate chemistry (Hassner et al. 1975; Newman et al. 1966).
Therefore, we explored other, milder activating reagents such as DCC, TosCl, CDI and TFAA in hope of changing the levulinate/lactone equilibrium, and/or enhancing reactivity towards cellulose hydroxyls (Scheme S2). DCC is a popular condensation reagent, which has been frequently applied to activate carboxylic acids and thereby prepare other types of cellulose esters. Acid anhydrides have been shown to be reactive intermediates in these reactions (Zhang and McCormick 1997). In our case, levulinic acid was allowed to react with DCC for 30 min under N2; this reaction mixture and DMAP were then added to cellulose DMAC/ LiCl solution, and the resulting mixture was allowed to react for 24 h.
TosCl is another useful carboxyl activation reagent. It has been shown that both mixed anhydrides and acid chlorides are formed when carboxylic acids were treated with TosCl (Heinze and Schaller 2000). Acylation of cellulose by TosCl activation of levulinate was also performed in two steps. TosCl and levulinic acid were stirred at 80 °C for 30 min and the resulting mixture was added slowly to cellulose solution.
Activation of the carboxylic acid with CDI provides another mild method for ester synthesis. The imidazolide (Staab 1962) of levulinic acid was firstly formed by reaction of levulinic acid with CDI at 80 °C for 30 min, which was then added to a cellulose solution. This method is of great interest for cellulose esterification, because the only by-products are readily removed CO2 and imidazole (Graebner et al. 2002).
Reaction of carboxylic acids with TFAA has been demonstrated to be an efficient esterification system for cellulose by producing a mixture of acid anhydrides (Morooka et al. 1984), even though the strongly acidic conditions may in some cases cause significant polymer chain degradation. In our case, levulinic acid and TFAA were stirred at 50 °C for 30 min. The pre-mixed solution was then added to the cellulose solution, followed by reaction for 24 h at 50 °C.
Preparation of cellulose levulinate with different DS values
Across the range of acylation approaches attempted, it quickly became clear that efficiency of acylation with levulinate is much inferior to that achievable with simple alkanoates like acetate or propionate; very high efficiencies are possible with simple alkanoyl chlorides or alkanoic anhydrides under the proper conditions (Edgar et al. 1995). In order to determine whether synthesis of high DS cellulose levulinates was possible, we explored preparation using different activating reagents using a range of reagent equiv/AGU at 80 °C for 24 h (Table 1). Reaction of cellulose with levulinic acid in the presence of DCC leads to the cellulose levulinates 1-3. At a molar ratio of 3 equiv each of levulinic acid and DCC per AGU, a rather small DSLev of 0.34 was reached within a reaction time of 24 h at 80 °C. Even treatment of cellulose with 6 equiv levulinic acid and DCC per AGU for 24 h at 80 °C afforded cellulose levulinate with a disappointing DSLev of 0.46. Fortunately, we found that 24 h reaction of cellulose with a large excess of both levulinic acid and DCC (20 equiv) afforded the potentially useful DSlev of 1.53 (generally for use of levulinate as a hydroxyl protecting group, DS 1.0 or 2.0 would be the target).
We sought to understand whether other activation methods could more efficiently afford cellulose levulinates with higher DS values, beginning by employing similar reaction conditions for TosCl-mediated activation of levulinic acid. Treating cellulose with 3 or 6 equiv TosCl-activated levulinate per AGU again provided a relatively low acylation extent, with DSLev below 0.5 (samples 4 and 5). Practical DSLev levels (1.51, sample 6) were again only achieved when cellulose was exposed to a large excess of reagent (20 equiv/AGU each of levulinic acid and TosCl, 24 h, 80 °C).
Somewhat disappointingly, even reaction of dissolved cellulose with levulinic acid imidazolide proceeded with rather low efficiency, leading to the corresponding cellulose levulinates with DSLev of 0.40, 0.48 and 1.64 at molar ratios of AGU/levulinic acid/CDI of 1/3/3, 1/6/6, and 1/20/20, respectively (24 h, 80 °C, samples 7–9). Thus, these three different activation methods (DCC, TosCl and CDI) all gave similar results: with fewer than 6 equiv of coupling reagents, low DS (below 0.5) cellulose levulinates were obtained; excess activation reagent (20 equiv) afforded cellulose levulinate with DS about 1.5–1.6. We experimented with lower temperatures and shorter reaction times, but under these conditions obtained cellulose levulinates with significantly lower DSLev. Higher temperatures and longer reaction times did not afford higher DS. At this point we do not know the extent to which activated levulinates arise from the ring open form (i) and to what extent from the lactone form (iv, Scheme 3). It is possible that low reactivity of the activated lactone form is the source of the low DS observed when using only moderate activated levulinic acid excess, even though activation of the hydroxyl group of the cyclic lactone form may also occur.

Equilibrium in solution between levulinic acid and its lactone form
Interestingly, activation by the (more acidic) TFAA method provided the highest DS. Only 3 equiv of reagent/AGU was enough to approach DSLev of 1 (0.96), while 6 equiv afforded DSLev 1.68 (24 h, 50 °C). Reaction of cellulose with 20 equiv/AGU TFAA and levulinic acid afforded the highest DSLev we observed under any reaction conditions, providing cellulose levulinate with DSLev of 2.42. It may be that higher DS values can be obtained using TFAA because, under acidic conditions, both activated levulinic acid and its lactone form react with cellulose. We suspect that the protonated hydroxyl group of 5-OH-γ-valerolactone may act as a leaving group, thereby creating a resonance-stabilized cation (vi). Subsequent attack by a cellulose hydroxyl group on the carbonyl carbon results in ring opening and affords cellulose levulinate (Scheme 4). We observed much more severe cellulose backbone degradation at 50 °C using the TFAA method, presumably due to the strongly acidic conditions (DP decreased from 260 to 96).

Proposed reaction of 5-OH-γ-valerolactone with cellulose in the presence of acid
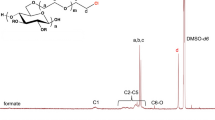
For determination of DSLev values, products 1–12 (see Table 1) were treated with propionic anhydride in pyridine in the presence of DMAP catalyst, yielding fully substituted cellulose levulinate propionates with simplified spectra. Completeness of perpropionylation was confirmed by the disappearance of the OH band in the FTIR spectrum (3,460 cm−1). The perpropionylated products are readily soluble in chloroform. Figure 1 shows the 1H NMR spectrum of cellulose levulinate 9 after perpropionylation in CDCl3. Proton peaks were assigned based on previous studies, 2D spectra, and long-range correlation spectra (Wang et al. 2005; Zheng et al. 2013a, b). Propionate and levulinate DS values were determined by means of 1H NMR spectroscopy, according to the equations in the Experimental section.

1H NMR spectrum of cellulose levulinate 9 after perpropionylation
It was important to determine regioselectivity, although our previous results with much more sterically demanding acylating reagents (pivaloyl chloride, adamantoyl chloride, and 2,4,6-trimethylbenzoyl chloride) suggested that high O-6 selectivity was not to be expected (Xu et al. 2011). Reaction of cellulose with 20 equiv/AGU CDI and levulinic acid at 50 °C for 24 h afforded a randomly substituted cellulose levulinate with DSLev at O-2/3 of 1.03 and DSLev at O-6 of 0.61. Partial DSPr values of cellulose propionate levulinate were determined directly by the ratio of partial propionate methyl resonances (O-2/3 1.10–1.25 ppm, O-6 0.95–1.10 ppm) to the integrals of the cellulose backbone protons (3.35–5.30 ppm). The partial DSLev value was subtracted from 1 (with successful perpropionylation there is a total DS of 1 at each position) to give the DSLev at that position.
Carbon signals (Fig. 2) were completely assigned by analogy to previous work (Wang et al. 2005). Signals at δ = 62.0 ppm (C-6), between 70.2 and 73.5 ppm (C-2, C-3 and C-5), at δ = 77.5 ppm (C-4), and at δ = 100.0 ppm (C-1) are the backbone carbons. The propionate methyl, methylene, and carbonyl carbons resonate at 8.9, 27.4, and 169.8–172.5 ppm, respectively. Two levulinate methylene carbons are at 28.8 and 39.9 ppm, and the levulinate methyl carbon is at 28.9 ppm. The levulinate ketone carbon and ester carbonyl carbons are at 206.8 and 173.2 ppm, respectively.

13C NMR spectrum of cellulose levulinate 9 after perpropionylation
Solubility of cellulose levulinates versus DS
Solubilities of cellulose levulinates with different DS values were investigated and the results are listed in Table 2. Compounds 4, 1, and 8 with DSLev below 0.5 were insoluble in any common organic solvents. Cellulose levulinate samples with DS values of 1.51 (sample 6) and 1.64 (sample 9) had better solubility in THF, DMSO and pyridine. Compound 12 with the highest DS of 2.42 was soluble in THF, chloroform, pyridine, and DMSO. Solubility of cellulose levulinates depends predictably (based on extensive literature precedent with simple cellulose alkanoates) on the DS values; higher DSLev leads to more organic solubility. Peracylation consistently improved solubility in organic solvents since the resulting cellulose alkanoate levulinates were more hydrophobic and not prone to intramolecular or intermolecular hydrogen bonding.
Selective deprotection of levulinate using hydrazine
The ability to selectively remove potential alcohol protecting groups is also an essential feature if they are to be useful. As we demonstrate above, protection of cellulose free hydroxyl groups as levulinate esters can be achieved, and accessibility of either DS 1.0 or DS 2.0 means that sufficient DS may be attainable for most practical situations. The small molecule literature indicates that levulinate can be removed by several reagents, including (a) Grignard reagents (Watanabe et al. 1992), (b) sodium bisulfite (Ono and Itoh 1988), (c) sodium borohydride (Hassner et al. 1975), and (d) hydrazine (Glushka and Perlin 1990; Ho and Wong 1975), among which, hydrazine was reported to selectively deprotect levulinate in the presence of other ester groups (O-acetyl, O-propionyl, O-benzoyl) (Ho and Wong 1975; Jeker and Tamm 1988). However, precedent for deacylation of cellulose alkanoates may not provide encouragement; Miyamato et al. has described (Miyamoto et al. 1985) that hydrazine catalyzes deacylation of cellulose acetate.
In order to investigate whether hydrazine can selectively deacylate cellulose levulinate esters in the presence of acetate or propionate esters, peracetylated or perpropionylated cellulose levulinates were treated with hydrazine in pyridine and propionic acid at room temperature for 24 h. The product was collected by precipitation in water and filtration, and was peracylated (with an alkanoate not present in the starting ester) to afford a well-resolved NMR spectrum for DS determination. Scheme S1 shows the reaction schemes.
The 13C NMR spectrum of the product of cellulose levulinate propionate 10 deacylation by hydrazine after peracetylation is shown in Fig. 3. No carbon signals of levulinate moieties are observed, confirming the complete removal of levulinate by hydrazine. DS values for the products of cellulose acetate levulinate or cellulose levulinate propionate hydrolysis by hydrazine after peracylation were determined by 1H NMR. As shown in Fig. S2, peaks between 1.80 and 2.15 ppm confirm acetate substitutions; peaks at 0.95–1.25 ppm and 2.18–2.42 ppm are propionate methyl protons and propionate methylene protons. Peaks for levulinate protons are absent. DSAc values were determined directly by the ratio of the acetyl resonances (1.80–2.08 ppm) to the integrals of the cellulose backbone protons (3.30–5.50 ppm). DSPr was calculated similarly by comparing integrals of the backbone protons with the propionyl methyl protons (0.95–1.25 ppm). As shown in Table S1, the starting DS acetyl or propionyl, respectively, remains the same after treatment with hydrazine, proving selective deacylation of levulinate by hydrazine in the presence of acetate and propionate.

13C NMR spectrum of the product of cellulose levulinate propionate 10 reaction with hydrazine, after peracetylation
Conclusions
Cellulose levulinates with a wide range of DS values (0.22–2.42) were successfully prepared by esterification of cellulose in DMAc/LiCl with levulinic acid in the presence of activating agents DCC, TosCl, CDI or TFAA. Comparing these four methods investigated, in situ activation of levulinic acid with TFAA provided the highest DSLev of 2.42 under comparable conditions. Efficiency is modest, but for the purposes of making cellulose ester homopolymers for initial structure-property investigations, is sufficient. 1H and 13C NMR spectra of cellulose levulinate propionate were assigned in detail based upon previous literature. Solubility of cellulose levulinate depends in predictable fashion on DSLev. Hydrazine can selectively cleave the levulinate groups of cellulose acetate levulinate and cellulose levulinate propionate while leaving acetates and propionates unscathed. With the availability of these methods for protection and mild deprotection of alcohols, levulinate shows great potential as a protecting group for synthesis of regioselectively substituted derivatives of cellulose and is likely to be of general use in polysaccharide chemistry. Further studies of subsequent modifications of the new cellulose esters and applications of levulinate as protecting group for regioselective synthesis of cellulose esters are under way.
Supporting information
The online version of this article contains supplementary material, which is available to authorized users, and includes the following: 1H and 13C NMR spectra of peracylated cellulose levulinate. Scheme of reaction of cellulose levulinate acetate or propionate with hydrazine, followed with perpropionylation or peracetylation. Table of DS values of the products of reaction of cellulose levulinate acetate or propionate with hydrazine after perpropionylation or peracetylation.
References
Bart HJ, Reidetschlager J, Schatka K, Lehmann A (1994) Kinetics of esterification of levulinic acid with n-butanol by homogeneous catalysis. Ind Eng Chem Res 33:21–25. doi:10.1021/ie00025a004
Buchanan CM, Donelson ME, Guzman-Morales E, Mackenzie PB, Wang B (2012) Cellulose ester optical films. US Pat Appl 13/409,735
Dharne S, Bokade VV (2011) Esterification of levulinic acid to n-butyl levulinate over heteropoly acid supported on acid-treated clay. J Nat Gas Chem 20:18–24. doi:10.1016/s1003-9953(10)60147-8
Edgar KJ, Arnold KM, Blount WW, Lawniczak JE, Lowman DW (1995) Synthesis and properties of cellulose acetoacetates. Macromolecules 28:4122–4128
Fernandes DR, Rocha AS, Mai EF, Mota CJA, Teixeira da Silva V (2012) Levulinic acid esterification with ethanol to ethyl levulinate production over solid acid catalysts. Appl Catal A Gen 425–426:199–204. doi:10.1016/j.apcata.2012.03.020
Fox SC, Li B, Xu D, Edgar KJ (2011) Regioselective esterification and etherification of cellulose: a review. Biomacromolecules 12:1956–1972. doi:10.1021/bm200260d
Glushka JN, Perlin AS (1990) Formation of disaccharides related to heparin and heparan sulfate by chemical modification of maltose. Carbohydr Res 205:305–321. doi:10.1016/0008-6215(90)80149-w
Graebner D, Liebert T, Heinze T (2002) Synthesis of novel adamantoyl cellulose using differently activated carboxylic acid derivatives. Cellulose 9:193–201
Greene TW, Wuts PGM (1999) Protective groups in organic synthesis, 3rd edn. Wiley, New York, pp 168–278
Hassner A, Strand G, Rubinstein M, Patchornik A (1975) Levulinic esters. Alcohol protecting group applicable to some nucleosides. J Am Chem Soc 97:1614–1615. doi:10.1021/ja00839a077
Hegner J, Pereira KC, DeBoef B, Lucht BL (2010) Conversion of cellulose to glucose and levulinic acid via solid-supported acid catalysis. Tetrahedron Lett 51:2356–2358. doi:10.1016/j.tetlet.2010.02.148
Heinze T, Schaller J (2000) New water soluble cellulose esters synthesized by an effective acylation procedure. Macromol Chem Phys 201:1214–1218. doi:10.1002/1521-3935(20000801)201:12<1214:aid-macp1214>3.0.co;2-9
Heinze T, Roettig K, Nehls I (1994) Synthesis of 2,3-O-carboxymethyl cellulose. Macromol Rapid Commun 15:311–317. doi:10.1002/marc.1994.030150403
Ho T-L, Wong CM (1975) Hydroxyl protection by levulinylation. Synth Commun 5:91–93. doi:10.1080/00397917508061437
Iwata T, Azuma J, Okamura K, Muramoto M, Chun B (1992) Preparation and NMR assignments of cellulose mixed esters regioselectively substituted by acetyl and propanoyl groups. Carbohydr Res 224:277–283. doi:10.1016/0008-6215(92)84113-7
Iwata T, Okamura K, Azuma J, Tanaka F (1996a) Molecular and crystal structure of cellulose acetate dipropanoate (CADP, 6-O-acetyl-2,3-O-propanoyl cellulose). Cellulose 3:107–124. doi:10.1007/bf02228794
Iwata T, Okamura K, Azuma J, Tanaka F (1996b) Molecular and crystal structure of cellulose propanoate diacetate (CPDA, 2,3-di-O-acetyl-6-O-propanoyl cellulose). Cellulose 3:91–106. doi:10.1007/bf02228793
Iwata T, Fukushima A, Okamura K, Azuma J-I (1997) DSC study on regioselectively substituted cellulose heteroesters. J Appl Polym Sci 65:1511–1515. doi:10.1002/(sici)1097-4628(19970822)65:8<1511:aid-app8>3.0.co;2-j
Jeker N, Tamm C (1988) Verrucarins and roridins. 46. Synthesis of new, unnatural macrocyclic trichothecenes: 3-isoverrucarin A ((1”-O)(3→4)abeo-verrucarin A), verrucinol, and verrucene. Helv Chim Acta 71:1895–1903. doi:10.1002/hlca.19880710808
Klemm D, Stein A (1995) Silylated cellulose materials in design of supramolecular structures of ultrathin cellulose films. J Macromol Sci Pure Appl Chem A32:899–904. doi:10.1080/10601329508010304
Koeners HJ, Verhoeven J, Van Boom JH (1980) Synthesis of oligosaccharides by using levulinic ester as an hydroxyl protecting group. Tetrahedron Lett 21:381–382. doi:10.1016/s0040-4039(01)85479-4
Kondo T (1994) Hydrogen bonds in regio-selectively substituted cellulose derivatives. J Polym Sci Part B Polym Phys 32:1229–1236. doi:10.1002/polb.1994.090320710
Koschella A, Klemm D (1997) Silylation of cellulose regiocontrolled by bulky reagents and dispersity in the reaction media. Macromol Symp 120:115–125. doi:10.1002/masy.19971200113
Koschella A, Heinze T, Klemm D (2001) First synthesis of 3-O-functionalized cellulose ethers via 2,6-di-O-protected silyl cellulose. Macromol Biosci 1:49–54. doi:10.1002/1616-5195(200101)1:1<49:aid-mabi49>3.0.co;2-c
Koschella A, Fenn D, Heinze T (2006) Water soluble 3-mono-O-ethyl cellulose: synthesis and characterization. Polym Bull 57:33–41. doi:10.1007/s00289-006-0534-2
Liebert T, Hussain MA, Heinze T (2005) Structure determination of cellulose esters via subsequent functionalization and NMR spectroscopy. Macromol Symp 223:79–91. doi:10.1002/masy.200550506
Melero JA, Morales G, Iglesias J, Paniagua M, Hernandez B, Penedo S (2013) Efficient conversion of levulinic acid into alkyl levulinates catalyzed by sulfonic mesostructured silicas. Appl Catal A 466:116–122. doi:10.1016/j.apcata.2013.06.035
Miyamoto T, Sato Y, Shibata T, Tanahashi M, Inagaki H (1985) Carbon-13 NMR spectral studies on the distribution of substituents in water-soluble cellulose acetate. J Polym Sci Polym Chem 23:1373–1381. doi:10.1002/pol.1985.170230511
Morooka T, Norimoto M, Yamada T, Shiraishi N (1984) Dielectric properties of cellulose acylates. J Appl Polym Sci 29:3981–3990. doi:10.1002/app.1984.070291230
Newman MS, Gill N, Darre B (1966) [3.2.1]-Bicyclic mechanism in the acyclic field. J Org Chem 31:2713–2714. doi:10.1021/jo01346a530
Ono M, Itoh I (1988) A new deprotection method for levulinyl protecting groups under neutral conditions. Chem Lett 585–588. doi:10.1246/cl.1988.585
Petzold K, Klemm D, Heublein B, Burchard W, Savin G (2004) Investigations on structure of regioselectively functionalized celluloses in solution exemplified by using 3-O-alkyl ethers and light scattering. Cellulose 11:177–193. doi:10.1023/B:CELL.0000025391.25835.e7
Philipp B et al (1995) Regioselective esterification and etherification of cellulose and cellulose derivatives. Part 1. Problems and description of the reaction systems Papier 49:3–7
Staab HA (1962) Syntheses with heterocyclic amides (azolides). Angew Chem 74:407–423
Tsukatani T, Inomata H, Ono H, Takemura A, Hori S, Isogai A, Yamada T (2003) Method for producing cellulose levulinate. Japanese Patent Application JP 2003082001
Vladimirova TV, Gal’braikh LS, Peker KS, Rogovin ZA (1965) Structure and properties of cellulose and its derivatives. CLXXII. Synthesis of keto-containing cellulose esters. Vysokomol Soedin 7:786–790
Wang YM, Ikeda A, Hori N, Takemura A, Ono H, Yamada T, Tsukatani T (2005) N,N-dicyclohexylcarbodiimide assisted synthesis and characterization of poly(vinyl alcohol-co-vinyl levulinate). Polymer 46:9793–9802. doi:10.1016/j.polymer.2005.07.088
Watanabe Y, Fujimoto T, Ozaki S (1992) A novel deacylation method using Grignard reagent without affecting the neighboring base-sensitive functional groups. J Chem Soc Chem Commun 681–683. doi:10.1039/c39920000681
Xu D, Edgar KJ (2012) TBAF and cellulose esters: unexpected deacylation with unexpected regioselectivity. Biomacromolecules 13:299–303. doi:10.1021/bm201724s
Xu D, Li B, Tate C, Edgar KJ (2011) Studies on regioselective acylation of cellulose with bulky acid chlorides. Cellulose 18:405–419. doi:10.1007/s10570-010-9476-9
Zhang ZB, McCormick CL (1997) Structopendant unsaturated cellulose esters via acylation in homogeneous lithium chloride/N,N-dimethylacetamide solutions. J Appl Polym Sci 66:293–305
Zheng X, Gandour RD, Edgar KJ (2013a) Probing the mechanism of TBAF-catalyzed deacylation of cellulose esters. Biomacromolecules 14:1388–1394. doi:10.1021/bm400041w
Zheng X, Gandour RD, Edgar KJ (2013b) TBAF-catalyzed deacylation of cellulose esters: reaction scope and influence of reaction parameters. Carbohydr Polym 98:692–698. doi:10.1016/j.carbpol.2013.06.010
Acknowledgments
This project was supported by a Grant from the U.S. Department of Agriculture (USDA, Grant Number 2011-67009-20090). We thank the Department of Sustainable Biomaterials, the Chemistry Department, and the Institute for Critical Technologies and Applied Science at Virginia Tech for their financial, facilities, and educational support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, X., Xu, D. & Edgar, K.J. Cellulose levulinate: a protecting group for cellulose that can be selectively removed in the presence of other ester groups. Cellulose 22, 301–311 (2015). https://doi.org/10.1007/s10570-014-0508-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0508-8