
Abstract
Marine organisms with a planktonic larval stage have the potential to be transported substantial distances, with the distance travelled depending on factors such as pelagic larval duration (PLD) and physical factors such as ocean currents and geographical barriers. The endemic New Zealand sea urchin, Evechinus chloroticus, is found throughout the North and South Islands, and with a PLD of approximately 30 days, is expected to show strong connectivity among all populations. Population connectivity and genetic differentiation were examined over both a geographically broad scale, throughout New Zealand, and on a fine scale (within the Hauraki Gulf on the North Island). Significant genetic differentiation was revealed through analysis of mitochondrial COI sequences (FST = 0.096 p < 0.01) and six microsatellite loci (FST = 0.0120 p < 0.008). This was consistent with a division between northern and southern regions located to the south of Cook Strait, at a phylogeographic barrier previously reported in other New Zealand benthic marine invertebrates. Fine-scale population differentiation was evident between the inner and outer Hauraki Gulf populations, and between the most northern populations and the remainder of the North Island. Together, this study suggests that strong coastal currents, upwelling in the Cook Strait region, and geographic distance (approximately 2000 km north to south) may all be acting to restrict gene flow and contribute to genetic divergence among populations of E. chloroticus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Larval dispersal has a major ecological and evolutionary role in shaping adult marine populations. Whether larvae are retained in the local population or are dispersed by coastal ocean currents to geographically distant populations can have a large affect on the population dynamics and genetic make-up of the mature and local population. Benthic marine organisms that exploit broadcast spawning as a mode of reproduction, and have a pelagic larval phase where embryos develop in the water column, are of particular interest in population connectivity studies, as pelagic larvae have the potential to travel extensive distances before settlement occurs (Hellberg 1996). Larval dispersal and population connectivity is a growing area of research and has large implications in designing Marine Protected Areas (MPA) and networks of MPAs (Botsford et al. 2001, 2009; Jones et al. 2007).
Many studies have investigated biotic and abiotic factors that have the potential to either facilitate or create a barrier to larval dispersal, and hence, affect the connectivity between populations. A correlation has been found between the time pelagic larvae spend in the water column, coined the pelagic larval duration (PLD), and the distance that is travelled by the larvae before recruitment and settlement into a population occurs (Shanks et al. 2003; Shanks 2009). Attributes of the hydrodynamic ocean environment such as persistent coastal ocean currents, eddies, and upwelling are also large components shaping marine populations, and may affect dispersal and gene flow by entraining or advecting larvae (Cowen et al. 2000; Banks et al. 2007; Treml et al. 2008; Rasmussen et al. 2009). Some have even argued that oceanic currents and upwelling play a larger role in determining levels of population connectivity than the PLD (Treml et al. 2008; White et al. 2010; Weersing and Toonen 2009). The development mode of pelagic larvae and large ocean currents do not necessarily preclude genetic homogeneity, as larval behavior may also play a role in creating genetic structure over a small spatial scale (Doherty et al. 1995; Leis and Carson-Ewart 2001; Levin 2006; Mercier et al. 2013).
Evechinus chloroticus, commonly known as kina, has a wide geographic distribution across the North and South Islands of New Zealand (Andrew 1988; Shears et al. 2008; Barker 2007), and broadcast spawns from late October to March. Spawning in general is synchronous in males and females and may potentially occur as mass spawning events in response to environmental cues such as temperature, tides, and lunar cycles (Lamare and Stewart 1998; Barker 2007). The planktonic larvae of E. chloroticus have a relatively long PLD of approximately 30 days (Dix 1970).
Previous population genetic studies have been conducted in E. chloroticus throughout New Zealand using both allozyme markers (Mladenov et al. 1997) and microsatellites (Perrin 2002), but not mitochondrial DNA (mtDNA). Both of these previous studies concentrated their sampling of populations in the South Island, with Perrin (2002) focused particularly in the southwestern Fiordland region. Mladenov et al. (1997) found little genetic differentiation and high levels of gene flow around New Zealand, apart from some detectable genetic subdivision among samples within Doubtful Sound, Fiordland. The microsatellite analyses (Perrin 2002) revealed further fine-scale differentiation among samples from Fiordland, with some significant differences observed at small scales (<100 km) within and among fiords. While the use of polymorphic microsatellite markers revealed greater differentiation between northern and southern populations, both studies had very limited sampling on the North Island (only two locations in each study). Due to these sampling regimes, it has been difficult to gain an overall picture of phylogeography and population connectivity estimates in E. chloroticus.
Studies investigating population differentiation have been conducted in numerous broadcast spawning intertidal species in New Zealand (Apte and Gardner 2002; Ayers and Waters 2005; Goldstien et al. 2006; Veale and Lavery 2011; Will et al. 2011; Wei et al. 2013a). Many of these studies documented a prominent phylogeographic break between the North and South Islands in the vicinity of Cook Strait (Ross et al. 2009). The previous sampling has not permitted an investigation of the existence of this north/south break among populations of E. chloroticus, and this study has extended sampling in this species to address this question.
An aspect of population structure in the marine realm that is often overlooked is the difference in genetic patterns that can be observed between small and large geographic scales. This was first brought to prominence by Johnson and Black (1982, 1984) who showed that in marine gastropod populations, significant small-scale genetic differentiation occurred among local populations, yet populations thousands of kilometers apart displayed similar levels of differentiation. This pattern is sometimes called chaotic genetic patchiness, and is most often explained by localized pulses of recruitment driving short-term small-scale differentiation, with the homogenizing effect of occasional long-distance dispersal resulting in long-term genetic differentiation only among very distant populations (Yearsley et al. 2013; Broquet et al. 2013). E. chloroticus has been shown to exhibit some patterns of subtle genetic differentiation at both small, within fjords, and large scales, throughout New Zealand (Mladenov et al. 1997; Perrin 2002). Although previous authors have largely attributed these patterns to the likely effects of local selective forces, more neutral forces of chaotic genetic patchiness may instead drive them. This alternative can only be tested through more extensive sampling at both small and large scales.
The research presented here aimed to (1) investigate scales of population connectivity in a broadcast spawning benthic marine invertebrate using two classes of genetic markers, mtDNA and microsatellites, over both a broad geographic scale (approximately 2000 km) and over a fine geographic scale (three populations within the Hauraki Gulf on the North Island, approximately 60 km apart), and (2) to assess if the previously described phylogeographic barrier between the North and South Islands was present among populations of E. chloroticus.
Materials and methods
Sampling and DNA extraction
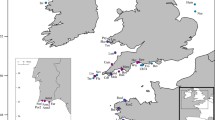
E. chloroticus samples were collected between November 2009 and August 2010 from locations across New Zealand by snorkel in 2–5 m of water at all locations except Barney’s Rock, Kaikoura, where samples were collected by SCUBA at depths of 12–15 m, (Fig. 1; Table 1). Twenty-five to fifty samples were collected from each location, except Barney’s Rock, Kaikoura, where only seven individuals could be collected. 5–10 spines, with the associated muscle tissue, were removed from each individual using forceps and preserved in 1.5 ml micro-centrifuge tubes filled with 95 % ethanol. The sampled tissue was stored at −20 °C within 2 weeks of being collected. For DNA extraction, a small amount of tissue was macerated using flame-sterilized forceps and scalpel, and the phenol chloroform isoamyl alcohol (PCI) protocol, modified from Hoelzel and Green (1998) was used. Re-suspended DNA was stored at −20 °C until polymerase chain reaction (PCR) was preformed (Birt 2000; Hoelzel and Green 1998).
PCR and sequencing
Mitochondrial marker COI
A previously developed primer set (Echino F1 and Echino R1; (Ward et al. 2008) was used to amplify a partial fragment of the mitochondrial gene Cytochrome Oxidase Subunit I (COI). The 25 μL PCR consisted of 2 μL of 15 ng/μL DNA template, 1 μL 10× reaction buffer, 0.5 μM of each primer, 0.2 mM dNTPs, 0.25 U of Taq DNA polymerase (Platinum Taq, Invitrogen), 2.5 mM of magnesium chloride (MgCl2), and 0.5 mg/μL of bovine serum albumin (BSA). The optimized thermal cycling protocol consisted of an initial denaturing step of 4 min at 94 °C, 35 cycles of 94 °C for 20 s, 48 °C for 15 s, 72 °C for 15 s, followed by a final extension of 72 °C for 10 min. All PCR amplifications were performed on Biometra and Applied Biosystems thermocyclers. PCR products were run on a 1.6 % agarose gel and stained with ethidium bromide. The amplified fragment length was approximately 650 bp.
The COI PCR products from 253 individuals were purified of excess dNTP’s and single stranded primers using a shrimp alkaline phosphate (SAP), exonuclease-I (Exo) digestion. PCR products plus the SAPEX mixture (10 U/mL SAP and 10 U/ml Exo) were incubated at 37 °C for 30 min, 80 °C for 15 min, and 20 °C for 15 min. The purified products were used in a cycle sequencing protocol using a 1/8 dilution of BigDye™ Dye Terminator chemistry v3.1 (Applied Biosystems Inc.). The products were purified with CleanSEQ™ using a SPRI™ paramagnetic bead tray (Agencourt Bioscience Corporation) following the prescribed protocol. The purified products were then sequenced on an Applied Biosystems 3130XL capillary DNA sequencer.
Microsatellite loci
Six microsatellite loci developed for E. chloroticus by Perrin and Roy (2000) were used in this study: A34, AAT42, B14, C1, C29, and G29 (Perrin and Roy 2000). Fluorescent-labeled primers were used for four loci (A34, AAT42, B14 and C1), and a CAG-tail method (Schuelke 2000) of attaching a fluorescent probe to the 5’ end of the PCR product was used for loci C29 and G29. The PCRs for the loci amplified with fluorescent-labeled primers were run at a total volume of 10 μL consisting of 15 ng/μL DNA template, 1.25 μL 10× reaction buffer, 0.5 μM fluorescent labeled forward primer, 0.5 μM of the reverse primer, 0.2 mM dNTPs, 0.25 U Taq DNA polymerase (Platinum Taq, Invitrogen), 2.5 mM of magnesium ion (Mg2+), and 1.25 mg/μL of BSA. In reactions for CAG-tail loci, the fluorescent-labeled forward primer was replaced with 0.05 μM of the forward CAG-tailed primer plus 0.5 μM of a matching generic fluorescent-labeled primer.
The PCR thermocycler temperature profile programs for each locus are as follows: program (1) 94 °C for 4 min, 35 cycles of 20 s at 94 °C, 53 °C for 15 s, 72 °C for 15 s, with a final extension period of 7 m at 72 °C (2) 94 °C for 4 min, 25 cycles of 94 °C for 1 m, 55 °C for 1 m, 72 °C for 1 m, 8 cycles of 94 °C for 1 m, 53 °C for 1 m, 72 °C for 1 m, with a final extension period of 7 m at 72 °C, programs (1) and (2) were modified from Perrin and Roy (2000). Program (3) 94 °C for 4 min, 25 cycles of 94 °C for 15 s, 46 °C for 20 s, 72 °C for 20 s, 8 cycles of 94 °C for 15 s, 45 °C for 20 s, with a final extension period of 7 m at 72 °C. Program (1) was used for loci A34, AAT42, B14, and C1, program (2) was used for loci C1, and program (3) was used to attach a CAG-tail fluorescent probe to loci C29 and G29. The PCR products were run on a 1.6 % electrophoresis gel to check if amplification was successful (Holleley and Geerts 2009).
Products from successful amplifications were cleaned to remove excess salts, dNTP’s, and unincorporated primers, with Ampure ™ using a SPRI™ plate following the manufacturer’s protocol. The cleaned products were multiplexed for each individual sea urchin. Two multiplex schemes were used from Multiplex Manager (Holleley and Geerts 2009). Loci B14, C1, C29, and G29 were grouped in multiplex A, and loci A34 and AAT42 were grouped in multiplex B. 3–4 μL of the pooled PCR products (1:4 dilution for each loci in multiplex A and a 1:2 dilution for each loci in multiplex B) were added to 10 μL of HiDi Formamide and 0.3 μL of LIZ 600 size standard (Applied Biosystems). This mix was then heat shocked at 95° for 5 min before being run on an ABI 3130 Genetic Analyzer.
Data analysis
Mitochondrial DNA
Haplotypes from the mitochondrial sequencing were identified using alignments in Geneious Pro 5.1.0™(Drummond et al. 2010). Genbank accession numbers are KP980809–KP981060. Haplotype diversity was calculated using gene diversity and nucleotide diversity measures following (Nei 1978) in ARLEQUIN v 3.5 (Excoffier and Lischer 2010).
Microsatellite markers
Genotypes from each individual were identified using GENEMAPPER v 3.7 (Applied Biosystems) and then checked manually for accuracy and consistency. Multiple PCRs of individuals, and multiple runs on the Genetic Analyzer ABI 3130, were conducted to detect any genotyping errors between PCRs and different runs on the sequencing machine. Approximately 10 % of the total samples were re-run for quality control. The dataset was checked for genotyping errors with Microchecker, and tested for the presence of null alleles (Van Oosterhout et al. 2004). The table of allele frequencies by population is provided in Supplementary Table S1.
The observed and expected heterozygosity (HO and HE respectively) for all loci and each population were calculated with the program Genetic Data Analysis (GDA) v 1.1 (Lewis and Zaykin 2002). Departures from Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were calculated in Genepop v 4.2 (Raymond and Rousset 1995) using the Markov chain method with 1000 dememorization steps, 100 batches, and 1000 iterations per batch. The significance of HWE and LD was determined by applying sequential Bonferroni correction (Rice 1989). The inbreeding coefficient (Fisher) for each locus and each population were also calculated in Genepop. The effective number of alleles per locus (Ne) was calculated using Ne = 1/(Sum π2). Shannon’s Information index (I) was calculated in the program GenAlEx (Peakall and Smouse 2006, 2012) using I = −1× Sum (π × Ln (π)) as an additional measure of genetic diversity (Sherwin et al. 2006).
Population differentiation
Genetic differentiation between populations was assessed using mitochondrial haplotype frequencies and microsatellite allele frequencies through analyses of molecular variance (AMOVA) (Excoffier et al. 1992) in ARLEQUIN v 3.5 (Excoffier and Lischer 2010). The standardized measures of genetic variance (F) values (Wright 1951, 1965; Weir and Cockerham 1984) were calculated for microsatellite allele frequencies, and from these calculations the total variance was partitioned into the variance explained among populations (FST), among populations within regional groupings (FSC), and among regional groupings (FCT). Both standard and corrected F′-statistics were calculated (Meirmans 2006; Meirmans and Hedrick 2011). For mitochondrial data, analyses were conducted using either frequency data alone (standard F-statistics) or including the genetic distances between haplotypes (Φ–statistics). Genetic differentiation for the mtDNA was calculated using a Kimura 2-parameter corrected measure of nucleotide differentiation (Kimura 1980). The pairwise F-statistics (Weir and Cockerham 1984) were calculated using haplotype frequencies for mtDNA data, and for each microsatellite locus an unbiased probability (P), using the Fisher method of exact differentiation (Fisher 1935), was calculated and tested against the null hypothesis that alleles were drawn from the same distribution in all populations. Populations were assigned a priori to North and South regional groups in the hierarchical AMOVA analyses, which allowed us to test if the previously described phylogeographic barrier between the North and South Islands was evident in E. chloroticus. The statistical significance of population pairwise FST values was corrected for false discovery rate (Benjamini and Yekutieli 2001).
The FST values for both marker types were used to produce a multi-dimensional scaling (MDS) plot in PRIMER 6 using 1000 repetitions and a minimal stress level of 0.001 (Clarke and Gorely 2006).
To assess if there was an isolation-by-distance pattern in both the data sets, a Mantel test was carried out in IBDWS (Jensen et al. 2005). A Mantel test uses the geographic distances between pairs of populations and the pairwise FST values to test the significance between geographic distances and genetic distances. Regression analyses were then used to assess the strength of the relationship.
The program STRUCTURE 2.3.3 was used to assign individuals to a population based on their genotypes from multiple loci using Bayesian methods (Pritchard et al. 2000; Falush et al. 2003). Analyses were undertaken both using and ignoring prior geographic location (Locprior option). Four iterations of 1,000,000 Monte Carlo Markov Chain (MCMC) repetitions were run following a 500,000 burn-in period for 1 to 11 inferred populations. An ad hoc calculation, ∆K, a measure of the second order rate of change of the log probability [Ln P(D)] of the data in relation to the number of K (clusters), was used to infer the most likely number of K (Evanno et al. 2005).
Results
Mitochondrial DNA sequence variation
A fragment of the Cytochrome Oxidase subunit I (COI) gene was sequenced for 252 individuals from the 11 sampling locations spanning the whole of New Zealand, from Northland to Stewart Island (shown in Table 1). Successful sequences were obtained for 83 % of the total individuals sampled. The sequence fragment length used in alignments was approximately 650 base pairs in length, and thirty-six haplotypes were identified from the successfully sequenced mtDNA.
A network of E. chloroticus haplotypes revealed a star-like phylogeny, with a high level of haplotype variation, but low level of nucleotide variation (Fig. 2). Two dominant haplotypes were observed: 51 % were Hap A and 19 % were Hap B. The haplotype network failed to reveal any obvious patterns of phylogeographic structure. Haplotypes were grouped into two regions to identify if there were any unique haplotypes to the North Region (including Nelson, Fig. 1) or to the South Region. The haplotype network did reveal four haplotypes (Hap S, T, 4, and 10), which were found exclusively in individuals from the South Region. These haplotypes were represented by only one individual, with the exception of Hap T (n = 7). The haplotype frequencies for each population are shown in Fig. 3.


Haplotype network of relationships among mitochondrial COI sequences. Haplotypes were randomly assigned either a letter or a number. The size of the circle reflects relative haplotype frequencies, with crosshatches on connecting lines indicating the number of base substitutions. Haplotypes found in northern and southern regions are indicated by red & blue respectively

Mitochondrial haplotype distribution of E. chloroticus in New Zealand. Haplotypes with a relative frequency less than 10 % were pooled as “others” for presentation. The size of the pie chart indicates the relative number of individuals sampled. The populations (from North to South) are Piha, west coast (PH), Mahinepua Bay, Northland (NoL), Army Bay, Whangaparaoa Peninsula (WP), Oneroa Bay, Waiheke Island (WI), Puriri Bay, Great Barrier Island (GBI), Mahia Peninsula, Hawke’s Bay (MP), Kau Bay, Wellington (WG), Cable Bay, Nelson (NL), Barney’s Rock, Kaikoura (KK), Patterson Inlet, Stewart Island (STI), Easy Harbour, Stewart Island (SL). Major current systems are shown with arrows: East Auckland Current (EAC), East Cape Current (ECC), Southland Current (SC), Westland Current (WC), D’Urville Current (DU), and West Australian Current (WAC)
Microsatellite Neutrality Tests and Allelic Variation
All six microsatellite loci were highly polymorphic with a range of 2–23 alleles present for each locus (Table 2). The experimental error rate in identifying allele size was <1 % (estimated from replicated genotyping of more than 10 % of individuals (N = 25 to 31) for each locus). Microsatellite loci A34 and C1 had the greatest allelic variation of the six loci used, with 15 and 23 alleles, respectively. Additionally, the six loci revealed relatively high levels of heterozygosity with a range of 0.690–0.748 over all populations. Populations all showed similar levels of microsatellite variation across the range sampled (Table 3). Tests of Hardy–Weinberg equilibrium (HWE) revealed only two significant divergences within populations (locus B14 in the Northland population, and locus AAT42 in the Patterson Inlet, Stewart Island population. Tests in Micro-Checker revealed that null alleles might be present in these instances, but only at low frequency (<5 %). No loci were significantly diverged from HWE when averaged across populations, and no populations were significantly diverged from HWE when averaged across loci.
Population differentiation
A signal of population structure was evident in both the mitochondrial data and the microsatellite data from AMOVA analyses (Table 4A, B), pairwise comparisons of genetic differentiation (Table 5; Fig. 4) and the assignment tests using STRUCTURE for the microsatellite data (Fig. 5). An AMOVA analysis was first used to test an initial partitioning scheme of grouping the populations into North Island and South Island regional groups, and revealed there was moderately significant population differentiation between the two island groups in the mitochondrial COI data (ΦCT = 0.030, p < 0.01) but not in the microsatellite data (FCT = 0.0039, p > 0.05, Table 4).

Multi-Dimensional Scaling (MDS) plot of pairwise FST values for a mtDNA data, and b microsatellite data. The South Island populations are in blue, and the North Island populations in red, with the most northerly populations in darker red. Note that the Nelson (NL) population is most similar to the North Island populations

Bar plot from STRUCTURE when K = 2. Each bar represents the proportion of each individual’s genotype assigned to the respective groups. Population abbreviations are defined in Table 1
To test if there was evidence of a phylogeographic break, or major genetic discontinuity, at the north of the South Island, as previously described for other marine species (Ross et al. 2009), the Nelson population was placed into the Northern group. This grouping revealed significantly greater regional differentiation in both the mitochondrial COI data (FCT = 0.090, p < 0.01; ΦST = 0.056, p < 0.01) and the microsatellite data (FST = 0.0092, p < 0.008) (Table 5).
Population pairwise tests of genetic differentiation revealed similar results between the two marker types. The general pattern was that the most southern Stewart Island populations were the most genetically differentiated, with some differentiation among North Island populations (Table 5). Both the mitochondrial and microsatellite markers revealed moderate levels of differentiation between the Stewart Island populations and all other populations except for Kaikoura.
On a smaller scale, both mtDNA and microsatellite analyses (Figs. 3, 4; Table 5) revealed some significant, but less clearly interpreted differences among the northern populations sampled. These comparisons suggest that the most northern population (Northland, NoL) and inner Hauraki Gulf populations (Whangaparoa Peninsula, WP and Waiheke Island, WI) are somewhat distinct in their haplotype and allele frequencies from the remaining northern populations. For mtDNA, these three populations (NoL, WP & WI) all have a much lower frequency of the most common A haplotype (Fig. 3), compared with the outer Hauraki Gulf population (Great Barrier Island) and the remaining northern populations, and are significantly different in a number of pairwise comparisons (Table 5). The microsatellites also revealed a similar trend in that no significant differences were observed among the northernmost and inner Hauraki Gulf populations (NoL, WP & WI) (Table 5), but that these populations were often significantly different from outer Hauraki Gulf and more southern populations (Great Barrier Island, Mahia Peninsula, Wellington and Nelson) and also the only west coast population (Piha). Not all of these pairwise comparisons were significant after correcting for false discovery rate, and there was some inconsistency in the significant pairwise comparisons between the mitochondrial and microsatellite analyses. However, for the microsatellite data, the significant population pairwise comparisons were consistent across a variety of measures of population divergence (FST, F′ST and Dest), and across all individual loci analysed (Supplementary Table S2).
The MDS plots (Fig. 4), display spatially the genetic relationships among sampled populations. They show that for both mitochondrial and microsatellite data, the northern and southern region populations group together. The Nelson population clearly falls within the northern group, and that the Kaikoura population, south of the proposed phylogeographic break, is intermediate. Among the North Island populations, both mtDNA and microsatellite plots reveal that the most northern population (NoL) and inner Hauraki Gulf populations (WP and WI) group together, somewhat apart from the remaining North Island populations, with GBI being the most significantly different of these (Table 5).
The STRUCTURE analysis produced the highest value of ΔK at K = 2, with clear differences between the Northern and Southern regions, with the Kaikoura and both Stewart Island populations on the South Island having similar assignment patterns (Fig. 5). From the STRUCTURE analysis it is apparent that the Whangaparoa Peninsula population had an assignment pattern slightly different from the other North Island populations.
Isolation by distance plots (Fig. 6), show that there is a significant but not strong correlation for both the mitochondrial and microsatellite datasets, (R2 = 0.234, p = 0.003 and R2 = 0.151, p = 0.009 respectively). It is clear that a large proportion of the isolation by distance signal is driven by the long distance comparisons with the two Stewart Island populations (Fig. 6), as these relationships become non-significant if the pairwise comparisons from these two populations are removed (R2 = 0.025, p = 0.8290 for mtDNA; and R2 = 0.033, p = 0.154 for microsatellite data).

Plots of population pairwise genetic distance (FST) versus geographic distance (km) for a mtDNA sequences and b microsatellite genotypes. Results of Mantel tests of isolation by distance are provided for each data set. Pairwise comparisons between the Patterson Inlet and Easy Harbour, Stewart Island populations and each of the other nine populations are indicated by a black circle
Discussion
To date, this study is the most comprehensive effort at describing the genetic patterns and population connectivity in E. chloroticus. The main findings of this research are that (1) E. chloroticus is not as genetically uniform as suggested by previous allozyme analysis (Mladenov et al. 1997), and that (2) significant genetic structure is evident over both large and small spatial scales. These findings are supported by previous microsatellite study of fiord populations in the far southwest of New Zealand (Perrin 2002), which revealed some genetic differences over small spatial scales. The phylogeographic break at the north of the South Island that is described for many New Zealand invertebrate species also appears to be present in E. chloroticus.
Population connectivity over a broad geographic scale
Mitochondrial and microsatellite DNA data for E. chloroticus revealed that there was genetic differentiation between the northern and southern populations, with the genetic division between the two occurring in the vicinity of the previously described phylogeographic break. This differentiation was most clearly supported by the STRUCTURE analysis and hierarchical AMOVA analyses that grouped the population closest to this phylogeographic barrier (Kaikoura) with the southern group, and the nearby Nelson population with the northern group. These results suggest a prolonged or persistent barrier impacting larval dispersal across the broad geographic scale.
Although there is some support for a pattern of isolation-by-distance among the sampled populations, it is also very clear that most of the isolation-by-distance signal is driven by the most divergent Stewart Island populations, and that the pattern largely disappears if these populations are removed. The Kaikoura population is somewhat intermediate. However, the most powerful test for assigning populations to either region, the microsatellite STRUCTURE analysis, places this population in the southern group, while the geographically close Nelson population is assigned to the northern region. Unfortunately, only a small sample could be acquired from the Kaikoura population for this study, limiting the ability to make strong conclusions about the exact location of the phylogeographic break, but the importance of the sample demanded its inclusion in the analyses.
Population differentiation between the North and South Islands is one of the most prominent patterns of marine genetic structure previously described in New Zealand, and has been observed in a number, but by no means all, marine invertebrate species examined to date. The Cook Strait region is an area where ocean currents become complex, and are proposed to create an oceanographic barrier to gene flow (e.g., Ross et al. 2009). More intense sampling in this region has identified a genetic break that occurs on the northwest coast of the South Island near Farewell Spit, and on the northeast coast of the South Island near Cape Campbell in several invertebrate species with planktonic larvae (Veale and Lavery 2011): the green-lipped mussel Perna canaliculus (Apte and Gardner 2002; Wei et al. 2013a); the cushion sea-star Patiriella regularis (Waters and Roy 2004; Ayers and Waters 2005); two species of limpets Cellana ornata and C. radians (Goldstien et al. 2006); and the snakeskin chiton Sypharochiton pelliserpentis (Veale and Lavery 2011). Other studies have clearly identified that a genetic break occurs either north of the top of the South Island in species of seagrass (Zostera muelleri, (Jones et al. 2008), estuarine clams (Ross et al. 2012) and abalone (Will et al. 2011), or further south of the proposed barrier at Cape Campbell in two species of bull kelp (Durvillaea antarctica and D. willana, (Collins et al. 2010; Fraser et al. 2010). It is also important to remember that a number of studies report that no genetic break is evident around the Cook Strait region (e.g., two species of estuarine amphipod, Stevens and Hogg 2004; an intertidal crab, Hinnendael 2008; an anemone, Veale and Lavery 2012; and a number of species of intertidal fishes, e.g., Hickey et al. 2009), highlighting that, although it may not be an uncommon phenomenon, it is far from universal for coastal marine species with New Zealand-wide distributions.
In E. chloroticus 9 % of the total mtDNA variance is partitioned between Northern and Southern regional groups (FCT = 0.090, p = 0.006, Table 4). In comparison, this is a higher percentage than has been found previously in the sea star (Patiriella regularis) (6 %) between similar Northern and Southern groups (Ayers and Waters 2005). However, higher levels of mtDNA divergence between these groups were found in the greenshell mussel (Perna canaliculus) (16 %) (Apte and Gardner 2002) and in the chiton (Sypharochiton pelliserpentis) (47 %) (Veale and Lavery 2011). Thus, E. chloroticus displays an intermediate level of differentiation along this recognized phylogeographic break.
These broad-scale results are also supported by previous population genetic analyses of this species that were not as extensive in either geographic or marker sampling (Mladenov et al. 1997; Perrin 2002). The previous analyses indicated there were significant genetic differences between far northern and far southern sampling locations, but were not able to identify accurately the location or degree of those differences. The current study has greatly increased our understanding of these broad-scale differences in E. chloroticus.
In general, the present results for E. chloroticus are supportive of the hypothesis that upwelling around Cape Campbell is acting as a barrier to gene flow (Apte and Gardner 2002). The proposed genetic break at Cape Farewell on the west coast of the South Island was unable to be assessed directly in this study, as no samples were obtained from the west coast of the South Island.
The broad patterns of genetic differentiation found amongst populations of E. chloroticus, are not peculiar for a species with a pelagic larva that has a relatively long development period. Numerous studies of population connectivity in sea urchins have revealed that despite the long PLD, patterns of genetic differentiation or isolation by distance do occur over large spatial scales (Addison and Hart 2004; Palumbi et al. 1997; Olivares-Banuelos et al. 2008; Benham et al. 2012).
Population differentiation within the North Island
Despite previous predictions of genetic uniformity due to the relatively long PLD in E. chloroticus, we also found evidence of low to moderate population differentiation among North Island populations, even at a relatively small scale in the Hauraki Gulf. Not all the fine-scale differences between populations were observed in both types of molecular marker in this study. This could be due to the inherent nature of each marker and their power to detect differentiation (Avise 1994; Hedrick 1999), or simply stochastic variation among loci (Balloux and Lugon-Moulin 2002).
There is a small amount of evidence (from microsatellite data alone) of genetic differentiation between populations from the east and west coasts of the North Island, (which may have been suspected), although sampling is still insufficient to confirm this. In the Hauraki Gulf, our findings suggest that there may be low levels of genetic differentiation present, such that inner Gulf populations are more genetically similar to populations further north, while the outer Gulf population on Great Barrier Island appears more genetically similar to southern North Island populations (MP and WG). The exact geographic pattern of these fine-scale genetic differences are not clear-cut, due to some inconsistencies in the patterns of pairwise population differences. However, it is clear that the North Island populations are not genetically panmictic, and that there is evidence that inner and outer Hauraki Gulf populations differ genetically.
The existence of fine-scale genetic differences in this species is supported by the previous genetic analyses (Perrin 2002), which indicated that significant genetic differentiation occurs among the fiords of the far southwest of New Zealand. Other studies have also shown evidence for fine-scale differentiation in sea urchins (Banks et al. 2007; Zulliger et al. 2009; Penant et al. 2013).
Factors driving fine-scale population differentiation
Distinct fine-scale population structure can arise due to habitat discontinuity, local ocean currents, or adaptation to distinct environments (Kinlan et al. 2005; Sanford and Kelly 2011). Alternatively, fine-scale variation in allele and genotype frequencies has been attributed to more neutral factors, such as random genetic drift, and fluctuations in spawning and larval dispersal pulses (Hedgecock et al. 2007). All these factors are possible causes of the patterns observed in E. chloroticus, and have been proposed to explain similar patterns in other sea urchins and other New Zealand species with planktonic larvae.
The population structuring in Stronglyocentrotus franciscanus along the west coast of North America was attributed to local recruitment patterns due to local hydrodynamics, despite the species’ long pelagic larval duration (49–133 days) and the unidirectional California Current System (North to South) (Benham et al. 2012). The surprising level of fine-scale differentiation in Paracentrotus lividus populations within the Atlantic and Mediterranean was believed to be driven by varying environmental conditions (plankton blooms and seawater temperatures) necessary for larval survival and recruitment (Penant et al. 2013). In the New Zealand context, some support has been presented to suggest that genetic divergences among populations of the mussel Perna canaliculus are best explained by local adaptation to varying sea surface temperatures (Wei et al. 2013b).
More neutral, random or “chaotic” factors have also been supported in other similar species. A study of abalone (Piggott et al. 2008) identified fine-scale genetic differentiation among populations along the eastern coast of Australia, despite the documented ability of the larvae to disperse large distances, and attributed the fine-scale genetic structure to random events of local larval retention. Similarly, significant genetic differences have been recorded between larval, juvenile and adult brittle stars within the same locality, strongly indicating that random recruitment events must play a factor (Muths et al. 2009).
In E. chloroticus, larval retention may play a major factor within the Hauraki Gulf, such that inner Gulf populations are more genetically similar to populations further north, while the outer Gulf population on Great Barrier Island appears more genetically similar to the Southern North Island populations. The major coastal current along the north-east of the North Island, the East Auckland Current, sweeps southward past Great Barrier Island in the outer Hauraki Gulf and around the East Cape, and is associated with large eddy systems (described in Chiswell 2003). Circulation in the inner Hauraki Gulf is somewhat isolated from the offshore East Auckland Current, and may act to segregate larval recruitment in the northern and inner Hauraki Gulf region from the remainder of the east coast by promoting retention of larvae.
Alternatively, the sub-tidal habitat, local hydrodynamics, and intermittent temporal persistence of urchin populations may be driving the fine-scale genetic patterns within the Hauraki Gulf populations. Adult E. chloroticus in the Hauraki Gulf have a patchy distribution and generally are found in low abundance in the inner Gulf, ranging to high abundance in the outer Gulf (Smith 2004). The patterns of abundance observed in the Gulf suggest that local hydrodynamics or environmental conditions, such as unsuitable settlement habitat, (like sheltered soft-sediment beaches and circulating sediment loads commonly found in the Gulf; Phillips and Shima 2006; Walker 2007), may limit recruitment and survival of larvae and juveniles in the inner Gulf to infrequent events.
Thus, the small levels of differentiation observed among the Hauraki Gulf populations of E. chloroticus could be a result of “chaotic patchiness” in recruitment (Johnson and Black 1982, 1984). Only a small proportion of the adult population may successfully spawn at a given location at any one time, leading to the successful local recruitment potentially coming largely from one cohort, and resulting in genetic variability among local sampling locations. Environmental differences may further differentiate local populations in the short-term through selective forces on the sensitive new recruits. Evidence of this phenomenon has been observed in other broadcast spawning marine invertebrates (e.g., Hedgecock et al. 2007; Hedgecock and Pudovkin 2011). Thus localized patterns of reproductive output and recruitment success may significantly impact the fine-scale population genetic structure of E. chloroticus at locations that are not subject to strong homogenizing water movements, such as the southern fiords and potentially the Hauraki Gulf.
Overall, the small-scale genetic differences between the inner and outer Hauraki Gulf may be due to purely local differences in cohort recruitment, or they may instead be part of a broader influence on larval selection and recruitment that separates the most northern and inner Gulf populations from those further south. This sets up several alternative hypotheses for more rigorous testing. In order to ascertain the true extent of fine-scale patterns of differentiation in this species, and whether they are persistent over time, additional sampling is needed over multiple years. We are currently in the process of undertaking this.
Conclusion
Although E. chloroticus is a species with high dispersal capabilities and the potential for high connectivity between geographically separated populations, the genetic analyses presented here reveal some significant spatial genetic heterogeneity at both large and small scales. The results from this research contrast with the early genetic research conducted on E. chloroticus, in which little genetic differentiation was reported among populations.
Overall, the genetic patterns observed in E. chloroticus were similar to those seen in some other marine broadcast spawning species in New Zealand. A north/south genetic divide is detected south of Cook Strait at low to moderate levels in E. chloroticus. Analysis of the present samples suggests that the genetic break lies near Cape Campbell on the East Coast of the South Island, although greater sampling is required to confirm this. There is also some evidence of small-scale genetic structure around the North Island at low to moderate levels, including low genetic divergence in both mtDNA and microsatellites between inner and outer Hauraki Gulf populations.
In summary, both broad-scale and fine-scale genetic patterns were evident in E. chloroticus. It appears that local and regional ocean currents and distinct geographical features may largely drive the broad genetic differentiation patterns observed, while local cohort recruitment and perhaps selection on recruits may drive the fine-scale patterns. The long PLD in this species is important in maintaining the species’ cohesion among populations within New Zealand, but does not prevent genetic divergence of populations at either large or small scales. The results from this study provide fertile hypotheses of specific spatial restrictions to gene flow or environmental-driven selection in this species. Additional sampling at varying geographic and temporal scales is being undertaken to further examine the genetic patterns of E. chloroticus presented here and to more rigorously test the proposed restrictions to connectivity.
References
Addison JA, Hart MW (2004) Analysis of population genetic structure of the green sea urchin (Strongylocentrotus droebachiensis) using microsatellites. Mar Biol 144:243–251. doi:10.1007/s00227-003-1193-6
Andrew NL (1988) Ecological aspects of the common sea-urchin, Evechinus chloroticus, in northern New Zealand—a review. N Z J Mar Freshwat Res 22:415–426
Apte S, Gardner JPA (2002) Population genetic subdivision in the New Zealand greenshell mussel (Perna canaliculus) inferred from single-strand conformation polymorphism analysis of mitochondrial DNA. Mol Ecol 11:1617–1628
Avise JC (1994) Molecular markers, natural history, and evolution. Chapman and Hall, New York
Ayers KL, Waters JM (2005) Marine biogeographic disjunction in central New Zealand. Mar Biol 147:1045–1052. doi:10.1007/s00227-005-1632-7
Balloux F, Lugon-Moulin N (2002) The estimation of population differentiation with microsatellite markers. Mol Ecol 11:155–165. doi:10.1046/j.0962-1083.2001.01436.x
Banks SC, Piggott MP, Williamson JE, Bove U, Holbrook NJ, Beheregaray LB (2007) Oceanic variability and coastal topography shape genetic structure in a long-dispersing sea urchin. Ecology 88:3055–3064
Barker M (2007) Chapter 16 ecology of Evechinus chloroticus. Dev Aquacult Fish Sci 37:319–338
Benham CE, Supernault KJ, Burton RS (2012) Genetic assessment of the population connectivity of the red urchin (Strongylocentrotus franciscanus). J Exp Mar Biol Ecol 432:47–54. doi:10.1016/j.jembe.2012.07.011
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Birt TP (2000) Polymerase chain reaction. In: Baker AJ (ed) Molecular methods in ecology. Blackwell Science, Oxford, pp 50–64
Botsford LW, Hastings A, Gaines SD (2001) Dependence of sustainability on the configuration of marine reserves and larval dispersal distance. Ecol Lett 4:144–150
Botsford LW, White JW, Coffroth MA, Paris CB, Planes S, Shearer TL, Thorrold SR, Jones GP (2009) Connectivity and resilience of coral reef metapopulations in marine protected areas: matching empirical efforts to predictive needs. Coral Reefs 28:327–337. doi:10.1007/s00338-009-0466-z
Broquet T, Viard F, Yearsley JM (2013) Genetic drift and collective dispersal can result in chaotic genetic patchiness. Evolution 67:1660–1675. doi:10.1111/j.1558-5646.2012.01826.x
Chiswell SM (2003) Circulation within the Wairarapa Eddy, New Zealand. N Z J Mar Freshwat Res 37:691–704
Clarke K, Gorely R (2006) Primer V6: User Manual/Tutorial.PRIMER-E, Plymouth
Collins CJ, Fraser CI, Ashcroft A, Waters JM (2010) Asymmetric dispersal of southern bull-kelp (Durvillaea antarctica) adults in coastal New Zealand: testing an oceanographic hypothesis. Mol Ecol 19:4572–4580. doi:10.1111/j.1365-294X.2010.04842.x
Cowen RK, Lwiza KMM, Sponaugle S, Paris CB, Olson DB (2000) Connectivity of marine populations: open or closed? Science 287:857–859. doi:10.1126/science.287.5454.857
Dix TG (1970) Biology of Evechinus chloroticus (Echinoidea:Echinometridae) from different localities. N Z J Mar Freshwat Res 4:385–405
Doherty PJ, Planes S, Mather P (1995) Gene flow and larval duration in 7 species of fish from the Great Barrier Reef. Ecology 76:2373–2391
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A (2010) Geneious v5.0. Available from www.geneious.com
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes—application to human mitochondrial-DNA restriction data. Genetics 131:479–491
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fisher RA (1935) The logic of inductive inference. J Royal Stat Soc 98:39–82
Fraser CI, Winter DJ, Spencer HG, Waters JM (2010) Multigene phylogeny of the southern bull-kelp genus Durvillaea (Phaeophyceae: Fucales). Mol Phylogen Evol 57:1301–1311. doi:10.1016/j.ympev.2010.10.011
Goldstien SJ, Schiel DR, Gemmell NJ (2006) Comparative phylogeography of coastal limpets across a marine disjunction in New Zealand. Mol Ecol 15:3259–3268. doi:10.1111/j.1365-294X.2006.02977.x
Hedgecock D, Pudovkin AI (2011) Sweepstakes reproductive success in highly fecund marine fish and shellfish: a review and commentary. Bull Mar Sci 87:971–1002. doi:10.5343/bms.2010.1051
Hedgecock D, Barber PH, Edmands S (2007) Genetic approaches to measuring connectivity. Oceanography 20:70–79
Hedrick PW (1999) Perspective: Highly variable loci and their interpretation in evolution and conservation. Evolution 53:313–318. doi:10.2307/2640768
Hellberg ME (1996) Dependence of gene flow on geographic distance in two solitary corals with different larval dispersal capabilities. Evolution 50:1167–1175. doi:10.2307/2410657
Hickey AJR, Lavery SD, Hannan DA, Baker CS, Clements KD (2009) New Zealand triplefin fishes (family Tripterygiidae): contrasting population structure and mtDNA diversity within a marine species flock. Mol Ecol 18:680–696. doi:10.1111/j.1365-294X.2008.04052.x
Hinnendael F (2008) Hemigrapsus crenulatus and H. sexdentatus. MSc. Leigh Marine Laboratory, Auckland
Hoelzel A, Green A (1998) PCR protocols and population analysis by direct DNA sequencing and PCR-based DNA fingerprinting. In: Hoelzel AR (ed) Molecular genetic analysis of populations: a practical approach. Oxford University Press, Oxford, pp 201–233
Holleley CE, Geerts PG (2009) Multiplex manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 46:511. doi:10.2144/000113156
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet. doi:10.1186/1471-2156-6-13
Johnson MS, Black R (1982) Chaotic genetic patchiness in an inter-tidal limpet, Siphonaria sp. Mar Biol 70:157–164
Johnson MS, Black R (1984) Pattern beneath the chaos—the effect of recruitment on genetic patchiness in an intertidal limpet. Evolution 38:1371–1383
Jones GP, Srinivasan M, Almany GR (2007) Population connectivity and conservation of marine biodiversity. Oceanography 20:100–111
Jones TC, Gemmill CEC, Pilditch CA (2008) Genetic variability of New Zealand seagrass (Zostera muelleri) assessed at multiple spatial scales. Aquat Bot 88:39–46. doi:10.1016/j.aquabot.2007.08.017
Kimura M (1980) A simple method for estimating evolutionary rates of base substitution through comparative studies of nucleotide sequences. Mol Evol 16:111–120
Kinlan BP, Gaines SD, Lester SE (2005) Propagule dispersal and the scales of marine community process. Divers Distrib 11:139–148. doi:10.1111/j.1366-9516.2005.00158.x
Lamare MD, Stewart BG (1998) Mass spawning by the sea urchin Evechinus chloroticus (Echinodermata: Echinoidea) in a New Zealand fiord. Mar Biol 132:135–140
Leis JM, Carson-Ewart BM (2001) Behaviour of pelagic larvae of four coral-reef fish species in the ocean and an atoll lagoon. Coral Reefs 19:247–257
Levin LA (2006) Recent progress in understanding larval dispersal: new directions and digressions. Integr Comp Biol 46:282–297. doi:10.1093/icb/024
Lewis PO, Zaykin D (2002) Genetic data analysis: computer program for the analysis of allelic data. Version 1.1. Free program distributed by the authors over the internet from http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php
Meirmans PG (2006) Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evol 60:2399–2402
Meirmans PG, Hedrick PW (2011) Assessing population structure: F(ST) and related measures. Mol Ecol Resour 11:5–18
Mercier A, Sewell MA, Hamel JF (2013) Pelagic propagule duration and developmental mode: reassessment of a fading link. Global Ecol Biogeogr 22:517–530. doi:10.1111/geb.12018
Mladenov PV, Allibone RM, Wallis GP (1997) Genetic differentiation in the New Zealand sea urchin Evechinus chloroticus (Echinodermata: Echinoidea). N Z J Mar Freshwat Res 31:261–269
Muths D, Jollivet D, Gentil F, Davoult D (2009) Large-scale genetic patchiness among NE Atlantic populations of the brittle star Ophiothrix fragilis. Aquat Biol 5:117–132. doi:10.3354/ab00138
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Olivares-Banuelos NC, Enriquez-Paredes LM, Ladah LB, De La Rosa-Velez J (2008) Population structure of purple sea urchin Strongylocentrotus purpuratus along the Baja California peninsula. Fish Sci 74:804–812. doi:10.1111/j.1444-2906.2008.01592.x
Palumbi SR, Grabowsky G, Duda T, Geyer L, Tachino N (1997) Speciation and population genetic structure in tropical Pacific Sea urchins. Evolution 51:1506–1517. doi:10.2307/2411203
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. doi:10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539 doi:10.1093/bioinformatics/bts460
Penant G, Aurelle D, Feral JP, Chenuil A (2013) Planktonic larvae do not ensure gene flow in the edible sea urchin Paracentrotus lividus. Mar Ecol Prog Ser 480:155. doi:10.3354/meps10194
Perrin C (2002) The effects of fiord hydography and environment on the population genetic structures of the sea urchin Evechinus chloroticus and the sea star Coscinasterias muricata in New Zealand. Marine Science and Zoology, Dunedin
Perrin C, Roy MS (2000) Rapid and efficient identification of microsatellite loci from the sea urchin, Evechinus chloroticus. Mol Ecol 9:2221–2223
Phillips NE, Shima JS (2006) Differential effects of suspended sediments on larval survival and settlement of New Zealand urchins Evechinus chloroticus and abalone Haliotis iris. Mar Ecol Prog Ser 314:149–158. doi:10.3354/meps314149
Piggott MP, Banks SC, Tung P, Beheregaray LB (2008) Genetic evidence for different scales of connectivity in a marine mollusc. Mar Ecol Prog Ser 365:127–136. doi:10.3354/meps07478
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rasmussen LL, Cornuelle BD, Levin LA, Largier JL, Di Lorenzo E (2009) Effects of small-scale features and local wind forcing on tracer dispersion and estimates of population connectivity in a regional scale circulation model. J Geophys Res Oceans. doi:10.1029/2008jc004777
Raymond M, Rousset F (1995) An exact test for population differentiation. Evolution 49:1280–1283
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225. doi:10.2307/2409177
Ross PM, Hogg ID, Pilditch CA, Lundquist CJ (2009) Phylogeography of New Zealand’s coastal benthos. N Z J Mar Freshwat Res 43:1009–1027
Ross PM, Hogg ID, Pilditch CA, Lundquist CJ, Wilkins RJ (2012) Population genetic structure of the New Zealand Estuarine Clam Austrovenus stutchburyi (Bivalvia: Veneridae) reveals population subdivision and partial congruence with biogeographic boundaries. Estuaries Coasts 35:143–154. doi:10.1007/s12237-011-9429-z
Sanford E, Kelly MW (2011) Local adaptation in marine invertebrates. In: Carlson CA, Giovannoni SJ (eds) Annual review of marine science, 3rd edn., pp 509–535. doi:10.1146/annurev-marine-120709-142756
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. doi:10.1038/72708
Shanks AL (2009) Pelagic larval duration and dispersal distance revisited. Biol Bull 216:373–385
Shanks AL, Grantham BA, Carr MH (2003) Propagule dispersal distance and the size and spacing of marine reserves. Ecol Appl 13:S159–S169
Shears NT, Smith F, Babcock RC, Duffy CAJ, Villouta E (2008) Evaluation of biogeographic classification schemes for conservation planning: application to New Zealand’s coastal marine environment. Conserv Biol 22:467–481. doi:10.1111/j.1523-1739.2008.00882.x
Sherwin WB, Jabot F, Rush R, Rossetto M (2006) Measurement of biological information with applications from genes to landscapes. Mol Ecol 15:2857–2869
Smith F (2004) Marine environment classification: physical influences on rocky reef assemblages in the Hauraki Gulf. Department of Conservation
Stevens MI, Hogg ID (2004) Population genetic structure of New Zealand’s endemic corophiid amphipods: evidence for allopatric speciation. Biol J Linn Soc 81:119–133. doi:10.1111/j.1095-8312.2004.00270.x
Treml EA, Halpin PN, Urban DL, Pratson LF (2008) Modeling population connectivity by ocean currents, a graph-theoretic approach for marine conservation. Landscape Ecol 23:19–36. doi:10.1007/s10980-007-9138-y
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. doi:10.1111/j.1471-8286.2004.00684.x
Veale AJ, Lavery SD (2011) Phylogeography of the snakeskin chiton Sypharochiton pelliserpentis (Mollusca: Polyplacophora) around New Zealand: are seasonal near-shore upwelling events a dynamic barrier to gene flow? Biol J Linn Soc 104:552–563. doi:10.1111/j.1095-8312.2011.01743.x
Veale AJ, Lavery SD (2012) The population genetic structure of the waratah anemone (Actinia tenebrosa) around New Zealand. N Z J Mar Freshwat Res 46:523–536. doi:10.1080/00288330.2012.730053
Ward RD, Holmes BH, O'Hara TD (2008) DNA barcoding discriminates echinoderm species. Mol Ecol Res 8:1202–1211. doi:10.1111/j.1755-0998.2008.02332.x
Walker JW (2007) Effects of fine sediments on settlement and survival of the sea urchin Evechinus chloroticus in northeastern New Zealand. Mar Ecol Prog Ser 331:109–118. doi:10.3354/meps331109
Waters JM, Roy MS (2004) Phylogeography of a high-dispersal New Zealand sea-star: does upwelling block gene-flow? Mol Ecol 13:2797–2806. doi:10.1111/j.1365-294X.2004.02282.x
Weersing K, Toonen RJ (2009) Population genetics, larval dispersal, and connectivity in marine systems. Marine Ecol Prog Ser 393:1–12. doi:10.3354/meps08287
Wei KJ, Wood AR, Gardner JPA (2013a) Population genetic variation in the New Zealand greenshell mussel: locus-dependent conflicting signals of weak structure and high gene flow balanced against pronounced structure and high self-recruitment. Mar Biol 160:931–949. doi:10.1007/s00227-012-2145-9
Wei KJ, Wood AR, Gardner JPA (2013b) Seascape genetics of the New Zealand greenshell mussel: sea surface temperature explains macrogeographic scale genetic variation. Mar Ecol Prog Ser 477:107–121. doi:10.3354/meps10158
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population-structure. Evolution 38:1358–1370
White C, Selkoe KA, Watson J, Siegel DA, Zacherl DC, Toonen RJ (2010) Ocean currents help explain population genetic structure. Proc Royal Soc B-Biol Sci 277:1685–1694. doi:10.1098/rspb.2009.2214
Will M, Hale ML, Schiel DR, Gemmell NJ (2011) Low to moderate levels of genetic differentiation detected across the distribution of the New Zealand abalone, Haliotis iris. Mar Biol 158:1417–1429. doi:10.1007/s00227-011-1659-x
Wright S (1951) The Genetical structure of populations. Annals of Eugenics 15:323–354
Wright S (1965) The interpretation of population-structure by F-statics with special regard to systems of mating. Evolution 19:395–420
Yearsley JM, Viard F, Broquet T (2013) The effect of collective dispersal on the genetic structure of a subdivided population. Evolution 67:1649–1659. doi:10.1111/evo.12111
Zulliger DE, Tanner S, Ruch M, Ribi G (2009) Genetic structure of the high dispersal Atlanto-Mediterreanean sea star Astropecten aranciacus revealed by mitochondrial DNA sequences and microsatellite loci. Mar Biol 156:597–610. doi:10.1007/s00227-008-1111-z
Acknowledgments
We would like to acknowledge all the assistance in collecting samples from the students and staff of the Molecular Ecology Laboratory and Marine Ecology Laboratory at the University of Auckland, thank you to S. Nagel, A. Veale, M. Hudson, R. Gallego, S. Knudson, E. Zarate, E. Baker, and S. Connell. We would also like to thank Dr. Mike Barker from the University of Otago, Dunedin and Phred Dobbins from the New Zealand Department of Conservation for obtaining samples from Stewart Island. This research was supported by the Faculty of Science, University of Auckland, and the Department of Conservation.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nagel, M.M., Sewell, M.A. & Lavery, S.D. Differences in population connectivity of a benthic marine invertebrate Evechinus chloroticus (Echinodermata: Echinoidea) across large and small spatial scales. Conserv Genet 16, 965–978 (2015). https://doi.org/10.1007/s10592-015-0716-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0716-2