
Abstract
Two regions of the mitochondrial genome (cytochrome oxidase I and ATPase 8–ATPase 6) were used to examine the population genetic structure of New Zealand’s endemic abalone (Haliotis iris). Samples were collected from 28 locations around New Zealand between January 2005 and February 2008. At least four phylogeographic breaks were present and occurred across the Chatham rise, in the western Cook Strait region, along the southeast coast of the South Island, and at East Cape in the North Island. Gene flow across the Chatham rise is probably limited due to infrequent dispersal across large geographic distances (~850 km), while factors limiting gene flow around the North and South Islands are less clear, and understanding these may require intense temporal and spatial sampling in complex hydrographic regions. High genetic diversity and weak genetic structure may be a general feature of abalone potentially reflecting large and/or ancient populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The interaction between a variety of extrinsic and intrinsic factors has been proposed to shape the genetic structure of marine invertebrates with bipartite life histories (reviewed in Palumbi 1994; Hellberg et al. 2002; Sponaugle et al. 2002). The biology of a species can help predict how it will respond to a particular environment, while past and present environmental features can help predict potential barriers and corridors to gene flow. However, neither biology nor New Zealand’s oceanography offer clear-cut predictions for the genetic structure of New Zealand’s black-foot abalone or paua (Haliotis iris).
Paua are long-lived marine gastropods that inhabit the intertidal and subtidal rocky reefs surrounding mainland and offshore islands and the Chatham Islands (Fig. 1). The commercial and cultural importance of paua has warranted much research regarding its biology. Paua are highly fecund broadcast spawners with a larval duration under 10 days (Tong et al. 1992). Spawning events, larval survival, and juvenile settlement and recruitment are variable and influenced by many abiotic and biotic factors (Poore 1973; Sainsbury 1982; Hooker and Creese 1995; McShane and Naylor 1995; Naylor and McShane 1997; Naylor and McShane 2001; Roberts et al. 2004; Phillips and Shima 2006).

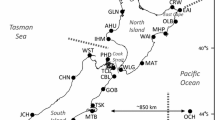
Cook Strait region and sampling locations. The box is an enlargement of the Cook Strait region. Shown are areas of upwelling proposed by Veale (2007), locations mentioned in text, and currents around the Cook Strait region. The flow and direction of the East Cape and Southland Currents are from Fig. 4 in Barnes (1985). The map of New Zealand shows the 28 sampling localities listed in Table 2. The location of the Chatham Islands sample (OCH; 44.01° S, 176.21° W) is not to scale and lies about 850 km off the east coast of New Zealand. The dashed lines labeled A, B, C, and D mark areas of genetic discontinuities identified using Barrier 2.2 (Manni et al. 2004). Thickness of the dashed lines denotes the percentage of resampled matrices identifying the barrier (supplementary information)
The numerous factors affecting spawning, larval survival, and juvenile settlement and survival result in variable recruitment over time and space, as observed by Sainsbury (1982). Potentially, this could result in patterns of genetic patchiness. On the other hand, the larvae are considered passive and as a result can be influenced by the local hydrodynamic environment (McShane 1992; Bohonak 1999). In general, population genetic research on abalone species has found either panmixia or slight differentiation, which was detectable with highly variable markers or a number of independent loci (e.g., Withler et al. 2003; Hara and Sekino 2005; Gruenthal et al. 2007). In several cases where pronounced differentiation was detectable with mitochondrial markers, phylogeographic breaks corresponded with present and past oceanography (e.g., Evans et al. 2004; Imron et al. 2007).
New Zealand’s marine environment is complex. Although New Zealand has six major offshore currents (summarized in Laing and Chiswell 2003), coastal circulation patterns are complicated with features like coastal topography, upwelling, eddies, and river plumes that could inhibit dispersal and/or promote local retention of larvae (Sponaugle et al. 2002; Schiel 2004). Arguably, the two most persistent features associated with genetic patterns of coastal marine invertebrates around New Zealand are (1) the large geographic distance (about 850 km) between the Chatham Islands and the mainland (North and South Islands; e.g., Goldstien et al. 2009) and (2) the Cook Strait region, an area encompassing the waterway separating the North and South Islands (e.g., Apte and Gardner 2002; Waters and Roy 2004; Goldstien et al. 2006; Veale 2007).
Although gene flow between the Chatham Islands and the mainland is most likely limited due to isolation by distance (Chiswell 2009), factors limiting gene flow across the Cook Strait region are unknown. Cook Strait was submerged 10,000–5,000 ya with the rise of sea levels at the end of the last ice age (Stevens et al. 1995). The present-day hydrography around the Cook Strait region (Fig. 1) is complex and involves the convergence of three offshore currents that vary in temperature and salinity (Heath 1970; Heath 1985), river discharge (Harris 1990), strong tidal flows and large amounts of tidal mixing (Heath 1978; Hume et al. 1992), and upwelling (Heath 1972; Bowman et al. 1983; Barnes 1985; Shirtcliffe et al. 1990). Potentially, present-day upwelling could limit larval dispersal and, therefore, gene flow (Star et al. 2003; Waters and Roy 2004; Veale 2007). Unfortunately whether upwelling really limits larval dispersal is debatable (Roughgarden et al. 1988; Poulin et al. 2002; Shanks and Brink 2005), and the upwelling hypothesis also does not reconcile well with attempts to date genetic disjunctions across the Cook Strait region (Apte and Gardner 2002; Goldstien et al. 2006).
Previous genetic studies on paua support differentiation between the Chatham Islands and the mainland (Frusin 1982; Smith and McVeagh 2006). However, genetic structure around the mainland remains inconclusive, in part due to a limited number of samples (<4), which do not span the geographic range of paua, and small sample sizes (<11). To elucidate paua genetic structure further, this study analyzed variation in two regions of the mitochondrial genome (COI and ATPase8–ATPase6) for samples from 28 locations around New Zealand to identify if genetic structure exists and, if it does, to determine the pattern, specifically genetic differentiation between the Chatham Islands and the mainland and between the North and South Islands.
Materials and methods
Samples
Between January 2005 and February 2008, foot or mantle tissue was collected from 13–24 paua from 28 locations (Fig. 1, Table 1). Paua were collected by a variety of means, including confiscated illegal catch, commercial catch, recreational catch, and scientific catch, and as a result, different tissues were available for different samples. Individual paua varied in size and therefore age (Naylor et al. 2007). All individuals were mature (i.e. had shell lengths ≥44 mm; Hooker and Creese 1995); however, not all individuals were larger than the minimum legal catch size (≥125 mm).
DNA extraction, PCR amplification, and sequencing
DNA was extracted from individuals collected at 27 of the 28 sites using Qiagen’s DNEasy® Blood & Tissue Kit. Abalone DNA from the remaining site (OPT) was extracted using a modified LiCl protocol (Gemmell and Akiyama 1996). Two regions of the mitochondrial genome were amplified: cytochrome oxidase I (mtCOI) and ATPase8–ATPase6. These are separated by 1526 bp in H. rubra (Maynard et al. 2005). Initially, a 581-bp fragment of mtCOI was amplified with primers F1 and R1 (Metz et al. 1998). However, due to inconsistent amplifications, two new internal mtCOI primers (mtCOI_F2 (5′-TTTAGGGGACGACCAACTGTA-3′) and mtCOI_R2 (5′-TACGGTCGGTTAGGAGCATT-3′)) were designed for paua using Primer3 (Rozen and Skaletsky 2000). These modified mtCOI primers amplified a 540-bp fragment. A 723-bp fragment of the ATPase8–ATPase6 region was amplified using primers COIIcons-F and H22-R1 (Maynard et al. 2005).
Both mtCOI and ATPase8–ATPase6 were amplified in 25 μL reaction volumes containing 1–40 ng of genomic DNA, 200 μM of dNTPs, 0.4 μM of each primer, 1.5 mM MgCl2, 1X NH4 Reaction Buffer (160 mM (NH4)2SO4, 670 mM Tris–HCl (pH 8.8 at 25°C), and 0.1% Tween-20), and 0.5 units BIOTAQ™ (Bioline). Thermal cycling profiles consisted of denaturation at 96°C for 2 min, 35 cycles of 96°C/20 s, 55°C (mtCOI) and 60°C (ATPase8–ATPase6)/30 s, 72°C/30 s (mtCOI) and 45 s (ATPase8–ATPase6), and a final cycle elongation step at 72°C for 7 min. Successful amplifications were purified according to manufacturer’s instructions using either a vacuum method with Eppendorf Perfectprep® PCR Cleanup 96 plates or a centrifugation method with PALL® AcroPrep™ 96-well Filter Plates.
Purified amplicons were directly sequenced with ABI Prism® Big Dye® Terminator v. 3.1 Cycle Sequencing Kit as per the manufacturer’s instructions but used at 0.125 the suggested volume of Big Dye® Terminator. Sequence products were purified using Sephadex™ GS-50 gel filtration (Amersham Bioscience) and run on an ABI3100 Genetic Analyzer at the University of Canterbury. Sequences were edited with Sequencher™ 4.2.2 (Gene Codes Corporation). Sequence alignment was done by hand using Se-Al v2.0a11 (Rambaut 2002), and all variable sites were confirmed by visual inspection of chromatograms. A total of 459 bp of mtCOI and 597 bp of ATPase8–ATPase6 were obtained from 534 out of 538 individuals.
Analyses
The mitochondrial regions were concatenated giving a total of 1056 bp for analyses. Sequence variation within samples was assessed with standard molecular indices calculated in Arlequin 3.5 (Excoffier et al. 2005). To evaluate similarity and differences among haplotypes for each fragment, percent divergences between haplotype pairs were calculated using maximum likelihood settings in Paup* (Swofford 1998). Maximum likelihood parameters were established separately for each mitochondrial region in jModelTest (Guindon and Gascuel 2003; Posada 2008). According to Akaike information criterion (Posada and Buckley 2004), the most appropriate model of sequence evolution was the Tamura and Nei (1993) model (TrN) with the proportion of invariant sites (I) equal to 0.7800 for the mtCOI region and the general time reversible model (GTR) with I equal to 0.5560 for ATPase 8–ATPase 6 region.
Haplotype networks were constructed to visually examine similarities and differences among haplotypes (Posada and Crandall 2001). Due to differences among network-building algorithms (Cassens et al. 2005), relationships between haplotypes were inferred with three frequently used network-building algorithms: median-spanning (Excoffier and Smouse 1994; implemented in Arlequin 3.5, Excoffier et al. 2005), median-joining (Bandelt et al. 1999; implemented in Network 4.2.0.1, Fluxus Technology Ltd.), and statistical parsimony (Templeton et al. 1992; implemented in TCS, Clement et al. 2000). Differences among haplotype networks were minor, and thus, only the statistical parsimony network is presented.
To test for the presence of a genetic split between (1) the Chatham Islands and the mainland (North and South Island), (2) the North Island and the South Island, and (3) the areas north and south of the upwelling regions (Fig. 1), analyses of molecular variance (AMOVAs), based on the number of pairwise differences, were employed (Excoffier et al. 1992). AMOVAs were calculated in Arlequin 3.5 (Excoffier et al. 2005), and significance tests used 16002 permutations. To identify alternative patterns, associations between genetic distance (ΦST) and both geographic distance and spatial geometry were examined. First, isolation by distance was tested using a Mantel test (Mantel 1967) calculated in Arlequin 3.5 (Excoffier et al. 2005). Coastal distances between locations were determined using ArcMap™ 9.1 (Environmental Systems Research Institute, Inc.) and Google™ Earth 5.2. Second, barriers representing areas of large genetic discontinuities between sampling locations were identified using Monmonier’s (1973) maximum difference algorithm implemented in Barrier 2.2 (Manni et al. 2004). Barrier 2.2 connects adjacent sampling locations using Delaunay triangulation. The default triangulation was manipulated as much as possible to reflect sampling along the entire coast of the North and South Islands (supplementary information). To evaluate the robustness of the predicted barriers, Monmonier’s (1973) maximum difference algorithm was also run on 100 resampled distance matrices. SEQBOOT (Felsenstein 2004) was used to generate 100 bootstrap replicates of the original data set, and these replicates were used to create 100 genetic distance matrices in Arlequin 3.5 (Excoffier et al. 2005).
Additional information about processes affecting genetic structure was inferred through mismatch distributions and neutrality tests (implemented in Arlequin 3.5, Excoffier et al. 2005). Mismatch distributions compared the observed numbers of pairwise differences between haplotypes with simulated data under two models of expansion: pure demographic expansion (Slatkin and Hudson 1991; Schneider and Excoffier 1999) and spatial expansion with migration (Ray et al. 2003; Excoffier 2004). The significance of the sum of squared deviations between the observed and expected mismatch distributions and the raggedness index (Harpending et al. 1993) were assessed with 10,000 parametric bootstraps. The presence of geographic structure within all samples and within groups limited the application of mismatch distributions to only the sampling locations. Tajima’s D (Tajima 1989) and Fu’s Fs (Fu and Li 1993) were used to assess sequence neutrality and mutation-drift equilibrium. Tajima’s D examines the relationship between the number of segregating sites and nucleotide diversity to test for deleterious alleles and balancing selection under the assumption of population equilibrium (Tajima 1989), while Fu’s Fs tests for the excess of recent mutations (indicative of population growth, hitchhiking, and background selection) via the relationship between the mean number of nucleotide differences and the number of alleles (Fu 1997).
Results
A 459-bp fragment of mtCOI and a 597-bp fragment of ATPase8–ATPase6 were amplified in 534 paua from the 28 locations (GenBank accession numbers for unique mtCOI sequences: JF441275-JF4411316 and unique ATPase8–ATPase6 sequences: JF4411317-JF4411424). The mtCOI fragment and the ATPase8–ATPase6 fragment corresponded to base pairs 3504–3612 and base pairs 5582–6178, respectively, in the H. rubra mitochondrial genome (ACCN: NC_0059400). As separate fragments, ATPase8–ATPase6 was more variable than mtCOI: it had a larger number of polymorphic sites, higher haplotype diversity, and greater nucleotide diversity (supplementary information). The mtCOI contained 33 polymorphic sites of which 13 were parsimony informative, while ATPase8–ATPase6 contained 96 polymorphic sites of which three had indels and 40 were parsimony informative. Percent pairwise divergence between haplotypes calculated using a maximum likelihood approach ranged from 0.22–1.31% for mtCOI and 0.17–2.02% for ATPase8–ATPase6.
The concatenated sequences (bp = 1056) contained 129 polymorphic sites (122 transitions, 9 transversions, 3 indels) of which 53 were parsimony informative. The overall haplotype diversity was 0.900 ± 0.008; otherwise, haplotype diversity ranged from 0.5824 (DSD)–0.9810 (SPB) within sampling locations (supplementary information). The overall nucleotide diversity was 0.004 ± 0.002, while nucleotide diversity ranged from 0.001 (MTB)–0.006 (IHM) within sampling locations (Table 2).
A total of 147 haplotypes were identified (Fig. 2). Only 23 haplotypes were shared among locations, and the remaining 124 haplotypes were private. Four haplotypes (numbered 9, 11, 18, and 19, Fig. 2) were identified in 50 or more individuals. In general, haplotypes were closely related as expected according to the low nucleotide diversity.

Statistical parsimony network. The network was constructed in TCS (Clement et al. 2000). Black represents South Island samples. White represents North Island samples. Light gray represents South Island samples (TCL and PHD) that are north of the upwelling areas (Fig. 1). Dark gray represents Chatham Island (OCH) individuals. Dashed lines represent indels and black tick marks represent missing haplotypes
Significant population genetic structure existed among all samples (ΦST = 0.039, p < 0.001). Grouping the locations according to structures proposed by Smith and McVeagh (2006) showed that Chatham Islands paua were distinct from North and South Island paua (ΦCT = 0.127, p = 0.033; Table 2). In addition, the frequency of the most common shared haplotypes (9, 11, 18, and 19) differed between the Chatham Islands and the mainland samples. Haplotypes 9, 11, and 19 were common, and haplotype 18 was rare in the mainland samples, while haplotype 18 was as common as haplotype 9, 11, and 19 in the Chatham Islands sample (Fig. 2).
After removing the Chatham Islands sample, significant population genetic structure still existed among the mainland samples (ΦST = 0.033, p < 0.001; Table 2). To test whether this mainland genetic structure was related to the Cook Strait region, further AMOVAs tested two different divisions around Cook Strait: (1) a split across Cook Strait and (2) a split across upwelling regions as proposed in Veale (2007). Both scenarios resulted in similar significant ΦCT indices (Table 2). However, significant differentiation still occurred between samples within groups (ΦSC). In both cases, the variance within sampling locations was around 95%.
To interpret the AMOVA results better, molecular indices for the above groupings and pairwise comparisons of ΦST were inspected (Table 3, Fig. 3). Noticeably, the number of haplotypes, the number of private haplotypes, and the haplotype diversities were larger for groups that contained North Island samples (Table 3). In fact, haplotype diversities between northern and southern groups differed by more than two standard deviations. The proposed groups also differed in the frequency of the most common shared haplotypes 9, 11, 18, and 19 (Fig. 2). For example, haplotype 18 was absent at nine North Island sampling locations, while it was only absent at one South Island sampling location. The pairwise ΦST showed that MTB and TIM (South Island), IHM and GLN (North Island), and Chatham Islands were the most divergent samples (Fig. 3). A larger proportion of significant comparisons (32%) occurred between North and South Island samples than between samples within either island (19% for the North Island and 18% for the South Island).

Pairwise ΦST (below the diagonal) for 28 sampling locations around New Zealand. Above the diagonal, a + indicates p < 0.05 and an open circle (○) indicates significance after Bonferroni correction (p = 0.00014). Samples are arranged first according to island and then roughly north to south along the west coast and north to south along the east coast of each island
A Mantel test indicated that there was isolation by distance among all samples (r2 = 0.221, p = 0.019). This was most likely due to the inclusion of the Chatham Islands sample, and removing the Chatham Islands sample from the test resulted in no significant relationship between genetic divergence and geographic distance among mainland samples (r2 = 0.141, p = 0.081). Among mainland samples, isolation by distance was identified for North Island paua (r2 = 0.280, p = 0.027) but not for South Island paua (r2 = −0.131, p = 0.963).
At least four areas of genetic differentiation were consistently found across the original data set and the 100 resampled matrices using Barrier 2.2 (Fig. 1 and supplementary information; Manni et al. 2004). Area A separated the Chatham Islands (OCH) from the east coast of the South Island (MTB, TIM, GOB) and was identified first (contained the maximum pairwise ΦST) in 100% of the resampled matrices. Two other areas, B and C, were identified among the top five potential barriers in ≥ 98% of the replicate data sets. Area B separated the southwest coast of the North Island (IHM and AHU) from the northwest coast of the South Island (WST and PHD), while area C separated the southeast of the South Island (NPT) from the east coast of the South Island (TIM). Area D occupied the northeast corner of the North Island with potential barriers occurring between EAI and OLB (supported with 99% of the resampled matrices), OLB and CRW (supported with 62% of the resampled matrices), and CRW and EAI (supported with 51% of the resampled matrices, supplementary information).
For comparison to groupings divided by Cook Strait, the sampling locations were partitioned into three groups (1. samples north of areas A and D, 2. samples south of areas A and D, 3. EAI), based on the areas of genetic differentiation predicted with Barrier 2.2 (Manni et al. 2004). Analyses using this grouping showed significant differentiation between groups (ΦCT) and no significant differentiation within groups (ΦSC, Table 2). Haplotype diversity was larger for the group of sampling locations north of areas A and D (Table 3), and common haplotypes 18 and 19 were absent from all sampling locations north of areas A and D. No isolation by distance was detected in either group.
Mismatch analyses of individuals grouped according to sampling locations could not reject models of demographic and range expansions. Modes were in the range of 0–4 and 7–9 pairwise differences, consistent with common haplotypes (9, 11, 18 and 19) being either 1–2 or 7–9 mutations apart (Fig. 2), and as a result, population parameters estimated from the mismatch distributions varied. For instance, coalescence time in mutational time units (τ) ranged from 0.313–9.852 for demographic expansions and 0.504–8.069 for spatial expansions (supplementary information). Although mismatch analyses suggested patterns of expansions, significance of Tajima’s D and Fu’s Fs varied according to sampling location (supplementary information). Tajima’s D and Fu’s Fs were negative and significant when all individuals were treated as a single group and for all North, South, and Chatham Island groupings (Table 3). Given that the mismatch analyses for sampling locations all supported models of expansions, and the large excess of recent mutations indicated by large negative Fu’s Fs and the star-shaped region of the haplotype network (Fig. 2), these significant negative values could indicate population expansion instead of selection on mtDNA.
Discussion
Paua’s relatively short larval duration (Tong et al. 1992) and passive larval stage (McShane 1992) suggested its population genetic structure would be vulnerable to features associated with population genetic structure identified in other New Zealand coastal marine invertebrates (Ross et al. 2009). Paua samples collected from around New Zealand had highly significant but modest genetic structure. All AMOVAs resulted in significant structure, suggesting that the Chatham Islands sample was different from the mainland samples and the northern samples were different from southern samples. Monmonier’s maximum difference algorithm identified additional potential genetic breaks at the southeast of the South Island and the northeast of the North Island.
The Chatham Islands vs. North and South Islands
The separation of the Chatham Islands sample from the North and South Island samples was consistent with Smith and McVeagh’s (2006) preliminary genetic study of paua. The differentiation between Chatham Islands and mainland paua was also consistent with the few intraspecific studies of New Zealand coastal marine invertebrates that have incorporated samples from the Chatham Islands (Smith et al. 1989; Clarke 2001; Goldstien et al. 2009). Differentiation between paua from the mainland and paua from the Chatham Islands probably resulted from isolation by distance.
The combination of moderate genetic structure and shared haplotypes supports a conclusion of very limited gene flow between the Chatham Islands and the mainland. Limited gene flow may occur via chance dispersals due to delayed metamorphosis. Roberts and Lapworth (2001) reported that paua larvae that underwent metamorphosis at 26 and 30 days could survive, but at reduced rates compared to larvae that underwent metamorphosis in under 10 days. This borders the lower limits of a 30–50 day period estimated for simulated passive planktonic larvae (or particles) traveling from the South Island to the Chatham Islands (in fact, the 10−4 percentile dispersal time was 27 days; Chiswell 2009).
North Island vs. South Island
The inclusion of more sampling locations, more individuals, and/or an additional 459 bp of mtCOI resulted in the detection of highly significant genetic structure among mainland samples that was not previously identified with mtDNA in Smith and McVeagh’s (2006) preliminary genetic study. AMOVAs rejected the hypothesis of homogeneity around the Cook Strait region, but unlike other New Zealand invertebrates, such as Sypharochiton pelliserpentis (Veale 2007), Cellana ornata (Goldstien et al. 2006), Patiriella regularis (Waters and Roy 2004; Ayers and Waters 2005), and Perna canaliculus (Apte and Garder 2002; Star et al. 2003), the structuring did not necessarily correspond to known regions of upwelling as proposed by Veale (2007).
The lack of a strong partition in pairwise ΦST and the low level of divergence (ΦST = 0.039) emphasized that paua do not have an obvious genetic structure, as compared to the level and pattern of genetic structures identified in C. ornata (ΦST = 0.829, Goldstien et al. 2006) and S. pelliserpentis (ΦST = 0.45, Veale 2007). Although ΦST values are not directly comparable, the level of differentiation in paua was more similar to species like P. regularis with a 9–10-week pelagic larval stage (ΦST = 0.072, Waters and Roy 2004) or P. canaliculus with a greater than 4-week pelagic larval stage (ΦST = 0.162, Apte and Gardner 2002). The lack of concordance in pattern and level of population differentiation among New Zealand coastal invertebrates suggest that the effects of potential barriers to gene flow in the Cook Strait are species-specific. Differences in larval behavior and life-history characteristics (Hedgecock1986; Bohonak 1999; Ross et al. 2009), ecology (Reid et al. 2006) and/or demography may limit how and when potential barriers of the Cook Strait region influence population genetic structure.
Additionally, a north–south split may not be an accurate description of paua genetic structure. At least three areas of reduced gene flow were evident around the mainland and only Area B corresponds with a portion of the Cook Strait region (Fig. 1). Area D, at the eastern promontory of the North Island, corresponds with the offshore divergence of the East Auckland Current and a series of offshore eddies that can impinge on the coastline to the south (Heath 1985, Chiswell and Roemmich 1998). Potentially, these features could sweep larvae offshore or entrain larvae beyond survival and, thereby, limit gene flow. Such a pattern of limited gene flow has been identified in two amphipods (Paracorophium spp., Stevens and Hogg 2004) and an anemone (Actinia tenebrosa, Veale 2007). Further, the presence of a barrier in this location is also supported by biogeographic data, with a strong biogeographic boundary observed in a range of taxa around East Cape (Powell 1961; Pawson 1961; Moore 1961; Francis 1996). Unlike Areas B and D, Area C does not correspond to an obvious hydrographic barrier, nor has it been identified in other organisms as a potential barrier to gene flow. The significant differentiation of samples MTB and TIM in this area from a large number of other samples may indicate the role of other processes such as local retention of larvae in shaping the genetic structure of paua.
Note, Monmonier’s (1973) maximum differentiation algorithm is considerably less accurate at identifying true population structure in instances of high levels of gene flow and/or low ratios of within to between group gene flow (Dupanloup et al. 2002), both or either of which may be the case with paua. Also although the correspondence of genetic breaks and contemporary hydrography has been emphasized, we cannot exclude other explanations for genetic structure such as past hydrography (e.g., Barber et al. 2006), the effects (i.e., genetic patchiness) of repeated variability of new recruits (Sainsbury 1982, Hedgecock 1994), and/or the potential non-neutrality (which could not be excluded using Tajima’s D and Fu’s Fs) mtDNA fragments (Meiklejohn et al. 2007). Testing concordance of the structure found here to that obtained with multiple neutral nuclear markers can help to address these alternatives.
Furthermore, issues such as small sample sizes, temporal variation in sampling, and the paua fishery may confound results. Small sample sizes may reflect statistical sampling errors in which the samples do not capture the true variation in a population (Holsinger and Weir 2009). Collecting a larger number of individuals from these locations would reduce these errors, and given the small pairwise ΦST values between sampling locations considerably larger samples (e.g., N = 100) would be needed (Kalinowski 2005). The paua were collected in just over a three-year period (January 2005–February 2008), so variation between samples may reflect temporal variation (e.g., due to variability in recruitment) rather than or in addition to spatial variation (e.g., Lee and Boulding 2007, 2009). Unfortunately, we do not have temporally varying samples from the same location to untangle temporal vs. spatial variation. Based on pairwise ΦST results, the effect of temporal variation seems small. The five locations with the earliest sample dates (GOB, DSD, CCB, NPT, MAT; sampled in January 2005) were not significantly different from the four locations (JCH, CHN, WAI, MHP) sampled over 2.5 years later. A clear temporal pattern could not be identified among significant comparisons that did not fit one of the north–south genetic splits examined in this paper. For example, the South Island sample MTB was significantly divergent from other South Island samples collected within a month (CCB, GOB, and NPT) to samples collected within 2.5 years (JCH), while it was not divergent from samples collected within 1 month (DSD and GOB) to samples collected within 2.75 years (CHN). The fact that the samples most likely contained a range of cohorts may dampen the effect of temporal variation resulting from variable recruitment (Hedgecock 1994). Finally, prolonged periods of intense fishing results in reduced genetic diversity and fishery-induced selection (Hauser et al. 2002; Allendorf et al. 2008). The amount of commercial fishing around New Zealand varies according to the estimated stock size of different regions and could potentially alter the genetic make-up of some samples. Teasing apart the influence of all these factors on the main pattern found here requires significant additional work that includes collecting a large number of samples from commercially fished, non-commercially fished, and marine preserves over a long time frame.
Noticeably, northern (lower latitude) samples had a larger number of private haplotypes and higher haplotype diversity; a pattern that has also been identified in the endemic cushion star (P. regularis, Ayers and Waters 2005). Such a pattern, combined with significant neutrality tests, suggests population expansion and mirrors a pattern indicative of climate-driven expansions observed in the northern hemisphere (Maggs et al. 2008). Although mismatch distributions support demographic and spatial expansion, they cannot clearly identify a common expansion time (supplementary information).
Similar to other abalone species (e.g., H. cracherodii, Gruenthal and Burton 2008; H. rufescens, Burton and Tegner 2000; H. midae, Evans et al. 2004; and H. rubra, Conod et al. 2002), paua mtDNA variation was characterized by high haplotype diversity resulting from a large number of rare haplotypes with few nucleotide differences (or low nucleotide diversity). Large levels of genetic variation can accumulate in populations that are ancient, occupy a diversity of niches, or have an increased mutation rate. Potentially, the large levels of diversity are indicative of large effective population sizes in abalone slowing the loss of genetic diversity due to genetic drift.
Conclusion
The mtDNA gene regions used in this study identified genetic differentiation among paua from around New Zealand. In general, the genetic splits between the Chatham Islands and the mainland and between northern and southern mainland samples are consistent with those identified in other coastal marine invertebrates (Ross et al. 2009), but the magnitude of differentiation in paua was lower. Although this may be a product of marker choice, paua could potentially have higher levels of gene flow or may have larger population sizes buffering against the effects of genetic drift. Further spatial and temporal sampling, as well as studies of adult and larval movement, is needed to elucidate processes occurring in the Cook Strait region, the northeast of the North Island, and the southeast of the South Island. Additional genetic structure among mainland samples may also exist (see Smith and McVeigh 2006), but will only be identified with more variable markers (Waples 1998, Kalinowski 2002).
References
Allendorf FW, England PR, Luikart G, Ritchie PA, Ryman N (2008) Genetic effects of harvest on wild animal populations. Trends Ecol Evol 23:327–337
Apte S, Gardner JPA (2002) Population genetic subdivision in the New Zealand greenshell mussel (Perna canaliculus) inferred from single-strand conformation polymorphism analysis of mitochondrial DNA. Mol Ecol 11:1617–1628
Ayers KL, Waters JM (2005) Marine biogeographic disjunction in central New Zealand. Mar Biol 147:1045–1052
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Barber PH, Erdmann MV, Palumbi SR (2006) Comparative phylogeography of three codistributed stomatopods: origins and timing of regional lineage diversification in the coral triangle. Evolution 60:1825–1839
Barnes EJ (1985) Eastern Cook Strait region circulation inferred from satellite-derived, sea-surface, temperature data. N Z J Mar Freshwater Res 19:405–411
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74:21–45
Bowman MJ, Kibblewhite AC, Murtagh RA, Chiswell SM, Sanderson BG (1983) Circulation and mixing in greater Cook Strait, New Zealand. Oceanol Acta 6:383–391
Burton RS, Tegner MJ (2000) Enhancement of red abalone Haliotis rufescens stocks at San Miguel Island: reassessing a success story. Mar Ecol Prog Ser 202:303–308
Cassens I, Mardulyn P, Milinkovitch MC (2005) Evaluating intraspecific “Network” construction methods using simulated sequence data: do existing algorithms outperform the global maximum parsimony approach? Syst Biol 54:363–372
Chiswell SM (2009) Colonisation and connectivity by intertidal limpets among New Zealand, Chatham and Sub-Antarctic Islands. II. Oceanographic connections. Mar Ecol Prog Ser 388:121–135
Chiswell SM, Roemmich D (1998) The East Cape current and two eddies: a mechanism for larval retention? N Z J Mar Freshwater Res 32:385–397
Clarke CB (2001) Growth and survival of Haliotis iris in northern New Zealand. Dissertation, University of Auckland, Auckland, New Zealand
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Conod N, Bartlett JP, Evans BS, Elliott NG (2002) Comparison of mitochondrial and nuclear DNA analyses of population structure in the blacklip abalone Haliotis rubra Leach. Mar Freshw Res 53:711–718
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Evans BS, Sweijd NA, Bowie RCK, Cook PA, Elliott NG (2004) Population genetic structure of the perlemoen Haliotis midae in South Africa: evidence of range expansion and founder events. Mar Ecol Prog Ser 270:163–172
Excoffier L (2004) Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol 13:853–864
Excoffier L, Smouse PE (1994) Using allele frequencies and geographic subdivision to reconstruct gene trees within a species: molecular variance parsimony. Genetics 136:343–359
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Felsenstein J (2004) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle
Francis MP (1996) Geographic distribution of marine reef fishes in the New Zealand region. N Z J Mar Freshwater Res 30:35–55
Frusin A (1982) Electrophoretic study of some paua (Haliotis iris) proteins. Master Thesis, Victoria University, Wellington, New Zealand
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Gemmell NJ, Akiyama S (1996) An efficient method for the extraction of DNA from vertebrate tissues. Trends Genet 12:338–339
Goldstien SJ, Schiel DR, Gemmell NJ (2006) Comparative phylogeography of coastal limpets across a marine disjunction in New Zealand. Mol Ecol 15:3259–3268
Goldstien SJ, Gemmell NJ, Schiel DR (2009) Colonisation and connectivity by intertidal limpets among New Zealand, Chatham and Sub-Antarctic Islands. I. Genetic connections. Mar Ecol Prog Ser 388:111–119
Gruenthal KM, Burton RS (2008) Genetic structure of natural populations of the California black abalone (Haliotis cracherodii Leach, 1814), a candidate for endangered species status. J Exp Mar Biol Ecol 355:47–58
Gruenthal KM, Acheson LK, Burton RS (2007) Genetic structure of natural populations of California red abalone (Haliotis rufescens) using multiple genetic markers. Mar Biol 152:1237–1248
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hara M, Sekino M (2005) Genetic difference between Ezo-awabi Haliotis discus hannai and Kuro-awabi H. discus populations: microsatellite-based population analysis in Japanese abalone. Fish Sci 71:754–766
Harpending HC, Sherry ST, Rogers AR, Stoneking M (1993) The genetic structure of ancient human populations. Curr Anthropol 34:483–496
Harris TFW (1990) Greater Cook Strait: form and flow. New Zealand Oceanographic Institute: DSIR Marine and Freshwater, Wellington, New Zealand
Hauser L, Adcock GJ, Smith PJ, Ramirez JHB, Carvalho GR (2002) Loss of microsatellite diversity and low effective population size in an overexploited population of New Zealand snapper (Pagrus auratus). Proc Natl Acad Sci USA 99:11742–11747
Heath RA (1970) Hydrology and circulation in central and southern Cook Strait, New Zealand. N Z J Mar Freshwater Res 5:178–199
Heath RA (1972) Oceanic upwelling produced by northerly winds on the north Canterbury coast, New Zealand. N Z J Mar Freshwater Res 6:343–351
Heath RA (1978) Semidiurnal tides in cook strait. N Z J Mar Freshwater Res 12:87–97
Heath RA (1985) A review of the physical oceanography of the seas around New Zealand–1982. N Z J Mar Freshwater Res 19:79–124
Hedgecock D (1986) Is gene flow from pelagic larval dispersal important in the adaptation and evolution of marine invertebrates. Bull Mar Sci 39:550–564
Hedgecock D (1994) Does variance in reproductive success limit effective population size of marine organisms? In: Beaumont RA (ed) Genetics and evolution of aquatic organisms. Chapman & Hall, London, pp 122–134
Hellberg ME, Burton RS, Neigel JE, Palumbi SR (2002) Genetic assessment of connectivity among marine populations. Bull Mar Sci 70:273–290
Holsinger KE, Weir BS (2009) Genetics in geographically structured populations: defining, estimating and interpreting FST. Nature Rev Genet 10:639–650
Hooker SH, Creese RG (1995) Reproduction of Paua, Haliotis iris Gmelin 1791 (Mollusca, Gastropoda), in north-eastern New Zealand. Mar Freshw Res 46:617–622
Hume TM, Bell RG, Delange WP, Healy TR, Hicks DM, Kirk RM (1992) Coastal oceanography and sedimentology in New Zealand, 1967–91. N Z J Mar Freshwater Res 26:1–36
Imron, Jeffrey B, Hale P, Degnan BM, Degnan SM (2007) Pleistocene isolation and recent gene flow in Haliotis asinina, an Indo-Pacific vetigastropod with limited dispersal capacity. Mol Ecol 16:289–304
Kalinowski S (2002) How many alleles per locus should be used to estimate genetic distances? Heredity 88:62–65
Kalinowski S (2005) Do polymorphic loci require large sample sizes to estimate genetic distances? Heredity 94:33–36
Laing A, Chiswell S (2003) The ocean medium. In: Andrew N, Francis M (eds) The living reef: the ecology of New Zealand’s rocky reefs. Craig Potton Publishing, Nelson, New Zealand, pp 24–31
Lee HJ, Boulding EG (2007) Mitochondrial DNA variation in space and time in the northeastern Pacific gastropod, Littorina keenae. Mol Ecol 16:3084–3103
Lee HJ, Boulding EG (2009) Spatial and temporal population genetic structure of four northeastern Pacific littorinid gastropods: the effect of mode of larval development on variation at one mitochondrial and two nuclear DNA markers. Mol Ecol 18:2165–2184
Maggs CA, Castilho R, Foltz D, Henzler C, Jolly MT, Kelly J, Olsen J, Perez KE, Stam W, Vainola R, Viard F, Wares J (2008) Evaluating signatures of glacial refugia for North Atlantic benthic marine taxa. Ecology 89:S108–S122
Manni F, Guerard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol 76:173–190
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Maynard BT, Kerr LJ, McKiernan JM, Jansen ES, Hanna PJ (2005) Mitochondrial DNA sequence and gene organization in Australian backup abalone Haliotis rubra (Leach). Mar Biotechnol 7:645–658
McShane PE (1992) Early life history of abalone: a review. In: Shepherd SA, Tegner MJ, Guzman Del Proo SA (eds) Abalone of the world: biology, fisheries and culture. Fishing News Books, Oxford, pp 120–138
McShane PE, Naylor JR (1995) Depth can affect postsettlement survival of Haliotis iris (Mollusca, Gastropoda). J Exp Mar Biol Ecol 187:1–12
Meiklejohn CD, Montooth KL, Rand DM (2007) Positive and negative selection on the mitochondrial genome. Trends Genet 23:259–263
Metz EC, Robles-Sikisaka R, Vacquier VD (1998) Nonsynonymous substitution in abalone sperm fertilization genes exceeds substitution in introns and mitochondrial DNA. Proc Natl Acad Sci USA 95:10676–10681
Monmonier M (1973) Maximum-difference barriers: an alternative numerical regionalization method. Geogr Anal 5:245–261
Moore LB (1961) Distribution patterns of New Zealand seaweeds. Tuatara 9:18–23
Naylor JR, McShane PE (1997) Predation by polychaete worms on larval and post-settlement abalone Haliotis iris (Mollusca: Gastropoda). J Exp Mar Biol Ecol 214:283–290
Naylor JR, McShane PE (2001) Mortality of post-settlement abalone Haliotis iris caused by conspecific adults and wave exposure. N Z J Mar Freshwater Res 35:363–369
Naylor JR, Manighetti BM, Neil HL, Kim SW (2007) Validated estimation of growth and age in the New Zealand abalone Haliotis iris using stable oxygen isotopes. Mar Freshw Res 58:354–362
Palumbi SR (1994) Genetic-divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst 25:547–572
Pawson DL (1961) Distribution patterns of New Zealand echinoderms. Tuatara 9:9–18
Phillips NE, Shima JS (2006) Differential effects of suspended sediments on larval survival and settlement of New Zealand urchins Evechinus chloroticus and abalone Haliotis iris. Mar Ecol Prog Ser 314:149–158
Poore GCB (1973) Ecology of New Zealand abalones, Haliotis species (Mollusca: Gastropoda) 4. Reproduction. N Z J Mar Freshwater Res 7:67–84
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Posada D, Buckley TR (2004) Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol 53:793–808
Posada D, Crandall KA (2001) Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 16:37–45
Poulin E, Palma AT, Leiva G, Narvaez D, Pacheco R, Navarrete SA, Castilla JC (2002) Avoiding offshore transport of competent larvae during upwelling events: the case of the gastropod Concholepas concholepas in Central Chile. Limnol Oceanogr 47:1248–1255
Powell AWB (1961) New Zealand biotic provinces. Tuatara 9:1–8
Rambaut A (2002) Se-Al v2.0a11 Carbon. University of Oxford, Oxford, UK
Ray N, Currat M, Excoffier L (2003) Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol 20:76–86
Reid DG, Lal K, Mackenzie-Dodds J, Kaligis F, Littlewood DTJ, Williams ST (2006) Comparative phylogeography and species boundaries in Echinolittorina snails in the central Indo-West Pacific. J Biogeogr 33:990–1006
Roberts RD, Lapworth C (2001) Effect of delayed metamorphosis on larval competence, and post-larval survival and growth, in the abalone Haliotis iris Gmelin. J Exp Mar Biol Ecol 258:1–13
Roberts RD, Kaspar HF, Barker RJ (2004) Settlement of abalone (Haliotis iris) larvae in response to five species of coralline algae. J Shellfish Res 23:975–987
Ross PM, Hogg ID, Pilditch CA, Lundquist CJ (2009) Phylogeography of New Zealand’s coastal benthos. N Z J Mar Freshwater Res 43:1009–1027
Roughgarden J, Gaines S, Possingham H (1988) Recruitment dynamics in complex life-cycles. Science 241:1460–1466
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ, pp 365–386
Sainsbury KJ (1982) Population dynamics and fishery management of the Paua, Haliotis iris.1. Population structure, growth, reproduction, and mortality. N Z J Mar Freshwater Res 16:147–161
Schiel DR (2004) The structure and replenishment of rocky shore intertidal communities and biogeographic comparisons. J Exp Mar Biol Ecol 300:309–342
Schneider S, Excoffier L (1999) Estimation of demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Shanks AL, Brink L (2005) Upwelling, downwelling, and cross-shelf transport of bivalve larvae: test of a hypothesis. Mar Ecol Prog Ser 302:1–12
Shirtcliffe TGL, Moore MI, Cole AG, Viner AB, Baldwin R, Chapman B (1990) Dynamics of the Cape Farewell upwelling plume, New Zealand. N Z J Mar Freshwater Res 24:555–568
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial-DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Smith PJ, McVeagh MS (2006) Genetic population structure of black-foot paua. Research report for contract SAP2005–01, Ministry of Fisheries. Wellington, New Zealand
Smith PJ, Macarthur GJ, Michael KP (1989) Regional variation in electromorph frequencies in the tuatua, Paphies subtriangulata, around New Zealand. N Z J Mar Freshwater Res 23:27–33
Sponaugle S, Cowen RK, Shanks A, Morgan SG, Leis JM, Pineda JS, Boehlert GW, Kingsford MJ, Lindeman KC, Grimes C, Munro JL (2002) Predicting self-recruitment in marine populations: biophysical correlates and mechanisms. Bull Mar Sci 70:341–375
Star B, Apte S, Gardner JPA (2003) Genetic structuring among populations of the greenshell mussel Perna canaliculus revealed by analysis of randomly amplified polymorphic DNA. Mar Ecol Prog Ser 249:171–182
Stevens MI, Hogg ID (2004) Population genetic structure of New Zealand’s endemic corophiid amphipods: evidence for allopatric speciation. Biol J Linn Soc Lond 81:119–133
Stevens GR, McGlone M, McCulloch B (1995) Prehistoric New Zealand. Reed Publishing (NZ) Ltd., Auckland, New Zealand
Swofford DL (1998) PAUP*, version 4b10: phylogenetic analysis using parsimony (*and other methods). Sinauer Associates, Sunderland
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence Data. 3. Cladogram estimation. Genetics 132:619–633
Tong LJ, Moss GA, Redfearn P, Illingworth J (1992) A manual of techniques for culturing paua, Haliotis iris, through to the early juvenile stage. MAF Technical Report No. 31. Ministry of Fisheries, Wellington, New Zealand
Veale AJ (2007) Phylogeography of two intertidal benthic marine invertebrates around New Zealand, the waratah anemone (Actinia tenebrosa) and the snakeskin chiton (Sypharochiton pelliserpentis). Master Thesis, University of Auckland, Auckland, New Zealand
Waples RS (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89:438–450
Waters JM, Roy MS (2004) Phylogeography of a high-dispersal New Zealand sea star: does upwelling block gene-flow? Mol Ecol 13:2797–2806
Withler RE, Campbell A, Li SR, Brouwer D, Supernault KJ, Miller KM (2003) Implications of high levels of genetic diversity and weak population structure for the rebuilding of northern abalone in British Columbia, Canada. J Shellfish Res 22:839–847
Acknowledgments
This work was funded by the New Zealand Ministry of Fisheries Tender GEN2007/A, the Minority International Research Training Program, and the University of Canterbury’s Targeted and Canterbury Scholarships. Amplifications were sequenced at the University of Canterbury Sequencing and Genotyping service. Thanks to the many people and companies who assisted collecting samples (in alphabetical order): Ed Arron, Andy Basset, Paul Buisson, Joe Burke, Burkhart Fisheries Ltd, Ra Clay, Christine Conway, Jeremy Cooper, Department of Conservation, Paul Ferguson, Sharyn Goldstien, Steve Ham, Hal Hovell, Peter Keesing, Ra Kohere, Warrick Lyons, Peter Molloy, Erica Mendietta, Ministry of Fisheries, Riverton Fisheries Ltd, Tammy Steeves, Gerard Prendeville, Top Cat Abalone & Venison Products, and Martin Williams.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by S. Uthicke.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Will, M., Hale, M.L., Schiel, D.R. et al. Low to moderate levels of genetic differentiation detected across the distribution of the New Zealand abalone, Haliotis iris . Mar Biol 158, 1417–1429 (2011). https://doi.org/10.1007/s00227-011-1659-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-011-1659-x