
Abstract
We measured within- and among-population genetic variation in the green sea urchin (Strongylocentrotus droebachiensis) at 11 sites in the north Atlantic and northeast Pacific by using four-locus microsatellite genotypes. We found no differentiation among populations from Atlantic Canada, but strong differentiation across the north Atlantic and between the Atlantic and Pacific samples. High inbreeding coefficients at three loci are consistent with high variance in reproductive success. One population that was recently decimated by disease was strongly differentiated from some others, but there was little differentiation otherwise among populations in Atlantic Canada. On a larger scale, populations in Atlantic Canada were more similar to a population from the north Pacific than to populations in the northwest Atlantic. Differentiation among populations at this large spatial scale is consistent with biogeographical hypotheses of: (1) Pleistocene population reduction and isolation in the northeast Atlantic, but (2) extinction in the northwest Atlantic followed by extensive recolonization from the Pacific. In contrast to other recent studies of trans-Atlantic organisms, we found no evidence of extensive gene flow across the north Atlantic.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Green sea urchins, Strongylocentrotus droebachiensis, are ecologically and economically important members of the shallow-water, marine ecosystem of northern oceans. In many parts of this wide geographical range, the population biology of these animals is characterized by large fluctuations in adult abundance that are associated with intensive grazing of kelps, disease outbreaks, episodic recruitment of planktonic larvae, and commercial harvesting practices (Chapman 1981; Pringle et al. 1982; Miller 1985; Johnson and Mann 1988; Scheibling and Raymond 1990; Hatcher and Hatcher 1997). Green sea urchins have a high-dispersal life-history syndrome characterized by high fecundity, free spawning with external fertilization, and a planktonic larval stage that lasts from 4 to 21 weeks (Strathmann 1978; Hart and Scheibling 1988). The frequency and intensity of larval recruitment is strongly influence by this lengthy planktonic period. For example, such larvae spawned off the coast of Nova Scotia may be capable of travelling >1,000 km before settlement, even in relatively slow surface currents such as the Nova Scotian Current, which moves at 0.05–0.10 m s−1 (Petrie 1987). The majority of larvae in the northwest Atlantic settle in the summer months, but settlement is known to be highly variable in both space and time. Balch and Scheibling (2000) report differences of several orders of magnitude in the interannual settlement rates for S. droebachiensis in shallow waters off the Atlantic coast of Nova Scotia, and similar patterns have been observed in the Gulf of Maine (Harris et al. 1994) and the Bay of Fundy (Balch et al. 1998). While settlement pulses have been shown to correlate with high sea-surface temperatures (Hart and Scheibling 1988), Balch and Scheibling (2000) suggest that reproductive output and larval survival may be more important than environmental factors (e.g. temperature) in the generation of annual variation in settlement rates.
Species with the high-dispersal life-history syndrome are expected to generally fit a model of panmixia, with little genetic divergence among populations compared to species with limited dispersal ability (see reviews by Burton 1983; Bohonak 1999). However, allozyme studies of marine invertebrates have demonstrated small but statistically significant levels of population subdivision without isolation by distance (e.g. limpets: Johnson and Black 1982, 1984a, 1984b; crown-of-thorns starfish: Benzie and Stoddart 1992; sea urchins: Marcus 1977, Watts et al. 1990; zebra mussels: Lewis et al. 2000). In these cases the genetic differences between neighboring populations often exceed those between more distant populations. Population genetic studies of both S. purpuratus using allozyme and mitochondrial DNA (mtDNA) (Edmands et al. 1996; Flowers et al. 2002) and S. franciscanus using allozymes (Edmands et al. 1996) and nuclear DNA sequence data (Debenham et al. 2000) have revealed similar patterns. The larger data sets analyzed in these studies improved the resolution of genetic differentiation and revealed patterns that were undetectable in previous analyses (e.g. Palumbi and Wilson 1990; Palumbi 1995).
Although the prolonged larval stage of S. droebachiensis is expected to facilitate high levels of gene flow, it is possible that other physical and biological features of the northwest Atlantic ecosystem may promote population subdivision. For example, commercial harvesting (Hatcher and Hatcher 1997) and disease outbreaks caused by the protozooan Paramoeba invadens (Scheibling and Hennigar 1997) can both cause severe local population reduction in shallow waters. These localized population bottlenecks are followed by recruitment of planktonic larvae, and such episodic changes in population size may have important population genetic consequences. One of our original goals was to characterize the population genetic effects (if any) of spatially and temporally patchy disease outbreaks followed by variable patterns of larval recruitment. Spatially and temporally variable recruitment patterns are one potential source of unexpected population genetic differentiation in species with lengthy planktonic larval stages (Johnson and Black 1984a).
Here, we present frequency data of four-locus microsatellite alleles for 11 S. droebachiensis populations spanning >6,000 km of the species' range. We focus on populations from the northwest Atlantic, because knowledge of the population biology and ecological interactions of sea urchins is particularly well studied in this region. We compare differentiation among populations in Atlantic Canada to differentiation across the north Atlantic and between the north Atlantic and the north Pacific. Unlike other studies of Strongylocentrotus population genetics, we found evidence of strong population differentiation only at the largest spatial scales. This evidence is consistent with biogeographic hypotheses of Pleistocene range expansion into the north Atlantic from the north Pacific.
Materials and methods
Field sampling
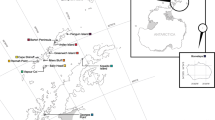
Tube foot or gonad tissue was obtained from Strongylocentrotus droebachiensis collected between 1999 and 2002 from ten sites throughout the north Atlantic (Fig. 1) and one site from the northeast Pacific. Sample sites were chosen to cover a wide portion of the species range in Atlantic Canada; specific sample locations were chosen based on accessibility and local knowledge of sea urchin occurrence. Two populations (at Bear Cove, west of Halifax) and East Jeddore (east of Halifax) were chosen because they experienced complete and nearly complete mortality, respectively, in 1995, as a result of disease outbreaks (Scheibling and Hennigar 1997). These two sites are <50 km apart (straight line distance).

Strongylocentrotus droebachiensis. Sampling locations of shallow-water populations in the northwest Atlantic Ocean. Arrows indicate average annual current directions and line thickness indicates average flow velocity (<0.005, 0.05–0.10, 2.5 m s−1). Populations are: Conception Bay (CBY), Bonne Bay (BBY), Harve St. Pierre (HSP), Miramichi Bay (MBY), Main-a-dieu (MAD), East Jeddore (JED), Bear Cove (BCV), and Digby (DBY). See Table 1 for exact population locations (Adapted with permission from the Nova Scotia Museum (http://museum.gov.ns.ca/mnh/nature/nhns/index.htm)
All samples were collected from depths ranging from 3 to 15 m using SCUBA. Sea urchins from San Juan Channel, Washington, were collected from about 50 m depth by dredge. Tissue samples were stored in 95% ethanol. The test diameters of individual sea urchins ranged from a minimum of 20 mm to a maximum 100 mm. We sampled a range of test diameters in order to sample sea urchins of different ages (and recruitment events). However, variation in test growth rate and asymptotic growth in older individuals led to a low correlation between test diameter and age within and among some populations (Meidel and Scheibling 1998).
Molecular analysis
Genomic DNA from some sea urchins was extracted from tube feet by incubating a single sucker in 20 μl of distilled deionized water (ddH2O) with 10 μg proteinase K (Qiagen) for 1 h at 65°C. Samples were then incubated for 5 min at 80°C, and 1 μl of this digest was used as template for each polymerase chain reaction (PCR). For most sea urchins, genomic DNA was isolated from gonad tissue using a cetyltrimethylammonium bromide (CTAB) buffer and phenol-chloroform extraction (Grosberg 1996). The two different extraction methods produced identical microsatellite genotypes. A total of 966 sea urchins (mean=88 per population; range=37–110) were characterized with the four microsatellite markers described by Addison and Hart (2002). The PCR products were amplified using the Stratagene Robocycler and MJ PTC-100 thermal cyclers. Fragment sizes were resolved on a Li-Cor DNA 4200, following the methodology of Addison and Hart (2002).
Data analysis
Allele frequencies, observed (H O) and expected heterozygosity (H E), and exact tests for the conformance to Hardy–Weinberg expectations (HWE) were calculated using the web version of GENEPOP (version 3.1; Raymond and Rousset 1995). Linkage disequilibrium was calculated using Fisher's exact test by permuting all two-locus genotypes within all populations using Genetic Data Analysis (GDA version 1.0; Lewis and Zaykin 2001). Allelic richness was calculated for each population using FSTAT (Goudet 2001) for the three loci in which no missing values were found.
To assess the levels of genetic differentiation within and among populations F-statistics (F IS, F IT, F ST) were calculated for the eight northwest Atlantic populations and again for all 11 populations throughout the species range using GDA; 95% confidence intervals were calculated for the F-statistics by bootstrapping across loci (number of replications=10,000) using GDA. Pairwise F ST values were computed for all northwest Atlantic populations. A dendogram based on coancestry distances computed by GDA was drawn using PAUP 4.0b10 (Swofford 2002). Pairwise tests of multi-locus genotypic differentiation were conducted for all northwest Atlantic populations with randomization procedures and G-tests for differentiation using FSTAT.
Results
Genetic variation within populations
All northwest Atlantic populations and the one Pacific population had high levels of genetic variability, with many alleles at each locus and high observed heterozygosities (Table 1). The two northeast Atlantic populations (Iceland and Norway) had fewer alleles at each locus, and the locus Sd76 failed to amplify in all individuals from these two populations. Allelic richness in Iceland and Norway varied across loci from 3.69 to 7.19 (allelic richness was not calculated for Sd76 because no alleles were amplified from northeast Atlantic samples). Allelic richness in all other population samples was 10.04–18.68. Significant departures from Hardy–Weinberg expectations were detected in 22 of the 42 possible tests (Table 1), and 18 of these were statistically significant after a tablewide Bonferroni correction for multiple tests (Rice 1989). Positive F IS values indicate the departure from Hardy–Weinberg expectations are a result of an excess in homozygosity. Jackknifing over all populations indicates that departures from Hardy–Weinberg expectations (F IS±SD) were significant for Sd156 (0.1878±0.0237), Sd76 (0.2302±0.0226), and Sd63 (0.1654±0.0322), but not for Sd121 (0.0095±0.0144). We have detected null alleles segregating in laboratory families at Sd156, but not at other loci (J. Ford, unpublished data). However, the widespread departures from Hardy–Weinberg expectations are not thought to be caused by null alleles because null/null homozygotes were conspicuously rare in this data set. Homozygote and heterozygote genotypes were easy to distinguish on gel images. Only one individual (from Digby) failed to amplify at one locus (Sd76), but PCR products were obtained for all remaining individuals in the northwest Atlantic. Failure rates were higher for all loci in the Pacific samples, but this could be due to the poor quality and limited quantity of these tissue samples. Null alleles were probably not common in this Pacific population, because we detected significant heterozygote deficits at only one of four loci (compared to two or three significant deficits for most northwest Atlantic populations in which null homozygotes were completely absent). No linkage disequilibrium was detected between any loci in any of the populations (data not shown).
Genetic variation among populations
We found no statistically significant genetic variation among populations in the northwest Atlantic (F ST=0.0016, P>0.05; Table 2). When we included all 11 populations sampled throughout the Atlantic and Pacific Oceans, the mean F ST increased to 0.0870 (P<0.05; Table 2). This value was significantly different from zero, and its magnitude suggests a moderate amount of genetic subdivision at this large (>6,000 km) spatial scale. When we removed Sd76 from the analysis, the resulting mean F ST based on the three-locus genotypes was reduced to 0.0625 (P<0.05; Table 2).
In addition to these quantitative population differences, the failure of Sd76 to amplify in the northeast Atlantic populations suggests that there has been little recent gene flow across the north Atlantic Ocean; all northeast Atlantic individuals were null homozygotes at this locus, and we found only one null homozygote at this locus in all other populations. Any significant flux of larvae from west to east should introduce amplifiable Sd76 alleles into Iceland and Norway. Migration from east to west (Wares and Cunningham 2001) could introduce null alleles into northwest Atlantic populations, and it is possible that such null alleles contribute to the deficit of Sd76 heterozygotes in Atlantic Canada (see "Discussion"). However, such migration does not appear to explain heterozygote deficits at other loci in the same populations.
The UPGMA (unweighted pair-group method using arithmetic averages) tree based on pairwise F ST values among all 11 populations clearly indicates the genetic subdivision between the northwest and northeast Atlantic populations of sea urchins, and the close relationship between the Pacific sample and those from the northwest Atlantic (Fig. 2). Pairwise tests of genotypic differentiation revealed three cases in which two populations had significantly different genotype frequencies after a Bonferroni correction (Table 3). These three cases all involved the population sampled from East Jeddore, which experienced nearly complete mortality as a result of a disease epidemic in 1995 (Scheibling and Hennigar 1997), and pairwise differences were marginally non-significant in three of the other four comparisons involving this population. No similar patterns were observed for samples collected at Bear Cove, which experienced complete mortality in 1995. These differences, therefore, provide no consistent evidence of an effect of disease-induced population fluctuations on population genetic differentiation.

Strongylocentrotus droebachiensis. UPGMA (unweighted pair-group methods using arithmetic averages) dendrogram of all 11 populations based on four-locus microsatellite coancestry distances (pairwise F ST), calculated using GDA (Genetic Data Analysis). A dendrogram based on Nei's D has the same topology and similar branch lengths
Discussion
Variation within the northwest Atlantic
The populations of green sea urchins (Strongylocentrotus droebachiensis) sampled throughout the northwest Atlantic Ocean are genetically homogeneous (F ST is not significantly different from zero). This is consistent with a high level of gene flow or a low rate of genetic drift, and suggests that populations on this spatial scale act as one large interbreeding unit. Samples collected from disease-affected populations (East Jeddore and Bear Cove) only 50 km apart had the highest pairwise genetic differences among all the comparisons made. These patterns are similar to the "chaotic genetic patchiness" observed for a variety of benthic marine invertebrates with the high-dispersal life-history syndrome (e.g. Johnson and Black 1982; see review by Hellberg et al. 2002). In these cases populations were genetically homogeneous over a large spatial scale, but included some statistically significant small-scale genetic heterogeneity. In our study, one population with a recent history of disease outbreak and population decline (East Jeddore) was genotypically distinct in pairwise comparisons, but others (e.g. Bear Cove) were not (Scheibling and Hennigar 1997). Some strong differences among populations recently affected by disease may reflect sporadic and unpredictable recruitment (Harris et al. 1994; Balch et al. 1998) and lack of older individuals at these sites. However, our data so far do not strongly support the hypothesized effect of population fluctuation on local population genetic differentiation. The effect of population decline and subsequent larval recruitment on genetic differentiation requires more detailed studies of the genetic composition of new recruits over several recruitment events.
Although mtDNA and sperm bindin sequence polymorphisms suggest genetic homogeneity of Strongylocentrotus populations on the west coast of North America (Palumbi and Wilson 1990; Debenham et al. 2000; Flowers et al. 2002), the lack of genetic substructure in our study is surprising, because the allozyme studies of sea urchins sampled at similar spatial scales have revealed significant levels of genetic heterogeneity (Marcus 1977; Watts et al. 1990; Edmands et al. 1996; Mladenov et al. 1997; Moberg and Burton 2000) using genetic markers that were much less polymorphic. Moberg and Burton (2000) reported F ST=0.033 for S. franciscanus adults from the eastern Pacific coast of North America. Remarkably, Edmands et al. (1996) reported the same value (F ST=0.033) for S. purpuratus adults sampled from the same coastline. Both estimates were significantly greater than zero. The nominal level of subdivision in these two species is much larger than that found in our study (F ST=0.0016; Table 2). Because all three species share similar larval biology and ecology, we expected to detect similar levels of genetic substructure. Burton (1983) suggested that, where the potential for extensive larval dispersal exists, populations should not have the capacity to differentiate on the scale of typical larval dispersal, and thus significant genetic subdivision at or below this geographic scale must be driven by natural selection. Differences in oceanographic conditions and patterns of surface currents on each coast may be responsible for the differences in population structure, but the potential influence of natural selection in the allozyme studies or of non-equilibrium population processes in our study cannot be ruled out.
Population genetic studies of cod (Gadus morhua; Bentzen et al. 1996; Pogson et al. 2001; Beacham et al. 2002; Knutsen et al. 2003) and herring (Clupea harengus; McPherson et al. 2001) in the northwest Atlantic using microsatellite markers have also demonstrated small but significant levels of genetic subdivision. However, microsatellite analysis of scallops (Herbinger et al. 1998) and barnacles (Dufresne et al. 2002) throughout this same region have failed to reveal any significant genetic heterogeneity. The consistent lack of genetic structure among these studies and in our own study suggest that the passive larval dispersal and sedentary adult lifestyle of many marine invertebrates may limit the formation of population substructure. In contrast, behavioral associations in fish, such as schooling or spawning-site fidelity, may be responsible for the formation of the genetically distinct assemblages that are frequently detected.
Heterozygote deficits
Populations sampled in the northwest Atlantic had high inbreeding coefficients (mean F IS=0.1499, P<0.05), and consistent heterozygote deficits were detected for three of the four loci used (Sd156, Sd76, and Sd63; Tables 1, 2). It is unlikely that null alleles are the cause of these deviations, because only one possible null homozygote was detected among 797 individuals screened. Inbreeding is also an unlikely explanation, because high dispersal is predicted to reduce the probably of mating among close relatives (reviewed by Bohonak 1999). While a correlation between allele frequencies at neutral loci and some allozyme alleles under selection has been demonstrated in acorn barnacles (Semibalanus balanoides) (Dufresne et al. 2002), possible selective forces that could act on linked loci in the green sea urchin across the wide geographic range sampled in this study are unknown. Selection could generate heterozygote deficiencies (e.g. Zouros and Foltz 1984), but selection is an unlikely explanation of the high inbreeding coefficients detected at multiple loci in green sea urchins.
An alternate explanation for these high inbreeding coefficients is based on the hypothesis that populations of marine free spawners experience a large variance in reproductive success (Hedgecock 1994). The chance matching of reproductive effort to oceanographic conditions that promote fertilization, growth, development, and settlement of larvae is likely a random process, in which the likelihood of reproductive success is independent of genotype. The two main predictions of this model are that cohorts should be genetically differentiated from each other, and each cohort should have reduced allelic diversity because few adults in the population contributed successfully to the resulting pool of offspring (Hedgecock 1994). If cohorts are temporally and spatially differentiated, then an age-structured population should demonstrate unrecognized temporal structure as a kind of Wahlund effect. Since the spawning populations sampled in our study are genetically homogeneous, the large deficits in heterozygotes are unlikely due to a spatial Wahlund effect. Instead, we suggest that our results are consistent with the prediction that large deficits in heterozygotes result from a temporal Wahlund effect caused by a large variance in reproductive success. Supporting evidence for this argument is the observation of small-scale genetic differentiation between geographically close populations in an otherwise panmictic population. It is unlikely that temporal variation in the sources of new recruits could alone account for small-scale genetic differentiation between East Jeddore and Bear Cove. The observation of equilibrium genotype frequencies in northeast Atlantic populations is not consistent with the hypothesis of reproductive variance, and suggests either that these populations experience reproductive conditions that promote low variance in reproductive success or that the temporal Wahlund effect is not a general explanation of within-population genetic variation in these and other sea urchins (e.g. Flowers et al. 2002).
Population structure across the species range
The significant F ST of 0.0870 (P<0.05) that was calculated among all 11 populations suggests that geographic subdivision in this species is manifest only on a very large spatial scale. The failure of Sd76 to amplify in the northeast Atlantic populations further suggests that these populations are not currently connected by gene flow to populations in the northwest Atlantic. The failure of this locus to amplify in some populations also highlights the potential pitfalls of employing hyper-variable markers such as microsatellites across a broad geographic range, in which there are potentially deep divergences between populations that are conspecific but genetically isolated from each other on large spatial and temporal scales.
Fossil records indicate that S. droebachiensis invaded the Atlantic from the Pacific after the Bering Seaway opened 3.5 million years ago (Durham and MacNeil 1967). It has been suggested that, as a result of more recent climate change, north Atlantic populations may have undergone recent extinctions or severe bottlenecks followed by reinvasion from the Pacific. Both Palumbi and Wilson (1990) and Palumbi and Kessing (1991) suggest recent, but not continuous, migration of S. droebachiensis from the Pacific to the northwest Atlantic in the last 300,000–90,000 years. Extensive outflow of freshwater along the Arctic coast of Siberia is thought to have restricted the trans-Arctic interchange to the Canadian Arctic but not the Eurasian Arctic (Gladenkov 1979). There is evidence that the magnitude of the extinctions caused by Pleistocene glacial advances were less severe in the northeast Atlantic than in the northwest Atlantic, because of the presence of rocky substratum offering glacial refugia to the south (Vermeij 1979). Our results are consistent with these hypotheses of historical phylogeography. The lack of amplification at Sd76, as well as the reduced number of alleles in the northeast Atlantic, suggests that these populations experienced a severe bottleneck (and became fixed for one or more null alleles at Sd76), but were not recently recolonized from the Pacific. In spite of the low genetic diversity within both of these populations at all loci (4–8 alleles, compared to 12–22 alleles in all other populations), we found no heterozygote deficits in the northeast Atlantic. Both of these patterns are consistent with a historical population reduction followed by restoration of equilibrium genotype frequencies in the absence of detectable gene flow from the Pacific or the northwest Atlantic population. In contrast, the relatively high polymorphism in the northwest Atlantic and genetic similarity to the Pacific population indicate either glacial refugia or extinction followed by more recent and extensive recolonization from the Pacific. The failure to find genetic evidence of glacial refugia in several other obligate rocky substratum inhabitants in the northwest Atlantic supports the local extinction argument (Wares and Cunningham 2001). However, contrary to Wares and Cunningham (2001), our results do not suggest recent recolonization from northeast Atlantic populations. Evidence of such a recolonization event would include lower diversity of alleles in the northwest Atlantic compared to the northeast Atlantic, a pattern opposite to our observations.
References
Addison JA, Hart MW (2002) Characterization of microsatellite loci in sea urchins (Strongylocentrotus spp.). Mol Ecol Notes 2:493–494
Balch T, Scheibling RE (2000) Temporal and spatial variability in settlement and recruitment of echinoderms in kelp beds and barrens in Nova Scotia. Mar Ecol Prog Ser 205:139–154
Balch T, Scheibling RE, Harris LG, Chester CM, Robinson SMC (1998) Variation in settlement of Strongylocentrotus droebachiensis in the northwest Atlantic: effects of spatial scale and sampling method. In: Mooi R, Telford M (eds) Echinoderms. Balkema, Rotterdam, pp 555–560
Beacham TD, Brattey J, Miller KM, Le KD, Withler RE (2002) Multiple stock structure of Atlantic cod (Gadus morhua) off Newfoundland and Labrador determined from genetic variation. ICES J Mar Sci 59:650–655
Bentzen P, Taggart CT, Ruzzante DE, Cook D (1996) Microsatellite polymorphism and the population structure of Atlantic cod (Gadus morhua) in the northwest Atlantic. Can J Fish Aquat Sci 53:2706–2721
Benzie JAH, Stoddart JA (1992) Genetic structure of outbreaking and non-outbreaking crown-of-thorns starfish (Acanthaster planci) populations on the Great Barrier Reef. Mar Biol 112:119–130
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74:21–45
Burton RS (1983) Protein polymorphisms and genetic differentiation of marine invertebrate populations. Mar Biol Lett 4:193–206
Chapman ARO (1981) Stability of sea urchin dominated barren grounds following destructive grazing of kelp in St. Margaret's Bay, eastern Canada. Mar Biol 62:307–311
Debenham P, Brzezinski M, Foltz K, Gaines S (2000) Genetic structure of populations of the red sea urchin, Strongylocentrotus franciscanus. J Exp Mar Biol Ecol 253:49–62
Dufresne F, Bourget E, Bernatchez L (2002) Differential patterns of spatial divergence in microsatellite and allozyme alleles: further evidence for locus-specific selection in the acorn barnacle, Semibalanus balanoides? Mol Ecol 11:113–123
Durham JW, MacNeil FS (1967) Cenozoic migrations of marine invertebrates through the Bering Strait region. In: Hopkins DM (ed) The Bering land bridge. Stanford University Press, Stanford, Calif., pp 326–349
Edmands S, Moberg PE, Burton RS (1996) Allozyme and mitochondrial DNA evidence of population subdivision in the purple sea urchin Strongylocentrotus purpuratus. Mar Biol 126:443–450
Flowers JM, Schroeter SC, Burton RS (2002) The recruitment sweepstakes has many winners: genetic evidence from the sea urchin Strongylocentrotus purpuratus. Evolution 56:1445–1453
Gladenkov YB (1979) Cenozoic molluscan assembledges in northern regions of the Atlantic and Pacific Oceans. Int Geol Rev 21:880–890
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html
Grosberg RK (1996) Simple extractions and RAPD-PCR protocols. In: Ferraris JD, Palumbi SR (eds) Molecular zoology: advances, strategies, and protocols. Wiley-Liss, New York, pp 470–473
Harris LG, Rice B, Nestler EC (1994) Settlement, early survival and growth in a southern Gulf of Maine population of Strongylocentrotus droebachiensis (Müller). In: David B, Guille A, Feral J-P, Roux M (eds) Echinoderms through time. Balkema, Rotterdam, pp 701–706
Hart MW, Scheibling RE (1988) Heat waves, baby booms, and the destruction of kelp beds by sea urchins. Mar Biol 99:167–176
Hatcher BG, Hatcher AI (1997) Research directions and management options for sea urchin culture in Nova Scotia. Bull Aquacult Assoc Can 97:62–65
Hedgecock D (1994) Does variance in reproductive success limit effective population sizes of marine organisms? In: Beaumont AR (ed) Genetics and evolution of aquatic organisms. Chapman and Hall, London, pp 122–134
Hellberg ME, Burton RS, Neigel JE, Palumbi SR (2002) Genetic assessment of connectivity among marine populations. Bull Mar Sci 70[Suppl]:273–290
Herbinger CM, Vercaemer BM, Gjetvaj B, O'Dor RK (1998) Absence of genetic differentiation among geographically close sea scallop (Placopecten magellanicus) beds with cDNA and microsatellite markers. J Shellfish Res 17:117–122
Johnson CR, Mann KH (1988) Diversity, patterns of adaptation, and stability of Nova Scotia kelp beds. Ecol Monogr 58:129–154
Johnson MS, Black R (1982) Chaotic genetic patchiness in an intertidal limpet, Siphonaria sp. Mar Biol 70:157–164
Johnson MS, Black R (1984a) Pattern beneath the chaos: the effect of recruitment on genetic patchiness in an intertidal limpet. Evolution 38:1371–1383
Johnson MS, Black R (1984b) The Wahlund effect and the geographical scale of variation in the intertidal limpet Siphonaria sp. Mar Biol 79:295–302
Knutsen H, Jorde PE, Andre C, Stenseth NC (2003) Fine-scaled geographical population structuring in a highly mobile marine species: the Atlantic cod. Mol Ecol 12:385–394
Lewis KM, Feder JL, Lamberti GA (2000) Population genetics of the zebra mussel, Dreissena polymorpha (Pallas): local allozyme differentiation within midwestern lakes and streams. Can J Fish Aquat Sci 57:637–643
Lewis PO, Zaykin D (2001) Genetic Data Analysis: computer program for the analysis of allelic data, version 1.0 (d16c). Free program distributed by the authors via internet from http://lewis.eeb.uconn.edu/lewishome/software.html
Marcus NH (1977) Genetic variation within and between geographically separated populations of the sea urchin, Arbacia punctulata. Biol Bull (Woods Hole) 153:560–576
McPherson AA, Stephenson RL, O'Reilly PT, Jones MW, Taggart CT (2001) Genetic diversity of coastal northwest Atlantic herring populations: implications for management. J Fish Biol 59:356–370
Meidel SK, Scheibling RE (1998) Size and age structure of the sea urchin Strongylocentrotus droebachiensis in different habitats. In: Mooi R, Telford M (eds) Echinoderms. Balkema, Rotterdam, pp 737–742
Miller RJ (1985) Succession in sea urchin abundance in Nova Scotia, Canada. Mar Biol 84:275–286
Mladenov PV, Allibone RM, Wallis GP (1997) Genetic differentiation in the New Zealand sea urchin Evechinus chloroticus (Echinodermata: Echinoidea). NZ J Mar Freshw Res 31:261–269
Moberg PE, Burton RS (2000) Genetic heterogeneity among adult and recruit red sea urchins, Strongylocentrotus franciscanus. Mar Biol 136:773–784
Palumbi SR (1995) Using genetics as an indirect estimator of larval dispersal. In: McEdward LR (ed) Ecology of marine invertebrate larvae. CRC Press, Boca Raton, Fla., pp 369–387
Palumbi SR, Kessing BD (1991) Population biology of the trans-Arctic exchange: mtDNA sequence similarity between Pacific and Atlantic sea urchins. Evolution 45:1790–1805
Palumbi SR, Wilson AC (1990) Mitochondrial DNA diversity in the sea urchins Strongylocentrotus purpuratus and S. droebachiensis. Evolution 44:403–415
Petrie B (1987) Undulations of the Nova Scotia current. Atmos-Ocean 25:1–9
Pogson GH, Taggart CT, Mesa KA, Boutilier RG (2001) Isolation by distance in the Atlantic cod, Gadus morhua, at large and small spatial scales. Evolution 55:131–146
Pringle JD, Sharp GJ, Caddy JF (1982) Interactions in kelp bed ecosystems in the northwest Atlantic: review of a workshop. In: Mercer MC (ed) Multispecies approaches to fisheries management advice. Can Spec Publ Fish Aquat Sci 59:108–115
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rice WR (1989) Analysing tables of statistical tests. Evolution 43:223–225
Scheibling RE, Hennigar AW (1997) Recurrent outbreaks of disease in sea urchins Strongylocentrotus droebachiensis in Nova Scotia: evidence for a link with large-scale meteorologic and oceanographic events. Mar Ecol Prog Ser 152:155–165
Scheibling RE, Raymond BG (1990) Community dynamics on a subtidal cobble bed following mass mortalities of sea urchins. Mar Ecol Prog Ser 63:127–145
Strathmann RR (1978) Length of pelagic period in echinoderms with feeding larvae from the northwest Pacific. J Exp Mar Biol Ecol 34:23–27
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony (* and other methods), version 4. Sinauer, Sunderland, Mass.
Vermeij GJ (1979) Anatomy of an invasion: the trans-Arctic interchange. Paleobiology 17:281–307
Wares JP, Cunningham CW (2001) Phylogeography and historical ecology of the north Atlantic intertidal. Evolution 55:2455–2469
Watts RJ, Johnson MS, Black R (1990) Effects of recruitment on genetic patchiness in the urchin Echinometra mathaei in Western Australia. Mar Biol 105:145–151
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Zouros E, Foltz DW (1984) Possible explanations of heterozygote deficiency in bivalve molluscs. Malacologia 25:581–591
Acknowledgements
We thank T. Balch, C. Begin, N. Hagen, J. Svavarsson, and B. Hooper for providing samples; F. Harper, S. Watts, and J. Lindley for assistance collecting samples using SCUBA; D. Duggins and the Friday Harbor Laboratories for dredge collections; D. Cook for valuable technical support; J. Lake for laboratory help; and two anonymous reviewers for comments on the manuscript. This work was supported by the Natural Sciences and Engineering Research Council of Canada, Canada Foundation for Innovation, the Nova Scotia Department of Economic Development, and the Timiskaming First Nation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R.J. Thompson, St. John's
Rights and permissions
About this article
Cite this article
Addison, J.A., Hart, M.W. Analysis of population genetic structure of the green sea urchin (Strongylocentrotus droebachiensis) using microsatellites. Marine Biology 144, 243–251 (2004). https://doi.org/10.1007/s00227-003-1193-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00227-003-1193-6