
Abstract
An important application of DNA-based genetic techniques in conservation genetics is to establish the taxonomic identity of individuals whose ancestry is uncertain, especially those from declining populations. Massasauga rattlesnakes (Sistrurus catenatus) in Missouri currently persist in three isolated populations and have been classified as eastern massasauga rattlesnakes (S. c. catenatus), which is a candidate for listing as a Federally Threatened or Endangered species. However, previous morphological evidence has suggested that these snakes are either hybrids between the eastern massasauga (S. c. catenatus) and the non-threatened western massasauga (S. c. tergeminus) or are pure S. c. tergeminus. Here, we analyze microsatellite DNA and mtDNA variation to assess the taxonomic identity of individuals from two of the three populations specifically those found in Holt and Linn Counties, Missouri. Population and phylogenetic analyses strongly support the conclusion that snakes in both populations are genetically most similar to reference S. c. tergeminus individuals from Kansas and are not related to S. c. catenatus individuals from Illinois and Ohio. These results argue that the current classification of these snakes as endangered S. c. catenatus is unwarranted and hence that the overall range-wide numbers of S. c. catenatus are less than previously estimated. Nonetheless the Missouri populations, while not part of the federally listed taxon, are strongly deserving of protected status at the state level because of their rarity within the state.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
An important use of DNA-based genetic techniques in conservation genetics is to assess the taxonomic identity of individuals from declining populations whose ancestry is uncertain due to inadequate data (Frankham et al. 2002). This is an important task because if the taxonomic status is not correctly assigned then a number of significant yet erroneous conservation decisions can be made, including that unrecognized endangered species may be at risk of becoming extinct (Bowen and Avise 1996; Daugherty et al. 1990) but also that populations of common species or hybrids between species may be granted unwarranted protection (Emerson and Wallis 1994; Wayne 1996 (but see Wilson et al. 2000); Culver et al. 2000; Haig et al. 2001; Parham et al. 2001; Stuart and Parham 2007). While other considerations such as the ecological role or rarity in a specific locality may play a role in deciding the conservation value of a population or species, key conservation legislation (e.g., US Endangered Species Act) is often based on taxonomic identity. Thus, accurate identification of individuals that face an increased risk of extinction is an important step in developing appropriate conservation plans.
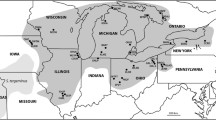
Populations of massasauga rattlesnakes (Sistrurus sp.) in northwestern and central Missouri present a case where genetic techniques could be fruitfully applied to assess the uncertain taxonomic status of a rare vertebrate. These snakes currently persist in only three locations in Holt, Linn, and Chariton counties (Missouri Department of Conservation 2000) despite being historically wide-spread in the northwestern and central parts of the state (see Fig. 1). Geographically, these populations are located at the interface between two currently recognized subspecies of Sistrurus catenatus, namely the eastern massasauga (S. c. catenatus) which is currently designated by the US Fish and Wildlife Service as a candidate for listing as a Federally Threatened or Endangered species USFWS (2008) and the western massasauga (S. c. tergeminus) which has no protected status. At present, snakes in all Missouri populations are classified as S. c. catenatus based on unpublished analyses of morphological and venom variation and patterns of habitat utilization (summarized in Szymanski 1998). In contrast, earlier research by Evans and Gloyd (1948) used variation in ventral scales, dorsal blotches, and overall coloration to argue that populations in Chariton and nearby counties were “intergrades” (=hybrids) between S. c. catenatus and S. c. tergeminus while snakes in Holt county were S. c. tergeminus. Multiple studies have shown that morphology can be a misleading indicator of phylogenetic relationships in snakes (e.g., Burbrink et al. 2000) and characteristics such as venom composition and habitat utilization are highly variable in these snakes (e.g., Gibbs et al. 2009). Clearly, application of genetic analyses using multiple markers and techniques would useful in clarifying the taxonomic status of Missouri populations and hence their conservation status as has been done for other threatened snakes (Keogh et al. 2003; Sumner et al. 2010).

Map of the range and localities of the massasauga rattlesnakes (Sistrurus catenatus) sampled to determine the species identity of Sistrurus sp. from Missouri. The dark gray shading represents the range of Sistrurus c. catenatus and the light gray range is that of Sistrurus c. tergeminus. Solid black circles are the localities from samples were collected. The open circle is a population of Sistrurus sp. in Missouri that was not sampled. Question marks highlight the unknown boundary of each species range in eastern and western Missouri. S. c. tergeminus populations: RC Russell County (lat: 38°53′23″N, long: 98°51′26″W); CB Cheyenne Bottoms (38°27′46.19″N, 98°39′9.97″W); EC Ellsworth County (37°17′4″N, 98°34′52″W); Missouri Sistrurus sp. populations: PSP Pershing State Park (39°45′32″N; 93°12′53″W); SCNWR Squaw Creek National Wildlife Refuge (40°04′08″N, 95°13′34″W); SLNWR Swan Lake National Wildlife Refuge (39°36′11″N, 93°10′44″W not sampled); Sistrurus c. catenatus populations: SSSP South Shore State Park (38°46′40″N, 89°13′37″W); PRF Prairie Road Fen (40°3′34″N, 83°47′44″W); KL Killdeer Plains State Wildlife Area (40°44′16″N, 83°15′1″W)
Recent phylogenetic analyses using multiple DNA loci have shown that all three subspecies of the currently recognized species S. catenatus (S. c. catenatus, S. c. tergeminus, and S. c. edwardsii) are taxonomically distinct from each other under a genealogical species concept although the evidence is strongest for the distinctiveness of S. c. catenatus from the other two subspecies (Kubatko et al. 2010). This means that comparisons of the genetic composition of snakes from the Missouri populations with reference samples from nearby S. c. catenatus and S. c. tergeminus population should allow the taxonomic identity of the Missouri snakes to be inferred. Specifically, if all Missouri snakes are S. c. catenatus, then phylogenetic and population-based analyses will show strong genetic similarities between known S. c. catenatus populations. Likewise, if some populations such as those in Holt County in western Missouri hence close to currently assumed range limit for S. c. tergeminus in fact consist of S. c. tergeminus individuals then they will be genetically similar to individuals from reference populations of this taxon. Finally, populations of hybrid individuals will show a mixture of genetic markers characteristic of each presumed parental taxa (S. c. catenatus and S. c. tergeminus).
Here, we assess these possibilities for individuals in two Missouri populations from Holt and Linn Counties. Specifically, we compare microsatellite DNA and mtDNA variation of Missouri snakes with the genetic composition of S. c. catenatus individuals from populations in Illinois and Ohio populations and S. c. tergeminus individuals from multiple populations in Kansas using a combination of population clustering techniques and phylogenetic analyses. A previous study compared levels of differentiation at three microsatellite loci among five Sistrurus populations in Missouri and Nebraska but did not make comparisons with unambiguously identified S. c. catenatus populations east of the Mississippi (Hart et al. 2008).
Methods
Population level analysis using microsatellites
For population analyses, we obtained genotypes for 17 microsatellite loci from 166 individuals collected from eight populations (see Fig. 1). We were interested in determining the taxonomic identity of individuals from two populations in Missouri: a population from Holt County located at Squaw Creek National Wildlife Refuge (n = 34) which had been classified as S. c. tergeminus by Evans and Gloyd (1948) and a population containing possible hybrids situated at Pershing State Park (n = 19), Linn County. Individuals from Pershing State Park were not analyzed by Evans and Gloyd (1948). However, due to the close proximity of this population (<15 km) to one (Swan Lake National Wildlife Refuge) that had been determined to contain putative hybrids, we assume that Pershing samples are similar in genetic makeup to Swan Lake individuals. For reference S. c. tergeminus individuals, we used microsatellite data generated by McCluskey et al. (unpublished) for samples collected from three locations in Kansas: Cheyenne Bottoms (n = 20); Ellsworth County (n = 11) and Russell County (n = 15). For representative S. c. catenatus genotypes, we used microsatellite data generated by Chiucchi and Gibbs (2010) for one population in southern Illinois (South Shore State Park: n = 18), and two populations in Ohio, Killdeer Plains Wildlife Area (n = 25) and Clark County (n = 21).
For the Missouri samples, we extracted DNA from frozen blood samples stored in lysis buffer (Seutin et al. 1991) using a standard phenol–chloroform protocol (Sambrook et al. 1989). We then genotyped all samples at 17 of the loci developed from an S. c. catenatus genomic library by Anderson et al. (2010) which also amplify microsatellite loci in S. c. tergeminus (McCluskey et al., unpublished). The loci used were Scu 200–214, 217, and 218. We followed the conditions described by Anderson et al. (2010) for PCR amplification protocols with the exception that we amplified Scu 210 and Scu 211 at an annealing temperature of 55°C. Amplicons were analyzed on an ABI 3100 Genetic Analyzer and we used the program GeneMapper v 3.7 score individual genotypes. To ensure that a consistent scoring criteria was used for all data we also ran select S. c. catenatus and S. c. tergeminus samples to compare the sizes of alleles at loci previously scored by Chiucchi and Gibbs (2010) and McCluskey et al. (unpublished) with the Missouri samples that were newly analyzed in this study.
Preliminary analyses indicated substantial genetic differentiation in microsatellite variation between reference samples of S. c. tergeminus from Kansas and S. c. catenatus from Illinois and Ohio (F st between pooled samples of each taxon = 0.21; Murphy et al., unpublished). We then used STRUCTURE (Pritchard et al. 2000), a cluster-based analysis program which uses a Bayesian approach to infer the genetic composition of individuals from both Missouri populations. Our explicit goal in this analysis was to determine the genetic identity of individuals in the Missouri populations and not to examine population genetic structure per se in our samples (see Chiucchi and Gibbs 2010 for such an analysis for a larger number of S. c. catenatus populations). Therefore, we first confirmed that the program clearly would separate S. c. catenatus and S. c. tergeminus genotypes into distinct genetic clusters by analyzing the reference populations for each taxon with the program set to the condition of identifying K = 2 genetic clusters (for an example see Kinziger et al. 2008). After confirming this we reanalyzed the data using all samples including samples from both Missouri populations. Under this condition, “pure” Missouri samples of each taxon would cluster with the appropriate reference samples whereas hybrids would be identified as individuals consisting of a combination of genotypes from each cluster. For each analysis, we ran the program with a burn in period of 100,000 repetitions followed by 1,000,000 Markov chain Monte Carlo (MCMC) generations. We used the admixture model with correlated allele frequencies and calculated the 95% probability intervals for the proportion of the genome of each putative hybrid individual made up by each of the reference genomes (q-value). Individuals who had q values with 95% probability intervals that contained a q-value of 1 were considered as genetically statistically indistinguishable from that reference subspecies and hence not hybrids. Finally, we observed the ln probability graph for a lack of large deviations to ensure the statistical analysis had run enough repetitions. The final run of K = 2 was repeated five times to ensure consistency of the results.
Phylogenetic analyses using mtDNA
We conducted phylogenetic analyses of mtDNA variation in a limited set of Missouri samples in relation to previously published sequences of the same gene region from all S. catenatus subspecies as described in Kubatko et al. (2010). Specifically, we sequenced a 665 bp region of the ATP 6–8 genes in all 19 samples from the Pershing State Park population using the primers and PCR conditions described by Gibbs and Diaz (2010). We also included two ATP sequences reported by Kubatko et al. (2010) for two individuals from the Squaw Creek population in NW Missouri. We choose to analyze variation in this gene region because it mirrors the phylogenetic patterns revealed by a more in-depth species tree analysis by Kubatko et al. (2010) based on gene trees generated from 18 nuclear DNA sequence-based loci and one mtDNA locus (the ATP 6–8 gene region described here) (see Gibbs and Diaz 2010 for details of the loci used).
Following Kubatko et al. (2010), we used the phylogenetic program BEAST (Drummond and Rambaut 2007) to estimate gene tree relationships among all distinct S. c. catenatus mtDNA haplotypes in a Bayesian framework. We used gene sequences described in Kubatko et al. (2010) from the same gene region from S. miliarius subspecies to root the gene tree. We used the online version of MODELTEST 3.7 (http://darwin.uvigo.es/software/modeltest.html—see Posada and Crandall 1998) in combination with PAUP 4.0b10 (Swofford 2000) to choose among different substitution models using an AIC criteria to determine the best-fit model for the ATP gene region and then used parameter estimates to set priors in BEAST. We ran the program for 20 million generations and discarded the first 200,000 trees as burnin. We used posterior probabilities for each clade as a measure of our statistical confidence in the phylogenetic grouping of specific haplotypes.
Results
Population clustering analyses based on microsatellites
Levels of variation at microsatellite loci are comparable across samples from reference S. c. catenatus and S. c. tergeminus populations and samples from each of the two Missouri populations analyzed here (see Supplemental information). For example, mean observed (H o) heterozygosities were 0.59 (range: 0–0.88), 0.65 (0.125–1.00), 0.55 (0.22–0.97), and 0.71 (0.19–0.95) in pooled S. c. catenatus, S. c. tergeminus reference samples, Squaw Creek, and Pershing populations, respectively.
Analysis of microsatellite genotypes from the two reference populations under a K = 2 criteria in Structure show clear segregation into two highly-distinct clusters that closely match each population (results not shown): the mean probability assignment (± 95%CI) of S. c. catenatus individuals to cluster 2 (q2) equals 0.997 ± 0.002 while the mean probability of assignment of S. c. tergeminus individuals to cluster 1 (q1) was 0.995 ± 0.004. When the analysis is repeated but with samples from both Missouri populations included, all Missouri samples cluster with the S. c. tergeminus samples (Fig. 2). Specifically, samples from Squaw Creek have a mean q1 ± 95% CI value of 0.995 ± 0.002 (range: 0.979–0.999) while samples from Pershing have a mean q1 value of 0.976 ± 0.02 (range: 0.711–0.999) (Table 1). A single individual in the Pershing population has a q1 value of 0.711 which is lower than q1 values for the other 19 individuals (all q1 values > 0.935). However, the 95% PI for q1 for this snake (0.442, 1.000) contains 1 and so we cannot reject the hypothesis that the snake is genetically a pure S. c. tergeminus. These results were consistent across five separate runs with nearly identical average q-values and 95% confidence intervals. The ln likelihood plot for all five trials rapidly went to fixation and remained fixed for the entire run suggesting that all runs converged (results not shown). In summary, the microsatellite analyses strongly support the idea that individuals from the two Missouri populations are genetically S. c. tergeminus.

Graphical representation of STRUCTURE results for K = 2 populations. The proportion of each massasauga’s genome attributed to Sistrurus c. catenatus is black, and the proportion of each massasauga’s genome attributed to Sistrurus c. tergeminus is gray
Phylogenetic analyses of mtDNA variation
The phylogenetic results corroborate the finding from the microsatellite data that individuals in both Missouri populations are genetically most similar to S. c. tergeminus. All 19 samples from Pershing State Park had the same mtDNA haplotype (ScMO-1) while the two samples from the Squaw Creek population had a different but similar haplotype (ScMO-2) (percent sequence divergence between haplotypes: 0.76%). Both were distinct from all other S. catenatus haplotypes described by Kubatko et al. (2010) but each was much more similar to other western and desert massasauga (S. c. edwardsii) haplotypes than to those from the candidate endangered S. c. catenatus individuals: The mean percent sequence divergence between Sc-MO1 and other S. c. tergenimus and S. c. edwardsii haplotypes was 1.1% whereas between Sc-MO1 and S. c. catenatus haplotypes the value was 11.4%. Similar values were found for the same comparisons involving the Squaw Creek-specific haplotype (0.8% vs. 11.4%).
As found by Kubatko et al. (2010), Modeltest identified the HKY + I substitution model as the best describing patterns of nucleotide substitution in this gene region. Using this model in the BEAST analyses, we generated a tree (Fig. 3) in which, consistent with results in Kubatko et al. (2010), there are two well-supported (Posterior Probabilities = 1.0) ingroup clades, one consisting of haplotypes from S. c. catenatus and the other consisting of haplotypes from S. c. tergeminus and S. c. edwardsii and all the Missouri haplotypes providing phylogenetic support for the idea that the Missouri snakes are not S. c. catenatus but are genetically most similar to S. c. tergeminus.

Maximum clade credibility tree from analysis of mtDNA ATP sequences in BEAST. This tree represents the posterior sample with the maximum sum of clade posterior probabilities at the internal nodes. Posterior probabilities of each clade are shown at each node. Sced-1 (S. c. edwardsii haplotype); Scter-1 (S. c. tergeminus haplotype); Scat-1 to Sca-4 (S. c. catenatus haplotypes); Smb (S. m. barbouri haplotye); Smc-Sms (haplotype found in both S. m. milarius and S. m. streckeri) (all from Kubatko et al. (2010)); ScMO-1 (Pershing Lake haplotype); ScMO-2 (Squaw Creek haplotype) (this study)
Discussion
Our major result is that two independent genetic analyses provide strong evidence that Sistrurus rattlesnakes in two Missouri populations that are currently classified as S. c. catenatus (a candidate for listing as an endangered taxon) are genetically more similar to S. c. tergeminus which currently do not have protected status. This result has both conservation and evolutionary implications which we discuss below.
We emphasize that our genetic evidence is strongest in terms of concluding that the Missouri snakes we analyzed are not S. c. catenatus rather than unambiguously assigning them on the basis of genetic evidence as either S. c. tergeminus or S. c. edwardsii. There are a number of reasons for this. First, in the population-level analysis, we did not include genotypes from any S. c. edwardsii populations and so the only “choice” the program had was assigning Missouri snakes to either S. c. catenatus or S. c. tergeminus. Second, in the phylogenetic analysis, the Missouri sequences were part of a clade containing haplotypes from both S. c. tergeminus and S. c. edwardsii individuals. This is not surprising because in their recent phylogenetic analysis of subspecies within the genus Sistrurus. As described above, Kubatko et al. (2010) found evidence for the taxonomic distinctiveness of all three subspecies of S. catenatus; but this evidence was strongest for S. c. catenatus being distinct from the other two subspecies and weaker for S. c. tergeminus and S. c. edwardsii being distinct from each other. This is likely due to the influence of differences in the ages of each clade (S. c. catenatus:~3.0 million ybp; S. c tergeminus and edwardsii:~0.5 million ybp) and its effect on the genetic distinctiveness of each taxa through the influence of retained ancestral polymorphism. Given the uncertainty of how taxonomically distinct the two western forms of S. catenatus are, on the basis of our genetic data, we are most comfortable in concluding that the Missouri snakes belong to a genetically distinct complex consisting of weakly differentiated S. c. tergeminus and S. c. edwardsii forms. The geographic proximity of the Missouri populations suggests they are more closely related to the former than the latter taxon.
The most significant conservation implication of these results is that Sistrurus rattlesnakes presently in Missouri should not be candidates for listing as a Federal Endangered Species, because the genetic evidence indicates that they are not S. c. catenatus but most likely S. c. tergeminus. We acknowledge that we have only analyzed samples from two of three extant populations in the state; however, because of the close geographic proximity of Pershing State Park to the unsampled Swan Lake population, it is highly likely that they are genetically similar to each other. It is also possible that S. c. catenatus was present in the state at some point in the past, particularly in now extinct populations in the eastern part of the state (Szymanski 1998). This possibility could be explored through genetic analyses of museum specimens collected from now extirpated populations (Friedman and DeSalle 2008). Finally, regardless of their federal status, we strongly feel that the overall rarity of these snakes in Missouri (three extant populations) and the fact they are geographically isolated from other populations of S. c. tergeminus means that they deserve continued protection at the state level as endangered species (Missouri Department of Conservation 2000).
This result also has implications in terms of the range-wide conservation status of S. c. catenatus in that the reclassification of Missouri populations from S. c. catenatus to S. c. tergeminus means a “loss” of a number of populations which were previously considered to be among the largest and most viable S. c. catenatus populations at a range-wide level (M. Redmer, personal comminication). This means that extant numbers of this endangered snake are even lower than previously estimated and hence this taxon is even more imperiled than previously thought.
Finally, from an evolutionary perspective, our genetic evidence demonstrates that Pershing individuals are not hybrids as previously suggested based on morphological analyses by Evans and Gloyd (1948). The one possible exception is the single individual out of 19 analyzed from the Pershing population had a point estimate for a q1 value (probability of assignment to S. c. tergeminus) of 0.711 suggesting that it contained a mixture of microsatellite genotypes found in reference populations of S. c. catenatus and S. c. tergeminus. However, as pointed out above the confidence interval for the point estimate does not exclude 1.0 (pure S. c. tergeminus) and additional evidence supports the idea that this low value compared to the other individuals surveyed is due to chance and not hybridization. Specifically,this individual was shown to have a S. c. tergeminus mtDNA haplotype, and an independent analyses of all snakes in the Pershing population using a subset of the nuclear DNA markers described in Kubatko et al. (2010) and phylogenetic methods for detecting hybrids (Meng and Kubatko 2009) find no evidence for hybridization (Gerard and Kubatko, unpublished). Finally the large gap that currently exists between the Pershing populations and extant S. c. catenatus populations in Illinois (Fig. 1) suggests that it is unlikely that recent hybridization is the source of this individual. Overall, our results provide evidence against hybridization commonly occurring in natural populations of these rattlesnakes despite observations of its occurrence in other genera of rattlesnakes (Campbell et al. 1989; Murphy and Crabtree 1988) and again provides an example of how, particularly in snakes, inferences of genetic relationships based on morphology alone can be misleading (Burbrink et al. 2000).
References
Anderson CS, Gibbs HL, Chiucchi J (2010) Nineteen polymorphic microsatellite loci isolated from the eastern massasauga rattlesnake, Sistrurus c. catenatus. Conserv Genet Resour. doi:10.1007/s12686-010-9253-y
Bowen BW, Avise JC (1996) Conservation genetics of marine turtles. In: Avise JC, Hamrick JL (eds) Conservation genetics: case histories from nature. Kluwer Academic Publishers, Norwell, MA, p 397
Burbrink FT, Lawson R, Slowinski JB (2000) Mitochondrial DNA phylogeography of the polytypic North American rat snake (Elaphe obsoleta): a critique of the subspecies concept. Evolution 54:2107–2118
Campbell JA, Brodie ED Jr, Barker DG, Price AH (1989) An apparent natural hybrid rattlesnake and Crotalus willardi (Viperidae) from the Peloncillo mountains of southwestern New Mexico. Herpetologica 45:344–349
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Chiucchi JE, Gibbs HL (2010) Similarity of contemporary and historical gene flow among highly-fragmented populations of an endangered rattlesnake. Mol Ecol. doi:10.1111/j.1365-294X.2010.04860.x
Culver M, Johnson WE, Pecon-Slattery J, O’Brien SJ (2000) Genomic ancestry of the American puma (Puma concolor). J Hered 91:186–197
Daugherty CE, Cree A, Hay J, Thompson MB (1990) Neglected taxonomy and continuing extinctions of tuatara (Sphenodon). Nature 347:177–179
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Emerson BC, Wallis GP (1994) Phylogenetic relationships of the Prodontria (Coleoptera; Scarabaeidae; Subfamily Melolonthinae), derived from sequence variation in the mitochondrial cytochrome oxidase II gene. Mol Phylogenet Evol 4:433–447
Evans PD, Gloyd HK (1948) The subspecies of the massasuaga, Sistrurus catenatus, in Missouri. Bull Chicago Acad Sci 8:225–232
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Friedman M, DeSalle R (2008) Mitochondrial DNA extraction and sequencing of formalin-fixed archival snake tissue. Mitochondrial DNA 19:433–437
Gibbs HL, Diaz J (2010) Identification of single copy nuclear DNA markers for North American pit vipers. Mol Ecol Resour 10:177–180
Gibbs HL, Sanz L, Calvete JJ (2009) Snake population venomics: Proteomics-based analyses of individual variation reveals gene regulation effects on venom protein expression in Sistrurus rattlesnakes. J Mol Evol 68:113–125
Haig S, Wadner RS, Forsman ED, Mullins TD (2001) Geographic variation and genetic structure of spotted owls. Conserv Genet 2:25–40
Hart SN, Durbian FE, Dillman CB, Eckdahl TT (2008) Assessment of allelic variation among massasauga rattlesnake populations via microsatellite analysis. Trans Missouri Acad Sci 42:1–9
Keogh SJ, Scott IAW, Fitzgerald M, Shine R (2003) Molecular phylogeny of the Australian venomous snake genus Hoplocephalus (Serpentes, Elapidae) and conservation genetics of the threatened H. stephensii. Conserv Genet 4:57–65
Kinziger AP, Loudenslager EJ, Hakin DG et al (2008) Hybridization between spring- and fall-run chinook salmon returning to the Trinity River, California. North Am J Fish Manag 28:1426–1438
Kubatko LS, Gibbs HL, Bloomquist E (2010) Inferring species-level phylogenies and taxonomic distinctiveness using multi-locus data in Sistrurus rattlesnakes. Syst Biol (in press)
Meng C, Kubatko LS (2009) Detecting hybrid speciation in the presence of incomplete lineage sorting using gene tree incongruence: a model. Theor Popul Biol 75:35–45
Missouri Department of Conservation (2000) Missouri Animals of Conservation Concern. Jefferson City, MO
Murphy RW, Crabtree CB (1988) Genetic identification of a natural hybrid rattlesnake: Crotalus scutulatus scutulatus × C. viridis viridis. Herpetologica 44:119–123
Parham JF, Simison WB, Kozak KH, Feldman CR, Shi H (2001) New Chinese turtles: Endangered or invalid? A reassessment of two species using mitochondrial DNA, allozyme electrophoresis, and known locality specimens. Anim Conserv 4:357–367
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sambrook J, Fritsch EF, Maniatis T (1989) Extraction with phenol:chloroform. In: Molecular Cloning: A laboratory manual, pp. E.3-E.4. Cold Spring Harbor Laboratory Press, Plainview
Seutin G, White BN, Boag PT (1991) Preservation of avian blood and tissue samples for DNA analyses. Can J Zool 69:82–90
Stuart BL, Parham JF (2007) Recent hybrid origin of three rare Chinese turtles. Conserv Genet 8:169–175
Sumner J, Webb JK, Shine R, Keogh JS (2010) Molecular and morphological assessment of Australia’s most endangered snake, Hoplocephalus bungaroides, reveals two evolutionarily significant units for conservation. Conserv Genet 11:747–758
Swofford, D (2000) PAUP* 4.0b10. Sunderland, Massachusetts, USA
Szymanski J (1998) Status assessment for eastern Massasauga (Sistrurus c. catenatus). U.S. Fish and Wildlife Service, Fort Snelling, Minnesota
U. S. Fish and Wildlife Service (2008) Endangered and threatened wildlife and plants; review of native species that are candidates for listing as endangered or threatened; annual notice of findings on resubmitted petitions; annual description of progress on listing actions; proposed rule. Federal Register 73:75175‐75244
Wayne R (1996) Conservation genetics in the Candidae. In: Avise JC, Hamrick JL (eds) Conservation genetics: case histories from nature. Chapman & Hall, New York, pp 75–118
Wilson PJ, Grewal S, Lawford JNM et al (2000) DNA profiles of the eastern Canadian wolf provide evidence for a common evolutionary history independent of the grey wolf. Can J Zool 78:2156–2166
Acknowledgments
We especially thank Trish Crabill, Frank Durbian, and Rich Seigel for much appreciated assistance in obtaining samples from Missouri, and David Bender, Travis Taggart, Curtis Schmidt, and Joe Collins for their generous hospitality and help in obtaining samples in Kansas. Jose Diaz gave advice in the lab, Katy Greenwald, Mike Redmer, and Rich Seigel commented on the manuscript, and The Ohio State University College of Arts and Sciences and College of Biological Sciences provided financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Gibbs, H.L., Murphy, M. & Chiucchi, J.E. Genetic identity of endangered massasauga rattlesnakes (Sistrurus sp.) in Missouri. Conserv Genet 12, 433–439 (2011). https://doi.org/10.1007/s10592-010-0151-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0151-3