
Abstract
More than 2 decades ago, the discovery of osteoprotegerin (OPG) as inhibitor of the receptor of activator of nuclear factor Kb (RANK) ligand (RANKL) revolutionized our understanding of bone biology and oncology. Besides acting as decoy receptor for RANKL, OPG acts as decoy receptor for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). OPG, RANKL, and TRAIL are ubiquitously expressed, stimulating per se pivotal signaling cascades implicated in cancer. In the context of cancer cell–bone cell interactions, cancer cells skew the OPG/RANKL/RANK (RANKL cognate receptor) balance towards bone destruction and tumor growth through favoring the RANKL/RANK interface, circumventing OPG. Numerous preclinical and clinical studies demonstrate the dual role of OPG in cancer: antitumor and tumor-promoting. OPG potentially conveys an antitumor signal through inhibiting the tumor-promoting RANKL signaling—both the osteoclast-dependent and the osteoclast-independent—and the tumor-promoting TRAIL signaling. On the other hand, the presumed tumor-promoting functions of OPG are: (i) abrogation of TRAIL-induced apoptosis of cancer cells; (ii) abrogation of RANKL-induced antitumor immunity; and (iii) stimulation of oncogenic and prometastatic signaling cascades downstream of the interaction of OPG with diverse proteins. The present review dissects the role of OPG in bone oncology. It presents the available preclinical and clinical data sustaining the dual role of OPG in cancer and focuses on the imbalanced RANKL/RANK/OPG interplay in the landmark “vicious cycle” of skeletal metastatic disease, osteosarcoma, and multiple myeloma. Finally, current challenges and future perspectives in exploiting OPG signaling in bone oncology therapeutics are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the context of the nomination of the inflammatory milieu as an “enabling characteristic” fostering the acquisition of “hallmarks of cancer” [1], cytokines—a fundamental feature of tumor microenvironment—have been conceived as orchestrators of cancer [2]. Osteoprotegerin (OPG) is a member of tumor necrosis factor (TNF) receptor superfamily, produced mainly by osteoblasts, assigned to ensure bone homeostasis. It acts as a decoy receptor for receptor activator of nuclear factor kB ligand (RANKL), disrupting the RANKL-induced bone resorption [3,4,5,6,7], and for TNF-related apoptosis inducing ligand (TRAIL)—the protagonist of tumor immune surveillance [8]. Implicit in cancer biology, OPG binds myriad proteins beyond RANKL and TRAIL, transducing diverse signals [9]. Consequently, the discovery of OPG and RANKL in 1997 [4,5,6,7] revolutionized the notion, conceived in the mid 1970’s, that osteoclasts are implicated in bone oncology and are modulated by an authentic bone cell—the osteoblast [10,11,12,13].
An in-depth understanding of the biology of bone oncology is a prerequisite for the refinement of current therapeutic strategies. Of paramount importance is the mitigation of skeletal related events (SREs)—bone pain, hypercalcemia, pathologic fractures, and neurologic complications—encountered in both primary and metastatic bone tumors, subverting the survival and the quality of life of patients [14]. The treatment of primary bone tumors, mainly of osteosarcoma and of giant cell tumor of bone, poses many challenges [15, 16]. Moreover, the compendium of osteotropic primary tumors has been enriched since the first report of bone metastases related with breast cancer [17], designating skeleton as the most common metastatic site. A high proportion of breast and prostate cancer patients—70%—harbor bone metastases, while thyroid, kidney, and bronchus carcinomas colonize the bone with an incidence hovering at 30–40% [14]. The present review provides a comprehensive overview on the role of OPG in bone oncology, which extends beyond the inhibition of RANKL and TRAIL, involving several signaling cascades. It focuses on the interplay among OPG, RANKL, and TRAIL that governs the initiation and progression of cancer, being implicated in both primary and metastatic bone disease. The rationale for exploration of the therapeutic implementation of OPG is discussed with emphasis on current and future perspectives.
OPG: a decoy receptor for RANKL and TRAIL
OPG owes its discovery to its function as a decoy receptor for RANKL, inhibiting RANKL—induced bone resorption, a property reflected in its name derived from the latin words os (bone) and protegere (to protect). The TNFRSF11B gene encodes the OPG protein comprising 401 amino acids (aa), 21 aa of which constitute a signal peptide. Cleavage of the signal peptide generates a 380 aa soluble glycoprotein of 55 kDa. The latter is homodimerized to form the dimeric OPG of 110 kDa [18]. OPG is predominantly expressed by bone marrow stromal cells, detected also in hematopoietic, immune, and vascular system [9, 19]. It exerts a multitude of functions, binding to myriad partners, such as syndecan-1, glycosaminoglycans (GAGs), proteoglycans, and Factor VIII von Willebrand Factor complex. Βeyond being the cornerstone of bone biology, OPG is also implicated in cancer, metabolism, cardiovascular system, and rheumatic diseases [9, 19].
RANKL is the official nomenclature for a 316 aa protein, member of TNF family, encoded by TNFSF11 gene, initially nominated as OPG ligand (OPGL) [4, 5]. RANKL is indispensable for differentiation of osteoclasts in the presence of macrophage colony stimulating factor (M-CSF) and orchestrates the osteoclasts-mediated bone resorption. Interestingly, research in the field of immunology had unraveled RANKL, prior to its discovery via OPG. RANKL is expressed on T cells and interacts with its receptor, RANK, on mature dendritic cells (DCs) in order to regulate the survival of the latter [20, 21]. Several isoforms of RANKL have been recognized [22]. The expression of RANKL in a wide spectrum of tissues, including lymph nodes, thymus, mammary gland, lung, spleen and bone marrow, highlights its pleiotropic effect [23]. RANKL exerts a pivotal role in immune system through mechanisms highly divergent [24]. RANKL signaling curtails the antitumor immunity, stimulating the mTEC-mediated self-tolerance towards cancer specific antigens and the proliferation of T regulatory cells (T regs). On the other hand, RANKL enhances the antitumor immunity through stimulating memory cytotoxic T-lymphocyte responses toward tumor-associated antigens and promoting the lymph nodes genesis, the antigen presentation, the survival of DCs and monocytes/macrophages [24].
RANKL exerts its functions mainly via its cognate receptor RANK, a type I homotrimeric transmembrane protein encoded by TNSF11A gene, initially identified on osteoclast precursors (OCPs), mature osteoclasts, and DCs [6, 19,20,21]. Crucial adaptor molecules downstream of the interaction RANKL/RANK, such as TNF receptor associated factor 6 (TRAF6), stimulate seven signaling cascades, activating NFkB, cjun/activator protein 1 (AP-1), c-Myc, calcineurin/nuclear factor of activated T cells [NFAT]c1 (NFATc1), Src, mitogen-activated protein kinase (MAPK)/p38, and extracellular signal-regulated kinase (ERK) [19, 23]. Consequently, a plethora of signals additionally to differentiation and activation of osteoclasts are transduced.
OPG acts also as a decoy receptor for TRAIL, a type II transmembrane protein, identified as a member of TNF family, nominated initially as APO2L. Produced by T cells and natural killers (NK) cells, TRAIL is a crucial player of tumor immune surveillance, stimulating the extrinsic pathway of apoptosis in cancer cells expressing TRAILR1 (DR4) and TRAILR2 (DR5), leaving normal cells unaffected [25,26,27,28]. Interestingly, a TRAIL-elicited inhibition of cancer cell migration has been reported, mediated through MAPK Activating Death Domain protein (MADD) and chemokine receptor 7 (CXCR7) in breast cancer cells [29].
Lessons from clinical studies concerning the role of OPG in cancer
A potential tumor-promoting and prometastatic role of OPG was reported as early as in 2006 in a rich repertoire of tumors. This role was based on in vitro (breast, prostate, gastric cancer) and in vivo (breast, prostate, gastric, bladder cancer) data, as well as on clinical data concerning serum OPG concentrations (breast, prostate, bladder cancer) and OPG expression on malignant tissue (breast, prostate, gastric cancer) [30]. Likewise, mRNA expression of OPG in human colorectal cancer tissues has been significantly correlated with aggressive tumor behavior [31].
In a recent clinical study evaluating serum OPG levels in melanoma patients, the serum OPG levels proved to be statistically significantly higher in the melanoma group (median = 355.2 pg/ml) compared to the healthy group (median = 234.1 pg/ml) (p < 0.0001). The researchers stratified the melanoma patients into four groups according to Breslow score, which describes the melanoma thickness (depth) assessed by a pathologist. Comparing OPG levels in the healthy group with OPG levels at each Breslow score stage, no statistically significant difference in the levels of OPG at first Breslow score (median = 287.6 pg/ml) compared to healthy group (median = 234.1 pg/ml) (p = 0.0239 pg/ml) was observed. Statistically significant elevation of OPG levels in comparison to healthy group was demonstrated starting from the second Breslow score stage as follows: (i) Breslow score 1–2 mm: OPG median = 323.9 pg/ml, p < 0.0001; (ii) Breslow score 2–4 mm: OPG median = 398.0 pg/ml, p = 0.0004; (iii) Breslow score > 4 mm: median = 500.7 pg/ml, p < 0.0001. Comparing the OPG levels in group of positive and negative sentinel node metastases, no statistically significant change was observed. Considering that the Breslow score stage is a good indicator of the risk of metastasis reflecting spread beyond the skin, it is reasonable to hypothesize that OPG levels could have a clinical utility for melanoma patients, but this remains to be proved. In this study there was no significant correlation of OPG with sentinel lymph node positivity; therefore, OPG was not anticipated to correlate significantly with disease stage according to American Joint Committee on Cancer (AJCC)/tumor size, lymph nodes affected, metastases (TNM) staging system. Most importantly, in this study there was no analysis of direct correlation of OPG levels with overall survival and response to therapy. Consequently, although OPG was suggested as a promising biomarker for early stages of melanoma, it could not be designated either as a prognostic or a predictive biomarker; this perspective warrants further investigation [32].
Notably, an antitumor role of OPG is indicated by other clinical studies [33, 34]. According to Tromsø study, which recruited 6279 subjects from general population without prior cancer, OPG serum levels correlated with reduced risk of incident cancer at all sites and breast cancer [33]. Moreover, Parker et al. demonstrated a positive relation of tissue OPG expression with favorable clinicopathological characteristics of breast cancer, in particular small tumor size, absence of lymph node metastases, and low Ki-67 [34].
To interpret the discrepant clinical evidence, it is mandatory to dissect the direct and indirect role of OPG in cancer. The direct role of OPG in cancer is considered tumor-promoting—especially protumorigenic, prometastatic, and proangiogenic. The indirect role of OPG is defined by the distinct RANKL and TRAIL signaling that abrogates, given that OPG acts as decoy receptor for RANKL and TRAIL, which exert per se both tumor-promoting and antitumor effects. In other terms, OPG plays an indirect antitumor role through inhibition of tumor-promoting RANKL and TRAIL signaling, while it plays an indirect tumor-promoting role when abrogating the antitumor RANKL and TRAIL signaling.
The protumorigenic/prometastatic effect of OPG
A plethora of experimental and preclinical data provide the mechanistic underpinnings of the protumorigenic role of OPG. The landmark study of Goswami et al. shed more light into this role. In this study, administration of human recombinant OPG was shown to drive normal primary human mammary epithelial cells (HMEC) spheres towards a malignant phenotype. The OPG-mediated activation of various kinases, such as Aurora-A, Bub1, and BubR1, was demonstrated to induce aneuploidy in HMEC spheres. The reported pro-survival and pro-proliferative effect of OPG on HMEC spheres was ascribed to activation of AKT, glycogen synthase kinase 3b (GSK3b), p44/42, p65, and b-catenin. Moreover, the OPG-mediated upregulation of cluster of differentiation (CD) 24 (CD24) in HMEC spheres was incriminated in the OPG-induced enhancement of cancer cell proliferation, invasion, and migration [35]. This study demonstrated for the first time the immunohistochemical OPG expression in the invasive and the inflammatory breast cancer [35]. Recently, it was demonstrated that addition of human recombinant OPG (500 pg/ml) in sphere cultures of breast cancer cells SUM149PT, SUM1315MO2, inflammatory breast cancer tissue from patients, and primary human mammary epithelial cells (HMEC) spheres resulted in amplification of the DNA copy numbers of well-established oncogenes—serine-threonine kinase (AKT1), Aurora-A kinase (AURK1), epidermal growth factor receptor (EGFR), Myc, Serine/threonine-protein kinase (PAK1), cyclin dependent kinase 4 (CDK4), erb-b2 receptor tyrosine kinase 2 (ERBB2)—while downregulated tumor suppressive genes—cyclin-dependent kinase inhibitor 2A (CDKN2A), phosphatase and tensin homolog (PTEN), DNA topoisomerase II alpha (TOP2A), and retinoblastoma (RB) 1 (RB1) [36]. The protumorigenic and prometastatic role of OPG has been currently revolutionized by a mass spectrometry analysis in inflammatory and aggressive breast cancer cells, unveiling several proteins-partners of OPG which orchestrate the initiation and progression of cancer [36]. Moreover Weichhaus et al. observed reduced expression of proteases cathepsin D and Matrix Metalloproteinase-2 following OPG knockdown in a chick embryo model, suggesting that the prometastatic effect of OPG may be ascribed to regulation of protease expression and invasion [37]. From a different viewpoint, Heymann et al. speculated that OPG expressed on endothelial and immune cells at metastatic lymph nodes of human medullary thyroid carcinoma, detected by immunohistochemistry, acts as a chemoattractant for cancer cells, guiding the migration thereof [38]. Holen et al. underscored the anti-apoptotic role of OPG, demonstrating that OPG expression in breast cancer cells MDA-MB 436/231 inhibits TRAIL-mediated apoptosis, providing cancer cells with a survival advantage [39]. Benslimane-Ahmim et al. demonstrated that the OPG-induced expression of stromal cell-derived factor (SDF) 1 (SDF-1) by endothelial colony forming cells (ECFCs) guides the migration of MNNG-HOS human osteosarcoma cells and DU145 prostate cancer cells [40].
Recent studies designate OPG as a pivotal player of the inflammation-induced breast cancer progression in vitro and ex vivo. The interplay of OPG with fatty acid synthase (FASN)—a multifaceted enzyme critical for lipid biosynthesis—and with cyclooxygenase-2 (COX2)—a critical player in inflammatory component of tumor microenvironment—has been implicated in the inflammatory cascades in highly invasive inflammatory breast cancer, inducing survival kinase signaling, essential for breast cancer progression [41]. In vitro and ex vivo data reported by Chung et al. indicated OPG as mediator of the prometastatic signaling of interleukin (IL) 1β (IL-1β) in breast cancer. In fact, an autocrine loop was revealed in which OPG secretion was controlled by IL-1β via p38- and p42/44-mediated pathways and induced per se both the IL-1β expression and the IL-1β—mediated breast cancer cell invasion and matrix metallopeptidase (MMP) 3 upregulation [42].
To the best of our knowledge, so far, there are no straightforward analyses with statistically significant results addressing the protumorigenic/prometastatic effects of OPG described in model systems in the settings of human cancer. Moreover, it is acknowledged that the expression of molecules does not equate to function. More studies to resolve these issues are needed.
The proangiogenic effect of OPG
Since the first report of the role of OPG in the survival of endothelial cells and the formation of cord-like structures in vitro in humans [43], immense research has sustained the proangiogenic OPG signaling. The data of proteomics are consistent, showing that OPG pretreatment modifies human endothelial colony-forming cells (ECFCs) protein expression via down-regulating 23 spots and upregulating 6 spots, reflecting proteins regulating cell motility, adhesion, signal transduction, and apoptosis [44].
The proangiogenic role of OPG appears to be exerted through numerous sophisticated mechanisms, involving the kinases ERK and protein kinase B (AKT), as well as inhibition of RANKL, which inhibits angiogenesis, according to the study of Mc Conigle et al. in the rat aortic ring model of angiogenesis [45]. Moreover, OPG has been shown to elicit cytoskeletal reorganization and to activate FAK, Src, and ERK signaling in human dermal microvascular endothelial cells (HuDMECs) in vitro [46]. The in vitro binding of OPG with thrombospondin (TSP-1) in Weibel–Palade bodies (WPB) of human vascular endothelial cells has pointed to the OPG-mediated inhibition of the antiangiogenetic function of TSP-1 [47].
A signaling pathway initiated by the alpha(v)beta3 integrin has been shown in vitro and in vivo to activate NFkB, leading to upregulation of OPG, which in turn acts as a survival factor for endothelial cells [48]. The recruitment of endothelial cells by cancer cells has been demonstrated in vitro to orchestrate the avβ3-mediated increase of OPG in HuDMECs [49]. In bone, osteopontin has been demonstrated in vitro to induce the avβ3 integrin-mediated OPG expression, providing a survival advantage to rat aortic endothelial cells during bone resorption [50]. Moreover, a proangiogenic effect of OPG exerted by fibroblast growth factor (FGF)-2 has also been observed in vivo in human ECFCs [51].
To the best of our knowledge, so far, there are no straightforward analyses with statistically significant results addressing the proangiogenic effects of OPG described in model systems in the settings of human cancer. Moreover, it is acknowledged that expression of molecules does not equate to function. More studies to resolve these issues are needed.
The RANKL-induced signaling transduction in cancer
The RANKL-induced signaling plays a key role in fundamental pathways of “osteoimmunology” (the integration of immunity into bone biology), osteoclastogenesis, carcinogenesis, and cancer progression, which cross-talk to define the fate of cancer cells [52]. Expressed both by cancer cells per se and by tumor microenvironment, RANKL appears to exert a tumor-promoting—protumorigenic and prometastatic—role in a wide array of malignancies, including breast, prostate, colorectal, lung, bladder, and gastric cancer [reviewed in 52].
The most convincing evidence supporting the protumorigenic role of RANKL is derived from the investigation of the progestin-driven mammary carcinogenesis in a mouse model, demonstrating that both the genetic [53] and the pharmacological [54] interception of RANKL halted the carcinogenic effect of the synergy of medroxyprogesterone acetate with 7, 12 dimethylbenzanthracene.
The signaling pathways activated downstream of RANKL/RANK interaction are implicated in epithelial mesenchymal transition (EMT), neo-angiogenesis, cancer cell migration, and invasion [52, 55,56,57,58,59]. RANKL expressed on T regs infiltrating breast cancer has been involved in generation of pulmonary metastasis [60]. Furthermore, RANKL has been considered to orchestrate the migration of cancer cells, acting as either a chemoattractant or a “soil” factor [61].
The leucine rich repeat containing G protein-coupled receptor 4 (LGR4) is a recently recognized receptor interacting with RANKL, inhibiting osteoclast differentiation and bone resorption. Recent data implicate the RANKL/LGR4 interface in cancer cells proliferation [62].
However, the prognostic value of RANKL expression in cancer is further complicated by the increasingly reported antitumor aspect of RANKL signaling [34, 63,64,65]. For instance, Park et al. designated immunohistochemical RANKL expression in breast cancer tissues as independent predictor of improved skeletal-disease free survival in breast cancer patients [34]. Bhatia et al. observed that absence of RANKL immunohistochemical expression on breast cancer tissues correlated with a metastatic breast cancer phenotype. The authors postulated that decrease or absence of RANKL expression on cancer cells may allow the interaction of RANK expressed on cancer cells with RANKL expressed on normal osteoblasts and stromal cells of the bone, guiding osteotropism, thereby orchestrating a metastatic phenotype [63]. Further, decreased RANKL transcript levels in breast cancer samples in comparison with normal tissue has been associated with increased risk of local recurrence, bone metastases or disease-related death in breast cancer [64]. Interestingly, an antitumor RANK-c isoform inhibiting RANK signaling has been recently discovered by Papanastasiou et al. in breast cancer. This isoform was upregulated in breast cancer cells lines and was inversely associated with tumor grade and proliferation index [65].
The TRAIL-induced signaling transduction in cancer
Despite the established role of TRAIL in initiation of apoptosis of cancer cells [25,26,27,28], the latter may acquire resistance to TRAIL-induced apoptotic signaling through a plethora of mechanisms, involving the non-signaling TRAIL receptors TRAILR3, TRAILR4, and OPG [27]. Non-canonical TRAIL-induced cascades, downstream of TRAIL/TRAILR interaction, have been demonstrated to involve pivotal kinases, such as JNK, p38, ERK, protein kinase C (PKC), and PI3K/AKT, resulting in a dual signal transduction: tumor-promoting and antitumor. Cancer cells that escape from TRAIL-induced tumor immune surveillance have been postulated to highjack, through mechanisms as yet unidentified, the aforementioned pathways and divert them towards pro-survival, pro-proliferative, and promigratory signal transduction [25,26,27,28].
The role of TRAIL in the context of bone oncology is underexplored and rather dual: both antitumor and tumor-promoting. TRAIL has been shown in vivo in an animal model to reduce tumor incidence and growth and prevent tumor-related osteolysis, increasing significantly animals’ survival [66]. This effect consolidated the in vitro antitumor role of TRAIL observed previously in Ewing’s sarcoma and osteosarcoma cell lines. On the other hand, TRAILR1 has been suggested as a novel circulating tumor cells (CTC) marker for giant cell tumor of bone (GCT). The subpopulation of circulating tumor stem-like cells (CTSC) that expressed TRAILR1 showed increased potential for tumor formation in vitro and in vivo, increased resistance to denosumab (monoclonal antibody against RANKL), more frequent detection in circulation, and increased induction of tumorigenesis after serial adoptive transplantation in comparison with GCT cells non expressing TRAILR1 [67].
Given the complex role of TRAIL in cancer, the OPG-induced inhibition of TRAIL signaling extends beyond being a cancer cell strategy against TRAIL-mediated apoptosis [68]. Moreover, it is conceivable that the clinical outcome of the interplay OPG/RANKL/TRAIL is defined by a dynamic equilibrium among RANKL, TRAIL, and OPG [68].
Could the OPG/RANKL/TRAIL interplay fertilize the bone to be a “soil” for metastasis?
Basic principles of bone homing and colonization
Intense research over the last decades has revealed the steps of the metastatic process: metastatic “seeding”, generation of pre-metastatic niches, dormancy and reactivation of disseminated tumor cells (DTC). The creation of an immunosuppressive landscape is a prerequisite for the entire process [69]. Despite the great body of theories that have been postulated to interpret the development of metastases, an outstanding question is why some cancer types predilect to metastasize to bone. One of the most challenging issues as regards bone homing and colonization is to decipher the interaction of bone microenvironment with the genetic profile of cancer cells that is specific for bone tropism.
Though not completely elucidated, the generation of bone metastases has been conceived as a stepwise process. Epithelial–mesenchymal transition (EMT) endows cancer cells with an invasive phenotype that enables detachment from primary tumor, intravasation, survival in the blood stream evading immune surveillance and sheer stress of the blood flow, extravasation, homing to bone, and colonization of bone. Some of the bone colonizing cells adapt to bone microenvironment and grow immediately competing with hematopoietic stem cells (HSC) for the endosteal niche, while others enter a dormancy state. Reactivation of DTC gives rise to bone metastases [70].
Refining the prevailing “seed and soil” hypothesis of Paget, Sleeman et al. provided a comprehensive insight into the metastatic process, suggesting the “stromal progression hypothesis”. The authors postulated that the interplay between the “genetic signature” of cancer cells and the inflammatory tumor stroma, governs not only the early stages of carcinogenesis in primary sites but also the metastatic process resulting in creation of metastatic niches [71]. In that respect, the process of bone homing and colonization is determined by the cross-talk between DTC and bone microenvironment. This cross-talk is facilitated by the unique characteristics of bone microenvironment, principally: (i) the high vascularity of red marrow; (ii) the direct cancer cell–mesenchymal stromal cells interactions; (iii) the secretion of growth factors, angiogenic factors, and bone-resorbing factors from a multitude of cells in bone marrow (BM) [72].
Fundamental cells of BM are considered orchestrators of bone homing and colonization: (i) the bone mesenchymal stem cells (BMSCs), which constitute multipotent cells capable of differentiating into osteoblasts, chondrocytes, and adipocytes; (ii) the pericytes, (iii) the fibroblasts; and (iv) the osteoblasts.
Recently, bone marrow adipocytes (BMAs) have been brought into spotlight as crucial determinants of the predilection sites of bone metastasis, being abundant near the endosteal surface of diaphysis and the trabecular bone of epiphysis and metaphysis [73]. In 2015, the innovative study of Scheller et al. suggested an explanation for the proclivity of bone metastases for regions such as proximal femur, hip, and lumbar spine. The authors demonstrated in vivo that the “regulated bone marrow adipocytes” (rBMAs), which prevail in these sites, provide cancer cells with a survival advantage in challenging conditions, contrary to the stable “constitutive bone marrow adipocytes” [74]. BMA (especially the rBMA) play a tumor-promoting role through supplying free fat acids as a fuel for cancer cells, increasing leptin/adiponectin ratio, upregulating IL-6, and inducing the differentiation and activation of osteoclasts [73]. In vitro experiments have demonstrated that the parathyroid hormone related protein (PTHrP)-induced expression of thermogenic genes, such as uncoupling protein 1 (UCP1) and iodothyronine deiodinase 2 (Dio2), stimulates lipolysis in mature adipocytes, thereby providing cancer cells with energy [75]. This effect may contribute to the prometastatic effect of PTHrP for bone, but not visceral, metastases that has been demonstrated in vivo [76].
The molecular mechanisms underlying bone metastases constitute an evolving field of research, as extensively discussed elsewhere [77,78,79,80,81]. The present review highlights the evidence that sustains the implication of the RANKL/RANK/OPG axis in bone metastases. RANKL/RANK/OPG axis is expressed on cancer cells and in components of tumor as well as bone microenvironment.
The RANKL/RANK/OPG axis in bone homing and colonization
Bone is a metabolically active organ governed by a continuous remodeling cycle, finely balanced between bone formation and resorption; therefore, it is conceivable that “osteomimicry”, i.e. the acquisition from cancer cells of a bone cell–predominantly an osteoblast-like—phenotype, is mandatory for bone colonization [82].
Although osteomimicry is not completely understood, it clearly entails the presence of an “osteoblast-like gene signature” in osteotropic cancer cells, which encompasses several bone related genes (BRG) encoding proteins-hallmark of osteoblasts [83]. Bone metastatic breast cancer tissues have been shown to highly co-express a constellation of BRG that encode: (i) bone matrix proteins (e.g. asporin [ASPN], cartilage oligomeric matrix protein [COMP], secreted protein acidic and cysteine rich [SPARC]); (ii) extracellular matrix-degrading enzymes (e.g. matrix metallopeptidases MMPs, metalloendopeptidases [ADAMs], urokinase-type plasminogen activator [PLAU/uPA]); (iii) tissue inhibitors of metalloproteinases [TIMPs]; (iv) osteoblast cadherin [OB-cadherin/CDH11]; (v) osteoblast transcription factor (e.g. Runt-related transcription factor 2 [Runx2]); and (vi) cytokines (e.g. Stem Cell Growth Factor 1 [SCGFI]). Runx2 was shown in vitro to mediate the cancer-associated fibroblasts (CAF)/BMP2-induced co-expression of BRG in breast cancer cells through direct and indirect regulation of their transcription. The BRG expression was demonstrated in vitro to be implicated in chemotaxis, adhesion, anchorage-independent growth and proliferation of breast cancer cells colonizing the bone microenvironment [84]. The Wnt and the RANKL/RANK/OPG pathways are integrated in the osteoblast-like osteotropic profile of bone colonizing cancer cells. The Wnt signaling acts autocrinally to trigger cancer cell proliferation and osteotropism and paracrinally to favor osteoblast differentiation.
The in vivo study of Chu et al. revealed that RANKL expressed by prostate cancer cells in the setting of osteomimicry contributed to establishment of a pre-metastatic niche stimulating a “feed forward loop”. Especially, RANK/RANKL signaling induced the reprogramming of cancer cell and a prometastatic signal transduction through activation of crucial transcription factors, such as c-Myc/Max and AP4. The latter were shown to regulate the expression of genes implicated in EMT (Twist1, Slug, Zeb1, Zeb2), stem cell characteristics (Sox2, Myc, Oct3/4, and Nanog), neuroendocrine differentiation (Sox 9, HIF-1α and FoxA2), and osteoblast-like phenotype (c-Myc/Max, Sox2, Sox9, HIF1α and Runx2) [85].
It is known that signaling cascades required for EMT, invasion, motility, adaptation to bone microenvironment, and reactivation of dormant cancer cells involve multitude molecules, such as cell adhesion molecules, proteolytic enzymes (e.g. MMPs, cathepsins, CD26/DPPIV and uPA), chemokine receptors, integrins, and kinases indispensable for pro-proliferative and pro-survival signals (e.g. ERK1/2, p38, FAK/AKT), most of which being regulated by or interacting with RANKL/RANK/OPG axis [69, 86]. For instance, RANKL has been shown to favor EMT in mammary epithelial cells, in head and neck squamous carcinoma, in endometrial cancer, and in prostate cancer cells [57, 59, 87, 88]. An interesting mechanism through which RANKL induces EMT has been demonstrated in vitro in breast cancer cells, implicating increase of expression of vimentin, N-cadherin, Snail, and Twist and decrease of E-cadherin expression, as well as activation of NF-κB [89].
Moreover, bone homing and colonization appears to recapitulate a cross-talk between tumor and immune cells in which RANKL prevails. In particular, RANKL—expressed not only in osteoblasts but also in T and B cells—interfaces with its receptors on DCs, monocytes, macrophages, and tumor cells to create a “bridge” between immune system and bone remodeling [78]. Convincing evidence sustains this hypothesis. DCs, also known as antigen-presenting cells (APC), play a key role in regulation of cytotoxic T cell immune response activation [90]. In vitro and in vivo studies have shown that the interaction of RANK expressed on DCs with RANKL overexpressed on activated T cells fosters the survival of T cells, thereby priming T cells. FOXP3+ T regs (CD4+ T cells), which are well established immune suppressors, have been demonstrated as a major source of RANKL implicated in homing of cancer cells to bone [59]. Th17 cells, another crucial subset of CD4+ T cells, have been postulated to reinforce the osteoclastic activity and thus the osteolytic bone disease via the RANKL pathway [90].
Consolidating the hypothesis that the local chemokine milieu in bone microenvironment attracts preferentially certain cancer cells, Jones et al., in 2006, provided in vitro and in vivo evidence concerning the role of RANKL as a “soil” factor, which stimulates cytoskeletal changes, facilitating the migration of several human epithelial RANK expressing cancer cells. Interestingly, this study showed that RANKL could foster the migration of normal, non-transformed, cells as well [91].
RANKL and M-CSF—both indispensable for osteoclasts differentiation and activation—have been shown to be released from BMA, stimulating the BMA-mediated induction of osteolysis [73]. Interestingly, in vivo data indicate that this function is a characteristic of BMA at a preadipocyte stage [92, 93] and it is not observed in white adipose tissue [93]. Takeshita et al. working on a mouse model showed that the early adipogenic transcription factors C/EBPβ and C/EBPδ, but not the late factor peroxisome proliferator-activated receptor γ, elicit the transcription of RANKL gene through binding to RANKL promoter [92]. Notably, early loss of parathyroid hormone 1 receptor (PTH1R) signaling in BMSCs in mice was shown in vivo to result in uncontrolled differentiation of these cells to BMA capable of producing high levels of RANKL, inducing bone resorption [93].
The precise role of OPG in the context of bone metastatic niche remains controversial. It is postulated that the anti-osteoclastogenic effect of OPG may contradict bone colonization; yet, the direct and indirect tumor-promoting effect of OPG could be consistent with bone colonization. More studies are needed to clarify this issue. Whether OPG interacts with EMT is underexplored. The possibility of a role of OPG in EMT either directly or indirectly through abrogating RANKL/RANK interaction is an appealing hypothesis that merits future investigation.
Finally, it should be mentioned that cancer cells are devoid of bone resorption properties, but they may fuse with OCPs or bone marrow-derived cells to create mature osteclasts. In fact, genes-landmark of osteoclasts, such as cathepsin K, are included in the “osteomimicry gene signature”.
Imbalance in the OPG/RANKL/RANK ratio as an orchestrator of cancer cell–bone cell interactions
The interrelationship of cancer cells with bone microenvironment spurs a “vicious cycle” wherein cancer cells activate the osteoclasts to degrade the bone, releasing growth factors stored in bone matrix, which in turn foster the growth of tumor. This “vicious cycle” is a hallmark of bone metastases of solid tumors, of primary lesions of bone tumors such as osteosarcoma, and of bone disease related to multiple myeloma (MM). Considering that the balance among OPG, RANKL and RANK is credited with maintenance of skeletal integrity, it appears rational that the imbalance thereof is fundamental in bone oncology [94].
Indeed, it has been postulated that cancer cells hijack the host equilibrium among OPG, RANKL and RANK, favoring bone destruction via upregulation of RANKL in concert with either downregulation of OPG or rarely upregulation—but to a lesser extent than RANK—of OPG [95]. In the context of a cross-talk between cancer cells, bone microenvironment and tumor microenvironment, cancer cells recruit bone stromal cells—fibroblasts, endothelial cells, immune cells, and osteoblasts—via secretion of osteoclastogenic cytokines and growth factors, including IL-1, IL-6, IL-8, IL-11, TNF-a, PTHrP, that act in a paracrine, autocrine and endocrine way to initiate a “vicious cycle”. The tumor-induced M-CSF and RANKL expressed on osteoblasts, mesenchymal stem cells (MSCs), and immune cells interact with c-fms and RANK on OCPs, in order to induce the differentiation and activation of osteoclasts. Accordingly, the osteoclasts-driven resorption of bone matrix releases growth factors, including transforming growth factor beta (TGF-b), insulin-like growth factor 1 (IGF-1), platelet-derived growth factor (PDGF), and FGF, which enhance the tumor proliferation, perpetuating the “vicious cycle”. In response to the stimulative effect of tumor microenvironment, cancer cells per se produce RANKL, indicating a reciprocal dialogue between tumor and bone microenvironment. Moreover, osteoclasts nurture the “vicious cycle” via producing protumorigenic and prometastatic factors that foster the growth of bone tumors and metastatic lesions. In parallel, activated T cells in tumor microenvironment contribute to production of RANKL [94, 96]. Intriguingly, prolactin has been shown to transduce osteoclastogenic signaling, mainly via sonic hedgehog (SHH), that in turn activates ERK, reinforcing the “vicious cycle” [96]. As noted earlier, in osteosarcoma, this “vicious cycle” has been shown in vivo to be inhibited by TRAIL, reducing both osteolysis and tumor growth [66].
More intricate is rendered the signaling in cancer-induced osteoblastic, and mixed—both osteolytic and osteoblastic—lesions, emphasizing the multifaceted role of TGF-b [69]. Beyond TGF-b, a plethora of tumor-derived pro-osteoblastic growth factors and cytokines released through the osteolytic process favor the osteoblastic function, amplifying the osteoblastic lesions.
The clinical relevance of OPG in the “vicious cycle” recapitulates the interrelation of its tumor-promoting role and its antitumor role. While OPG signaling per se harbors an inherent tumor-promoting potential, the OPG-mediated abrogation of RANKL—and TRAIL-induced signaling could be rendered both tumor-promoting and antitumor. Table 1 depicts the in vitro and in vivo evidence sustaining the tumor-promoting role of OPG. It is highlighted that the majority of preclinical and clinical data building on the role of OPG in cancer are derived from studies outside the bone, necessitating further evaluation in the setting of bone oncology. Moreover, most studies pursuing skeletal metastatic disease focus on breast and prostate cancer; thus, it is mandatory to further address bone metastases in the context of other cancer types.
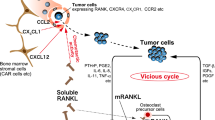
The RANKL/OPG axis in the context of the “vicious cycle” of the interplay among cancer cells, tumor microenvironment, and bone microenvironment is illustrated in Fig. 1.

The RANKL/OPG axis in the “vicious cycle” of the interplay among cancer cells, tumor microenvironment, and bone microenvironment (1) Cancer cells secrete cytokines and hormones/growth factors, including IL-1, IL-6, IL-8, IL-11, TNF-a, PTHrP, resulting in recruitment of bone stromal cells (e.g. osteoblasts, immune cells). (2) Bone stromal cells, mainly the osteoblasts and the immune cells, are induced by cancer cells to overexpress RANKL, which acts on osteoclast precursors. (3) RANKL promotes differentiation and activation of osteoclasts. (4) Increased osteoclastic activity leads to increased bone resorption, the hallmark of osteolytic lesions (5). Increased bone resorption, releases growth factors that result in tumor growth (6) and potentially increased osteoblastic function (7), leading to increased bone formation (8), the hallmark of osteoblastic lesions (9). (10) Osteoclasts-derived growth factors nurture the tumor. (11) Cancer cells per se produce RANKL. (12) Osteoblasts produce OPG which abrogates the RANKL-induced differentiation and activation of osteoclasts and potentially either fosters or inhibits tumor growth (13). (14) Bone metastases represent a continuum of dysregulated bone remodeling extending from predominantly osteolytic lesion to predominantly osteoblastic lesions. (15) Tumor growth perpetuates the “vicious cycle” aggravating the skeletal tumor burden. ↑: increase; ↓: decrease
The role of RANK/RANKL/OPG axis in osteosarcoma
Osteosarcoma is the most prevalent malignant pediatric primary bone tumor and the second most frequent cause of cancer-related death in pediatric population [97]. It usually affects the metaphysis of long bones and is diagnosed in the second decade of life [98]. The five-year survival rate is estimated 65% for localized disease, declining at 20% in case of lung metastases at initial diagnosis.
In 2007, Mori et al. reported for the first time the expression of RANK in osteosarcoma in vitro in human osteosarcoma cell lines and ex vivo in pathological tissues, showing also that the interaction RANKL/RANK phosphorylates the kinases ERK1/2, p38, and inhibitor of nuclear factor-Κb (IκB) [99]. Osteosarcoma, originated from osteoblast cell lineage, could provide the archetype of the deleterious “vicious cycle” orchestrated by RANKL/RANK interaction [97], resulting in mixed, osteolytic and osteoblastic, lesions.
Indeed, a great body of convincing evidence implicate RANK signaling in osteosarcoma. For instance, increased RANKL expression on cancer cells was observed in osteosarcoma-bearing mice in the study of Lamoureux et al. In this study, RANKL expression led to osteosarcoma related osteolysis, a process abrogated by OPG [100].
In 2010, Molyneux et al., working on a transgenic mouse model of osteosarcoma, showed that abundant expression of RANKL due to loss of the Protein Kinase CAMP-Dependent Type I Regulatory Subunit Alpha (Prkar1a) bone tumor suppressor gene enhanced tumorigenesis [97]. Furthermore, aberrant RANK expression and signaling has been noted in human [99] and mouse [101] osteosarcoma cell lines.
Prompted by this evidence, Beristain et al. selected RANK-positive mouse osteosarcoma and RANK-negative preosteoblastic MC3T3-E1 cells and subjected them to loss- and gain-of-RANK function analyses in order to explore the hypothesis that RANK, via an autocrine (homotypic) signaling cascade, incites tumorigenis in cells of both epithelial and mesenchymal origin. Among a wide array of tumorigenic properties examined, RANK homotypic signaling proved to exert an insignificant effect on cell proliferation, while favoring cell motility and anchorage-independent growth of osteosarcoma cells and preosteoblasts. On the other hand, RANK signaling in non-tumorigenic mammary epithelial NMuMG cells evoked their proliferation and anchorage-independent growth, while motility was not affected. Furthermore, RANK signaling activated kinase ERK1/2, a step pivotal for the anchorage-independent survival and invasion of osteoblastic cells, as well as the proliferation of mammary epithelial cells. The authors concluded that a cell-autonomous homotypic mechanism underlies the contribution of RANK signaling to tumorigenesis [102].
The landmark study of Chen et al. provided strong evidence on the efficacy of RANKL inhibition for the management of osteosarcoma in animal models. The researchers demonstrated that loss of a single Prkar1a allele due to increase of RANKL levels is a key mechanism in osteosarcoma genesis regardless of either RB or protein p53 status in genetically engineered mouse model (GEMM). Moreover, increased RANKL levels proved to be associated with increased osteosarcoma aggressiveness. The importance of this RANKL effect was highlighted by the observation that whole-body RANKL deletion completely abrogated tumorigenesis, while osteoclastic RANK deletion in GEMM led to attenuated tumor initiation and prolonged life span; this impact was related to inactivation of osteoclastogenesis and up-regulation of the tumor suppressor gene PTEN. To strengthen their observations, the authors treated GEMM with RANK-Fc, which resulted in a 50% prolongation of life span with 3 times decrease in lung metastases compared to non-RANK-Fc treated mice [103].
Recently, the seminal study of Navet et al. implicated intrinsic (tumoral) RANK signaling in the metastatic process of osteosarcoma. However, this prometastatic effect appeared not to be ascribed to direct promotion of tumor cell proliferation, but rather to chemoattraction of RANK expressing osteosarcoma cells by RANKL expressing cells in bone microenvironment (extrinsic) [98]. In this study, in vitro and in vivo experiments showed for the first time that RANK overexpressing osteosarcoma cells present increased migration capacity only in immune-deficient mice. Given that chemotherapy is known to induce immunosuppression, the authors postulated that in case of cancer resistant to treatment, RANK overexpressing cancer cells could contribute to a metastatic process mediated through RANKL/RANK signaling. Consequently, abrogation of this RANKL/RANK mediated metastatic effect via an antibody blocking RANKL was suggested by Navet et al. as a strategy to prevent metastasis in an immune-compromised context [98].
Furthermore, considering that RANKL proved to have no effect on proliferation but a positive effect on differentiation of RANK-expressing osteosarcoma cells, Navet et al. hypothesized that RANK expressed by osteosarcoma cells could be a factor of good prognosis. However, RANKL/RANK signaling was observed to exert a prometastatic effect fostering the development of lung metastases. As the background of this aggressive behavior was suggested the RANKL-induced expression of MMP on RANK expressing osteosarcoma cells, which could promote the escape of cancer cells from the initial site. Additionally, RANKL might also favor seeding of cancer cells to lungs through mechanisms still unknown, likely independently of RANK and OPG expression. Given that LGR4 is considered a third receptor of RANKL known to be involved in progression, migration and metastatic processes, the role of the RANKL/LGR4 interaction in the ontogenesis of osteosarcoma merits further exploration [98].
Navet et al. provided clinical data obtained through immunohistochemical detection of RANKL/RANK/OPG on biopsies of a cohort of osteosarcoma patients with or without metastases, showing no significant difference of intrinsic RANK expression between the two group of patients. In fact, RANKL availability in microenvironment proved to be the major determinant of metastatic dissemination. Decreased OPG/RANK ratio, observed in this study, was postulated to facilitate the interaction of RANKL available in the microenvironment with RANK expressed by osteosarcoma cells, thereby predisposing to metastatic dissemination [98].
The involvement of OPG/RANKL/RANK axis in the colonization of the metastatic organ is not a prerogative for bone-homing tumors. In fact, the prometastatic effect of RANKL/RANK interaction in the setting of lung-homing osteosarcoma is not unexpected, given that RANKL/RANK is implicated in bone resorption, metastasis, and immune surveillance evasion [103]. The correlation of decreased OPG/RANK ratio with the potential of osteosarcoma to metastasize to lungs, observed by Navet et al., may indicate an anti-metastatic effect of OPG. Due to the dual role of OPG in carcinomas and the limited knowledge concerning OPG in sarcomas, the comparison of the role of OPG in the metastatic potential of these two types of cancer is daunting. These intriguing issues could represent an appealing field of future research.
The role of RANKL/RANK/OPG axis in multiple myeloma
Multiple myeloma (MM) is a rare clonal plasma cell neoplasm with an age-standardized incidence rate of 2.1 per 100,000 persons globally in 2016 [104]. The majority of patients (≈ 90%) develop bone lesions, which result in serious SREs. Despite recent therapeutic advances, MM remains an incurable disease with substantial morbidity and mortality. Ongoing efforts for devising new therapeutic strategies capitalize on the cross-talk between MM cells and surrounding bone marrow components, which culminates in enhancement of growth of MM cells and bone destruction. Numerous studies have demonstrated that the BMSCs stimulated by adherent MM cells secrete plenty of cytokines that enhance the osteoclast-induced bone resorption, which in turn releases growth factors fostering the growth of MM cells [105]. The present article focuses on the key role of the RANKL/OPG interplay in the pathogenesis of MM related bone disease.
The hallmark of the MM related bone destruction is the MM cells induced increase of RANKL/OPG ratio, ascribed to upregulation of RANKL expression and downregulation of OPG expression. MM cells directly secrete RANKL or stimulate its secretion by T lymphocytes and apoptotic osteocytes [106]. The syndecan-1 expressed on MM cells has been shown in vitro to bind the heparin-binding domain of OPG, resulting in internalization and eventually degradation of OPG [30]. Certain MM-derived factors inhibit osteoblasts formation, leading to decreased OPG production, namely the sclerostin, the dickkopf-1 (DKK1), and the secreted Frizzled-related protein (sFRP)2/3 (inhibitors of the canonical Wnt pathway), as well as the IL-7, which downregulates Runx2 [106]. The chemokines macrophage inflammatory protein (MIP)1-a and MIP1-β, secreted by MM cells, interact with the integrin (α4β1) very late antigen-4 (VLA-4) and the vascular cell adhesion protein 1 (VCAM-1) on BMSCs to enhance both RANKL upregulation and OPG downregulation. Elevation of IL-3 in BM in MM patients acts in concert with RANKL and MIP1 to promote osteoclasts activation and bone destruction. Furthermore, activation of Notch signaling in MM cells and OCPs, evoked by BMSCs and MM cells, potentiates RANKL signaling [105].
Though beyond the scope of the present review, several additional mechanisms are implicated in the pathophysiology of MM, including: (i) overexpression of IL-6 by osteoclasts, BMSCs, and osteoblasts, resulting in MM cell expansion and bone destruction; (ii) production of tumor necrosis factor-a (TNF-a), B cell activating factor of TNF-a family (BAFF), and A proliferation inducing ligand (APRIL) by MM cells, boosting the survival thereof; (iii) production of numerous cytokines and growth factors by BMSC that foster MM cell proliferation, such as vascular endothelial growth factor (VEGF), SDF-1α, and IGF-1 [107, 108].
Seum RANKL/OPG ratio was demonstrated as a prognostic factor for MM in the study of Terpos et al.: Patients with ratio value less than 1 showed a 5-year probability of survival rate of 89%, while all patients with a ratio greater than 3 survived for less than 4 years [108].
Of clinical relevance is the elevation of RANKL and RANKL/OPG ratio in MM in parallel with the increase of the disease stage and the severity of bone lesions. Furthermore, serum RANKL levels and RANKL/OPG ratios have been correlated significantly with angiogenic cytokines (Hepatocyte growth factor [HGF] and VEGF), and factors of disease activity, such as IL-6, β2-microglobulin, and lactate dehydrogenase (LDH) [109].
Novel insights into the origin of RANKL in MM patients are provided by the clinical study of Spanoudakis et al. The authors demonstrated the contribution of invariant NKT (iNKT) TCRVα24 Jα18/Vβ11 cells—a subset of CD1d-restricted, glycosphingolipid (GSL)-specific immunoregulatory T cells regulating a variety of immune responses—to the increase of RANKL in MM. iNKT cells from peripheral blood (PB) and BM of MM patients were shown to express higher levels of RANKL than PB iNKT cells from normal subjects, whereas the expression of RANKL in BM iNKT cells of MM patients is higher than that observed in BM T cells or autologous PB iNKT and T cells. Importantly, a strong association of the activity of bone resorption with RANKL overexpressed on iNKT cells in MM patients was demonstrated through assessment of β-C-terminal telopeptide levels. Interestingly, enrichment of BM with iNKT cells compared to PB was designated as a specific feature of MM. Finally, in this study, iNKT cells in normal individuals proved to be a richer source of surface and likely soluble RANKL than conventional T cells both ex vivo and in vitro [110].
Pitari et al. demonstrated that the miR-21, an OPG-targeting miRNAs acting as a negative regulator of OPG gene expression, was upregulated in BM adherent to MM cells, downregulating OPG in HS-5 BM stromal cells. Consequently, the constitutive miR-21 inhibition significantly increased OPG production. Importantly, miR-21 was also shown to regulate RANKL production in BMSCs and osteoblasts via a feedback loop involving IL-6 and STAT3 signaling. Especially, miR-21 expression was stimulated by IL-6 in the presence of activated STAT3, while enhancing per se the phosphorylation of STAT3 via inhibition of PIAS3—a specific inhibitor of STAT3 phosphorylation. Phosphorylated STAT3 is known to stimulate the RANKL gene expression through IL-6 signaling; thus, the miR 21-induced STAT3 activation led to increased RANKL expression. Accordingly, the miR-21 inhibition is supposed to attenuate the osteolytic activity of BMSCs in a dual manner: increasing OPG, while decreasing RANKL production. This restoration of the aberrant RANKL/OPG ratio was shown to counteract the BM-related osteoclastic activity in vitro [111].
Additionally, silencing of miR-9718, which acts as a specific inhibitor of PIAS3, has been shown to attenuate RANKL-induced osteoclastogenesis in vivo [112].
In 2018, Food and Drug Administration (FDA) approved the administration of denosumab—a monoclonal antibody against RANKL—for prevention of skeletal-related events in MM patients, based on the landmark Phase III ‘482 study. Denosumab blocks pharmacologically the interface RANKL/RANK in a way reminiscent of the action of OPG [113]; yet, this issue is beyond the scope of the present review.
Exploration of the therapeutic potential of OPG
With the advent of current millennium, the development of the OPG construct OPG-Fc launched a new era in bone oncology, aiming at intercepting not only the cancer-related osteolysis but also the tumor growth. This construct was generated via removal of the signal peptide, the heparin binding domain and the death domain of native OPG, followed by fusion of the remaining peptide to the Fc domain of human IgG1. OPG-Fc comprises aa 22-194 of native OPG, maintains the potent dimeric form, and has a prolonged half-life compared to native OPG. It has been demonstrated to inhibit hypercalcemia and bone resorption induced by IL-1b, TNF-a, PTH, PTHrP, and 1, 25(OH)2 D3 [114].
Compelling evidence derived from several mouse models sustain the antitumor effect of OPG [114]. Administration of OPG-Fc or RANK-Fc in mouse models of breast, prostate, colon, and non-small lung cell (NSCLC) cancer, as well as epidermal carcinoma and melanoma has resulted in significant amelioration of overall survival, prevention of bone metastases, and decrease of tumor burden in bone [115]. Moreover, OPG-Fc has been proved efficient in treatment of humoral hypercalcemia of malignancy [116]. Additionally, OPG-Fc has been depicted to reduce the skeletal tumor burden in a syngeneic model and a nude mouse model, wherein osteolytic lesions were generated by, respectively, colon adenocarcinoma (col 26) cells and MDA-MB-231-breast cancer cells. This effect was ascribed to elimination of osteoclasts, while no extraosseous metastatic site was affected [117]. Interestingly, an inhibitory effect of recombinant OPG on prostate cancer-related skeletal tumor osteoblastic burden has been observed, highlighting that the osteoclastic function is inherent to osteoblastic lesions [118]. Finally, administration of OPG in a mouse model of bone cancer pain has been shown to blunt both peripheral and central sensitization, reducing the prohyperalgesic peptide DYN in spinal cordal and the c-Fos in deep dorsal horn. These alterations likely emerge from inhibition of osteoclastic activity [119].
The above-mentioned work preceded the FDA approval of denosumab in bone oncology in 2010 [113, 120], an issue beyond the scope of the present review. However, the advent of denosumab has not overshadowed the evolving research on the therapeutic potential of OPG, which continues to be very informative.
In 2015, Ottewell et al. demonstrated in vivo that the administration of OPG-Fc can counteract the growth of dormant MDA-MB-231 cells disseminated in bone, interrupting the RANKL/RANK interface [121].
Miller et al., in 2014, reported the effects of OPG-Fc in combination with docetaxel as regards tumor-induced osteolysis, tumor burden, and survival in the H1299Luc mouse model of NSCLC metastasis in bone. In mice with established NSCLC bone metastases, administration of OPG-Fc as monotherapy led to a 84.1% skeletal tumor growth abrogation compared with vehicle control at the end of the study (day 21). Docetaxel (35 mg/kg) as monotherapy led to a 96.5% reduction in skeletal tumor burden, while the combination of OPG-Fc and docetaxel (35 mg/kg) resulted in the most pronounced reduction in skeletal tumor burden at day 21 (99.7%) in comparison with either docetaxel alone (p < 0.001) or OPG-Fc alone (p < 0.001). The effect of docetaxel was attributed to reduced tumor burden, while the effect of OPG-Fc was ascribed to decreased osteoclastic activity. Moreover, RANKL inhibition via OPG-Fc completely averted the osteolytic bone lesions both as monotherapy and in combination with docetaxel [122]. Effective treatment of established bone metastases with an OPG-Fc armed conditionally replicating adenovirus (CRAd) in a murine model of osteolytic bone metastases of breast cancer has been reported by Cody et al., sustaining the antitumor potential of exogenous administration of OPG [123]. The combination of OPG-Fc with tamoxifen was reported to halt the skeletal metastatic bone disease in a mouse model of estrogen receptor-positive breast cancer, in 2012. In this model, OPG-Fc was shown to inhibit osteoclastic activity and prevent tumor-induced osteolysis, resulting in reduction of skeletal tumor burden. Tamoxifen as a single agent reduced MCF-7Luc tumor growth in the hind limbs. Combination of OPG-Fc with tamoxifen resulted in significantly enhanced inhibition of tumor growth compared with the inhibition observed with either single agent as monotherapy. OPG-Fc either as monotherapy or combined with tamoxifen, eliminated the osteolytic lesions [124]. Taken together, the above mentioned in vitro and in vivo data consolidate the indirect antitumor aspect of OPG ascribed to abrogation of RANKL.
Challenges in the therapeutic implementation of OPG
Despite the presumed antitumor effect of exogenous administration of OPG, the therapeutic implementation of OPG raises skepticism given the preclinical and clinical data sustaining the tumor-promoting effect of OPG. Indeed, the study of Zinonos et al. underscored the dual dynamics of OPG signaling, investigating the OPG overexpression in a xenogeneic murine model of osteolytic breast cancer: whereas OPG overexpression resulted in reduction of bone tumor burden and prevention of cancer-related osteolysis, it was correlated with increased propensity of cancer cells to colonize the lung [125].
However, it is highlighted that OPG-Fc is featured by absence of the death domain and the heparin binding domain of full length OPG. This discriminating feature might endow OPG-Fc with an antitumor potential, given that glycosaminoglycans binding to aa 195-401 domain of full length OPG (the region absent in OPG-Fc) have been implicated in the modulation of its bioactivity and its function [126]. Moreover, the proteins assigned to transduce tumor-promoting signals have been shown to bind the heparin binding domain of OPG; thus, the truncated form OPG-Fc (aa 1-194) is expected to be deprived of the tumor-promoting effects of full length OPG (aa 1-401).
The administration of the recombinant osteoprotegerin construct AMGN-0007 initially led to suppression of bone resorption in MM and breast carcinoma patients in a phase I study [127]; however, this construct was abandoned early due to fear of stimulating an immune response against endogenous OPG.
Interestingly, the scientific attention has been drawn to emerging innovative experimental strategies exploiting the antitumor potential of OPG, which worth further exploration. Firstly, Lamoureux et al. in a landmark study showed that OPG transgene expression in a rat osteosarcoma model can disrupt the osteosarcoma progression via inhibiting RANKL-induced osteolysis [98]. Secondly, as discussed earlier, inhibition of miR-21 in MM could be proven a therapeutic option for MM-related bone disease [111]. Thirdly, further research is warranted to investigate whether mesenchymal stromal cells (MSCs) transfected with adenovirus carrying the OPG gene could be endorsed as a novel treatment of osteosarcoma [128]. Finally, Higgs et al. attempted to harness the RANKL binding property of OPG averting the binding of TRAIL. The authors introduced a therapeutic approach based on genetically engineered MSCs expressing OPG mut Y49R and F107A that retain RANKL binding while abolishing TRAIL binding. This strategy proved to be effective in vivo, diminishing osteolytic tumor burden in a mouse model of tumor-induced osteolysis [129].
Conclusions and future perspectives
OPG has been revolutionizing the perception of bone biology for over 2 decades. A growing body of evidence points to the key role of OPG in bone oncology concerning both bone metastatic disease and primary bone tumors. The tumor-promoting aspect of OPG is synthesized by a direct—protumorigenic, proangiogenic, prometastatic—effect and an indirect effect exerted through abrogation of the antitumor potential of RANKL and TRAIL. The antitumor aspect of OPG signaling is exerted through inhibiting tumor-promoting potential of RANKL and TRAIL signaling.
More light should be shed into the role of OPG in giant cell tumor of bone (GCTB). GCTB is a primary bone tumor with a unique profile, arising from GCTB neoplastic cells—dysfunctional mesenchymal stromal-like cells—that express RANKL, which interacts with RANK expressed by multi-nucleated osteoclastic giant cells, promoting the osteolytic activity of the latter. This interaction is already targeted pharmaceutically by denosumab; however, OPG merits further exploration in the setting of this tumor [130, 131].
An overarching aim is to identify and modify the factors that determine whether the clinical outcome of the balance among OPG, RANKL, and TRAIL results in elimination or, on the contrary, progression of cancer. Translating biological insights concerning the multifaceted role of OPG in bone oncology into clinical settings raises some outstanding questions: (1) How does the simultaneous presence of biologically relevant concentrations of TRAIL and RANKL influence the outcome of OPG signaling, given that RANKL and TRAIL bind to OPG with equal affinity? (2) Do the ratios RANKL/OPG, RANKL/TRAIL, OPG/TRAIL, RANK/OPG determine the interplay OPG/RANKL/TRAIL? (3) Could OPG, RANKL, RANK serve as predictive and/or prognostic biomarkers of primary bone tumors and/or skeletal metastatic disease? (4) Is there any difference as regards the clinical significance between the serum concentrations of OPG, RANKL, RANK, TRAIL and the tissue expression thereof? (5) Does the diversity of methods used for evaluation of OPG expression in pertinent literature affect the interpretation of the results? (6) How is interpreted the discrepancy between in vitro and in vivo data concerning the OPG-induced abrogation of TRAIL apoptotic signal [132]?
Further research aiming to decipher the expanding repertoire of OPG ligands and downstream signaling cascades could unravel new druggable molecular pathways, empowering the individualization of metastatic and primary bone cancer therapeutics.
References
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
D’Amico L, Roato I (2015) The impact of immune system in regulating bone metastasis formation by osteotropic tumors. J Immunol Res. https://doi.org/10.1155/2015/143526
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319. https://doi.org/10.1016/S0092-8674(00)80209-3
Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, Morinaga T et al (1997) Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun 234:137–142. https://doi.org/10.1006/bbrc.1997.6603
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N et al (1998) Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 139:1329–1337. https://doi.org/10.1210/endo.139.3.5837
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T et al (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176. https://doi.org/10.1016/S0092-8674(00)81569-X
The American Society for Bone and Mineral Research President’s Committee on Nomenclature (2000) Proposed standard nomenclature for new tumor necrosis factor family members involved in the regulation of bone resorption. J Bone Miner Res 15:2293–2296. https://doi.org/10.1359/jbmr.2000.15.12.2293
Emery JG, McDonnel P, Burke MB, Deen KC, Lyn S, Silverman C et al (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273:14363–14367. https://doi.org/10.1074/jbc.273.23.14363
Baud’huin M, Duplomb L, Teletchea S, Lamoureux F, Ruiz-Velasco C, Maillasson M et al (2013) Osteoprotegerin: multiple partners for multiple functions. Cytokine Growth Factor Rev 24:401–409. https://doi.org/10.1016/j.cytogfr.2013.06.001
Mundy GR, Raisz LG, Cooper RA, Schechter GP, Salmon SE (1974) Evidence for the secretion of an osteoclast stimulating factor in myeloma. N Engl J Med 291:1041–1046. https://doi.org/10.1056/NEJM197411142912001
Galasko C (1976) Mechanisms of bone destruction in the development of skeletal metastases. Nature 263:507–508
Martin TJ (2013) Historically significant events in the discovery of RANK/RANKL/OPG. World J Orthoped 4:186–197. https://doi.org/10.5312/wjo.v4.i4.186
Chambers TJ (1980) The cellular basis of bone resorption. Clin Orthop Relat Res 151:283–293
Coleman RE (2006) Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res 12:6243s–6249s. https://doi.org/10.1158/1078-0432.CCR-06-0931
Dai X, Ma W, He X, Jha RK (2011) Review of therapeutic strategies for osteosarcoma, chondrosarcoma, and Ewing’s sarcoma. Med Sci Monit 17:177–190. https://doi.org/10.12659/MSM.881893
van Oosterwijk JG, Anninga JK, Gelderblom H, Cleton-Jansen AM, Bovée JV (2013) Update on targets and novel treatment options for high-grade osteosarcoma and chondrosarcoma. Hematol Oncol Clin North Am 27:1021–1048. https://doi.org/10.1016/j.hoc.2013.07.012
Paget S (1989) The distrubution of secondary growths in cancer of the breast, 1889. Cancer Metastasis Rev 8:98–101
Yamaguchi K, Kinosaki M, Goto M, Kobayashi F, Tsuda E, Morinaga T et al (1998) Characterization of structural domains of human osteoclastogenesis inhibitory factor. J Biol Chem 273:5117–5123. https://doi.org/10.1074/jbc.273.9.5117
Walsh MC, Choi Y (2014) Biology of the RANKL–RANK–OPG system in immunity, bone, and beyond. Front Immunol 5:511. https://doi.org/10.3389/fimmu.2014.00511
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER et al (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390:175–179. https://doi.org/10.1038/36593
Wong B, Josien R, Lee SY, Sauter B, Li HL, Steinman RM et al (1997) TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med 186:2075–2080
Ιkeda T, Kasai M, Utsuyama M, Hirokawa K (2001) Determination of three isoforms of the Receptor activator of nuclear factor-kappa B ligand and their differential expression in bone and thymus. Endocrinology 142:1419–1426. https://doi.org/10.1210/endo.142.4.8070
Boyce BF, Xing L (2007) Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther 9:S1. https://doi.org/10.1186/ar2165
Cheng ML, Fong L (2013) Effects of RANKL-targeted therapy in immunity and cancer. Front Oncol 3:329. https://doi.org/10.3389/fonc.2013.00329
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK et al (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3:673–682
Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A (1996) Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem 271:12687–12690. https://doi.org/10.1074/jbc.271.22.12687
Trivedi R, Mishra DP (2015) Trailing TRAIL resistance: novel targets for TRAIL sensitization in cancer cells. Front Oncol 5:69. https://doi.org/10.3389/fonc.2015.00069
Janssen EM, Droi NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD et al (2005) CD4 + T-cell help controls CD8 + T-cell memory via TRAIL-mediated activation-induced cell death. Nature 434:88–93. https://doi.org/10.1038/nature03337
Wang R, Li JC (2015) TRAIL suppresses human breast cancer cell migration via MADD/CXCR1. Asian Pac J Cancer Prev 16:2751–2756
Holen I, Shipman CM (2006) Role of osteoprotegerin (OPG) in cancer. Clin Sci 110:279–291. https://doi.org/10.1042/CS20050175
Tsukamoto S, Ishikawa T, Iida S, Ishiguro M, Mogushi K, Mizushima H et al (2011) Clinical significance of osteoprotegerin expression in human colorectal cancer. Clin Cancer Res 17:2444–4450. https://doi.org/10.1158/1078-0432.CCR-10-2884
Treskova I, Topolcan O, Windrichova J, Simanek V, Slouka D, Treska V, Kucera R (2018) OPG, OPN, EGF and VEGF levels at individual breslow score stages in malignant melanoma. Anticancer Res 38:4907–4911. https://doi.org/10.21873/anticanres.12806
Vik A, Brodin EE, Mathiesen EB, Brox J, Jørgensen L, Njølstad I et al (2015) Serum osteoprotegerin and future risk of cancer and cancer-related mortality in the general population: the Tromso study. Eur J Epidemiol 30:219–230. https://doi.org/10.1007/s10654-014-9975-3
Park HS, Lee A, Cha BJ, Bae JS, Song BJ, Jung SS (2014) Expression of receptor activator of nuclear factor kappa-B as a poor prognostic marker in breast cancer. J Surg Oncol 110:807–812. https://doi.org/10.1002/jso.23737
Goswami S, Sharma-Walia N (2015) Osteoprotegerin secreted by inflammatory and invasive breast cancer cells induces aneuploidy, cell proliferation and angiogenesis. BMC Cancer 15:935. https://doi.org/10.1186/s12885-015-1837-1
Goswami S, Sharma-Walia N (2016) Osteoprotegerin rich tumor microenvironment: implications in breast cancer. Oncotarget 7:42777–42791. https://doi.org/10.18632/oncotarget.8658
Weichhaus M, Segaran P, Renaud A, Geerts D, Connelly L (2014) Osteoprotegerin expression in triple-negative breast cancer cells promotes metastasis. Cancer Med 3:1112–1125. https://doi.org/10.1002/cam4.277
Heymann MF, Riet A, Le Goff B, Battaglia S, Paineau J, Heymann D (2008) OPG, RANK and RANK ligand expression in thyroid lesions. Regul Pept 148:46–53. https://doi.org/10.1016/j.regpep.2008.02.004
Holen I, Cross SS, Neville-Webbe HL, Cross NA, Balasubramanian SP, Croucher PI et al (2005) Osteoprotegerin OPG expression by breast cancer cells in vitro and breast tumors in vivo—a role in tumor cell survival? Breast Cancer Res Treat 92:207–215. https://doi.org/10.1007/s10549-005-2419-8
Benslimane-Ahmim Z, Pereira J, Lokajczyk A, Dizier B, Galy-Fauroux I, Fischer AM et al (2017) Osteoprotegerin regulates cancer cell migration through SDF-1/CXCR12 axis and promotes tumour development by increasing neovascularization. Cancer Lett 395:11–19. https://doi.org/10.1016/j.canlet.2017.02.032
Goswami S, Sharma-Walia N (2016) Crosstalk between osteoprotegerin (OPG), fatty acid synthase (FASN) and cycloxygenase-2 (COX-2) in breast cancer: implications in carcinogenesis. Oncotarget 7:58953–58974. https://doi.org/10.18632/oncotarget.9835
Chung ST, Geerts D, Roseman K, Renaud A (2017) Connelly L (2017) Osteoprotegerin mediates tumor-promoting effects of Interleukin-1 beta in breast cancer cells. Mol Cancer 16:27. https://doi.org/10.1186/s12943-017-0606-y
Cross SS, Yang Z, Brown NJ, Balasubramanian SP, Evans CA, Woodward JK et al (2006) Osteoprotegerin (OPG)—a potential new role in the regulation of endothelial cell phenotype and tumour angiogenesis? Int J Cancer 118:1901–1908. https://doi.org/10.1186/1476-4598-8-49
Benslimane-Ahmim Z, Poirier F, Delomenie C, Lokajczyk A, Grelac F, Galy-Fauroux I et al (2013) Mechanistic study of the proangiogenic effect of osteoprotegerin. Angiogenesis 16:575–593. https://doi.org/10.1007/s10456-013-9337-x
Mc Conigle JS, Giachelli CM, Scatena M (2009) Osteoprotegerin and RANKL differentially regulate angiogenesis and endothelial cell function. Angiogenesis 12:35–46
Kobayashi-Sakamoto M, Isogai E, Holen I (2010) Osteoprotegerin induces cytoskeletal reorganization and activates FAK, Src, and ERK signaling in endothelial cells. Eur J Haematol 85:26–35. https://doi.org/10.1111/j.1600-0609.2010.01446.x
Zannettino AC, Holding CA, Diamond P, Atkins GJ, Kostakis P, Farrugia A et al (2005) Osteoprotegerin (OPG) is localized to the Weibel-Palade bodies of human vascular endothelial cells and is physically associated with von Willebrand factor. J Cell Physiol 204:714–723. https://doi.org/10.1002/jcp.20354
Scatena M, Giachell C (2002) The alpha(v)beta3 integrin, NF-kappaB, osteoprotegerin endothelial cell survival pathway. Potential role in angiogenesis. Trends Cardiovasc Med 12:83–88. https://doi.org/10.1016/S1050-1738(01)00151-7
Reid PE, Brown NJ, Holen I (2009) Breast cancer cells stimulate osteoprotegerin (OPG) production by endothelial cells through direct cell contact. Mol Cancer 8:49. https://doi.org/10.1186/1476-4598-8-49
Malyankar UM, Scatena M, Suchland KL, Yun TJ, Vlark EA, Giachelli CM (2000) Osteoprotegerin in an αvβ5-induced NF-k B-dependent survival factor for endothelial cells. J Biol Chem 275:20959–20962. https://doi.org/10.1074/jbc.C000290200
Benslimane-Ahmim Z, Heymann D, Dizier B, Lokajczyk A, Brion R, Laurendeau I et al (2011) Osteoprotegerin, a new actor in vasculogenesis, stimulates endothelial colony-forming cells properties. J Thromb Haemost 9:834–843. https://doi.org/10.1111/j.1538-7836.2011.04207.x
Renema N, Navet B, Heymann MF, Lezot F, Heymann D (2016) RANK–RANKL signalling in cancer. Biosci Rep 36:e00366. https://doi.org/10.1042/BSR20160150
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee H et al (2010) Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468:98–102. https://doi.org/10.1038/nature09387
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R et al (2010) RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468:103–107. https://doi.org/10.1038/nature09495
Casimiro S, Mohammad KS, Pires R, Tato-Costa J, Alho I, Teixeira R et al (2013) RANKL/RANK/MMP-1 molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS ONE 8:e63153. https://doi.org/10.1371/journal.pone.0063153
Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL et al (2007) Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 446:690–694. https://doi.org/10.1038/nature05656
Odero-Marah VA, Wang R, Chu G, Zayzafoon M, Xu J, Shi C et al (2008) Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res 18:858–870. https://doi.org/10.1038/cr.2008.84
Chen LM, Kuo CH, Lai TY, Lin YM, Su CC, Hsu HH et al (2011) RANKL increases migration of human lung cancer cells through intercellular adhesion molecule-1 up-regulation. J Cell Biochem 112:933–941. https://doi.org/10.1002/jcb.23009
Yamada T, Tsuda M, Takahashi T, Totsuka Y, Shindoh M, Ohba Y (2011) RANKL expression specifically observed in vivo promotes epithelial mesenchymal transition and tumor progression. Am J Pathol 178:2845–2856. https://doi.org/10.1016/j.ajpath.2011.02.003
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM et al (2011) Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 470:548–553. https://doi.org/10.1038/nature09707
Hanada R, Hanada T, Sigl V, Schramek D, Penninger JM (2011) RANKL/RANK -beyond bones. J Mol Med 89:647–656. https://doi.org/10.1007/s00109-011-0749-z
Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G et al (2016) LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med 22:539–546. https://doi.org/10.1038/nm.4076
Bhatia P, Sanders MM, Hansen MF (2005) Expression of receptor activator of nuclear factor-kappaB is inversely correlated with metastatic phenotype in breast carcinoma. Clin Cancer Res 11:162–165
Owen S, Ye L, Sanders AJ, Mason MD, Jiang WG (2013) Expression profile of receptor activator of nuclear-κB (RANK), RANK ligand (RANKL) and osteoprotegerin (OPG) in breast cancer. Anticancer Res 33:199–206
Papanastasiou AD, Sirinian C, Kalofonos HP (2012) Identification of novel human receptor activator of nuclear factor-kB isoforms generated through alternative splicing: implications in breast cancer cell survival and migration. Breast Cancer Res 14:R112. https://doi.org/10.1186/bcr3234
Picarda G, Lamoureux F, Geffroy L, Delepine P, Montier T, Laud K et al (2010) Preclinical evidence that use of TRAIL in Ewing’s sarcoma and osteosarcoma therapy inhibits tumor growth, prevents osteolysis, and increases animal survival. Clin Cancer Res 16:2363–2374. https://doi.org/10.1158/1078-0432.CCR-09-1779
Liu JX, Zhang ZC, Shao ZW, Pu FF, Wang BC, Zhang YK et al (2017) TRAIL-R1 as a novel surface marker for circulating giant cell tumor of bone. Oncotarget 8:50724–50730. https://doi.org/10.18632/oncotarget.17042
Weichhaus M, Chung STM, Connelly L (2015) Osteoprotegerin in breast cancer: beyond bone remodeling. Mol Cancer 14:117. https://doi.org/10.1186/s12943-015-0390-5
Esposito M, Kang Y (2014) Targeting tumor-stromal interactions in bone metastasis. Pharmacol Ther 141:222–323. https://doi.org/10.1016/j.pharmthera.2013.10.006
Mishra A, Shiozawa Y, Pienta KJ, Taichman RS (2011) Homing of cancer cells to the bone. Cancer Microenviron 4:221–235. https://doi.org/10.1007/s12307-011-0083-6
Sleeman JP, Christofori G, Fodde R, Collard JG, Berx G, Decraene C et al (2012) Concepts of metastasis in flux: the stromal progression model. Semin Cancer Biol 22:174–186. https://doi.org/10.1016/j.semcancer.2012.02.007
Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM (2012) Bone marrow microenvironment in multiple myeloma progression. J Biomed Biotechnol. https://doi.org/10.1155/2012/157496
Luo G, He Y, Yu X (2018) Bone marrow adipocyte: an intimate partner with tumor cells in bone metastasis. Front Endocrinol (Lausanne) 9:339. https://doi.org/10.3389/fendo.2018.00339
Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B et al (2015) Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat Commun 6:7808
Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, Spiegelman BM (2014) Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513:100–104
Miki T, Yano S, Hanibuchi M, Kanematsu T, Muguruma H, Sone S (2004) Parathyroid hormone-related protein (PTHrP) is responsible for production of bone metastasis, but not visceral metastasis, by human small cell lung cancer SBC-5 cells in natural killer cell-depleted SCID mice. Int J Cancer 108:511
Buenrostro D, Mulcrone PL, Owens P, Sterling JA (2016) The bone microenvironment: a fertile soil for tumor growth. Curr Osteoporos Rep 14:151–158. https://doi.org/10.1007/s11914-016-0315-2
Wu MY, Li CJ, Yiang GT, Cheng YL, Tsai AP, Hou YT et al (2018) Molecular regulation of bone metastasis pathogenesis. Cell Physiol Biochem 46:1423–1438. https://doi.org/10.1159/000489184
Xiang L, Gilkes DM (2019) The contribution of the immune system in bone metastasis pathogenesis. Int J Mol Sci 20:E999. https://doi.org/10.3390/ijms20040999
Salvador F, Llorente A, Gomis RR (2019) From latency to overt bone metastasis in breast cancer: potential for treatment and prevention. J Pathol 249:6–18. https://doi.org/10.1002/path.5292
Graham N, Qian BZ (2018) Mesenchymal stromal cells: emerging roles in bone metastasis. Int J Mol Sci 19:E1121. https://doi.org/10.3390/ijms19041121
Rucci N, Teti A (2010) Osteomimicry: how tumor cells try to deceive the bone. Front Biosc (Schol Ed) 2:907–915. https://doi.org/10.2741/110
Rucci N, Teti A (2018) Osteomimicry: how the seed grows in the soil. Calcif Tissue Int 102:131–140. https://doi.org/10.1007/s00223-017-0365-1
Tan CC, Li GX, Tan LD, Du X, Li XQ, He R et al (2016) Breast cancer cells obtain an osteomimetic feature via epithelial-mesenchymal transition that have undergone BMP2/RUNX2 signaling pathway induction. Oncotarget 7:79688–79705. https://doi.org/10.18632/oncotarget.12939
Chu GC, Chung LW (2014) RANK-mediated signaling network and cancer metastasis. Cancer Metastasis Rev 33:497–509. https://doi.org/10.1007/s10555-013-9488-7
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14:611–622. https://doi.org/10.1038/nrc3793
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A et al (2012) RANK induces epithelial–mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res 72:2879–2888
Liu Y, Wang J, Ni T, Wang L, Wang Y, Sun X (2016) CCL20 mediates RANK/RANKL-induced epithelial–mesenchymaltransition in endometrial cancer cells. Oncotarget 7:25328–25339. https://doi.org/10.18632/oncotarget.8291
Tsubaki M, Komai M, Fujimoto S, Itoh T, Imano M, Sakamoto K et al (2013) Activation of NF-κB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J Exp Clin Cancer Res 32:62. https://doi.org/10.1186/1756-9966-32-62
Xiang L, Gilkes DM (2019) The contribution of the immune system in bone metastasis pathogenesis. Int J Mol Sci 20:999. https://doi.org/10.3390/ijms20040999
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I et al (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440:692–696
Takeshita S, Fumoto T, Naoe Y, Ikeda K (2014) Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem 289:16699–16710
Fan Y, Hanai JI, Le PT, Bi R, Maridas D, DeMambro V (2017) Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab 25:661–672
Dougall WC (2012) Molecular pathways: osteoclast-dependent and osteoclast-independent roles of the RANKL/RANK/OPG pathway in tumorigenesis and metastasis. Clin Cancer Res 18:326–335. https://doi.org/10.1158/1078-0432.CCR-10-2507
Mountzios G, Dimopoulos MA, Bamias A, Papadopoulos G, Kastritis E, Syrigos K et al (2007) Abnormal bone remodeling process is due to an imbalance in the receptor activator of nuclear factor–κB ligand (RANKL)/osteoprotegerin (OPG) axis in patients with solid tumors metastatic to the skeleton. Acta Oncol 46:221–229. https://doi.org/10.1080/02841860600635870
Shemanko CS, Cong Y, Forsyt A (2016) What is breast in the bone? Int J Mol Sci 17:E1764. https://doi.org/10.3390/ijms17101764
Molyneux SD, Di Grappa MA, Beristain AG, McKee TD, Wai DH, Paderova J et al (2010) Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J Clin Invest 120:3310–3325
Navet B, Ando K, Vargas-Franco JW, Brion R, Amiaud J, Mori K et al (2018) The intrinsic and extrinsic implications of RANKL/RANK signaling in osteosarcoma: from tumor initiation to lung metastases. Cancers (Basel) 10:E398. https://doi.org/10.3390/cancers10110398
Mori K, Le Goff B, Berreur M, Riet A, Moreau A, Blanchar F et al (2007) Human osteosarcoma cells express functional receptor activator of nuclear factor-kappa B. J Pathol 211:555–562. https://doi.org/10.1002/path.2140
Lamoureux F, Richard P, Wittrant Y, Battaglia S, Pilet P, Trichet V et al (2007) Therapeutic relevance of osteoprotegerin gene therapy in osteosarcoma: blockade of the vicious cycle between tumor cell proliferation and bone resorption. Cancer Res 67:7308–7318. https://doi.org/10.1158/0008-5472.CAN-06-4130
Wittrant Y, Lamoureux F, Mori K, Riet A, Kamijo A, Heymann D, Redini F (2006) RANKL directly induces bone morphogenetic protein-2 expression in RANK-expressing POS-1 osteosarcoma cells. Int J Oncol 28:261–269
Beristain AG, Narala SR, Di Grappa MA, Khokha R (2012) Homotypic RANK signaling differentially regulates proliferation, motility and cell survival in osteosarcoma and mammary epithelial cells. J Cell Sci 125:943–955. https://doi.org/10.1242/jcs.094029
Chen Y, Di Grappa MA, Molyneux SD, McKee TD, Waterhouse P, Penninger JM, Khokha R (2015) RANKL blockade prevents and treats aggressive osteosarcomas. Sci Transl Med 7:31. https://doi.org/10.1126/scitranslmed.aad0295
Cowan AJ, Allen C, Barac A et al (2018) Global burden of multiple myeloma: a systematic analysis for the global burden of disease study 2016. JAMA Oncol 4:1221–1227. https://doi.org/10.1001/jamaoncol.2018.2128
Raje NS, Bhatta S, Terpos E (2019) Role of the RANK/RANKL pathway in multiple myeloma. Clin Cancer Res 25:12–20. https://doi.org/10.1158/1078-0432.CCR-18-1537
Vallet S, Filzmoser JM, Pecherstorfer M, Podar K (2018) Myeloma bone disease: update on pathogenesis and novel treatment strategies. Pharmaceutics 10:E202. https://doi.org/10.3390/pharmaceutics10040202
Goranova-Marinova V, Goranov S, Pavlov P, Tzvetkova T (2007) Serum levels of OPG, RANKL and RANKL/OPG ratio in newly-diagnosed patients with multiple myeloma. Clin Correl Haematol 92:1000–1001
Terpos E, Szydlo R, Apperley JF, Hatjiharissi E, Politou M, Meletis J et al (2003) A Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood 102:1064–1069
Sfiridaki K, Pappa CA, Tsirakis G, Kanellou P, Kaparou M, Stratinaki M et al (2011) Angiogenesis-related cytokines, RANKL, and osteoprotegerin in multiple myeloma patients in relation to clinical features and response to treatment. Mediators Inflamm 2011:867576. https://doi.org/10.1155/2011/86757
Spanoudakis E, Papoutselis M, Terpos E, Dimopoulos MA, Tsatalas C, Margaritis D et al (2016) Overexpression of RANKL by invariant NKT cells enriched in the bone marrow of patients with multiple myeloma. Blood Cancer J 6:e500. https://doi.org/10.1038/bcj.2016.108
Pitari MR, Rossi M, Amodio N, Botta C, Morelli E, Federico C et al (2015) Inhibition of miR-21 restores RANKL/OPG ratio in multiple myeloma-derived bone marrow stromal cells and impairs the resorbing activity of mature osteoclasts. Oncotarget 6:27343–27358. https://doi.org/10.18632/oncotarget.4398
Liu T, Qin AP, Liao B, Shao HG, Guo LJ, Xie GQ, Yang L, Jiang TJ (2014) A novel microRNA regulates osteoclast differentiation via targeting protein inhibitor of activated STAT3 (PIAS3). Bone 67:156–165
https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm. Searched for Xgeva. Accessed in May 2019
Morony S, Capparelli C, Lee R, Shimamoto G, Boone T, Lace DL et al (1999) A chimeric form of osteoprotegerin inhibits hypercalcemia and bone resorption induced by IL-1β, TNF-α, PTH, PTHrP, and 1,25(OH)2D3. J Bone Miner Res 14:1478–1485. https://doi.org/10.1359/jbmr.1999.14.9.1478
de Groot AF, Appelman-Dijkstra NM, van der Burg SH, Kroep JR (2018) The anti-tumor effect of RANKL inhibition in malignant solid tumors—a systematic review. Cancer Treat Rev 62:18–28. https://doi.org/10.1016/j.ctrv.2017.10.010
Capparelli C, Kostenuik PJ, Morony S, Starnes C, Weimann B, Van G et al (2000) Osteoprotegerin prevents and reverses hypercalcemia in a murine model of humoral hypercalcemia of malignancy. Cancer Res 60:783–787
Morony S, Capparelli C, Sarosi I, Lacey DL, Dunstan CR (2001) Kostenuik PJ (2001) Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res 61:4432–4436
Yonou H, Kanomata N, Goya M, Kamij T, Yokose T, Hasebe T et al (2003) Osteoprotegerin/osteoclastogenesis inhibitory factor decreases human prostate cancer burden in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice. Cancer Res 63:2096–2102
Luger NM, Honore P, Sabino MA, Schwei MJ, Rogers SD, Mach DB et al (2001) Osteoprotegerin diminishes advanced bone cancer pain. Cancer Res 61:4038–4047
Castellano D, Sepulveda JM, García-Escobar I, Rodriguez-Antolín A, Sundlöv A, Cortes-Funes H (2011) The role of RANK-ligand inhibition in cancer: the story of denosumab. Oncologist 16:136–145. https://doi.org/10.1634/theoncologist.2010-0154
Ottewell PD, Wang N, Brown HK, Fowles CA, Croucher PI, Eaton CL, Holen I (2015) OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int J Cancer 137:968–977. https://doi.org/10.1002/ijc.29439
Miller RE, Jones JC, Tometsko M, Blake ML, Dougall WC (2014) RANKL inhibition blocks osteolytic lesions and reduces skeletal tumor burden in models of non-small-cell lung cancer bone metastases. J Thorac Oncol 9:345–354
Cody JJ, Rivera AA, Lyons GR, Yang SW, Wang M, Sarver DB et al (2010) Arming a replicating adenovirus with osteoprotegerin reduces the tumor burden in a murine model of osteolytic bone metastases of breast cancer. Cancer Gene Ther 17:893–905. https://doi.org/10.1038/cgt.2010.47
Canon J, Bryant R, Roudier M, Branstetter DG, Dougall WC (2012) RANKL inhibition combined with tamoxifen treatment increases anti-tumor efficacy and prevents tumor-induced bone destruction in an estrogen receptor-positive breast cancer bone metastasis model. Breast Cancer Res Treat 135:771–780. https://doi.org/10.1007/s10549-012-2222-2
Zinonos I, Luo K, Labrinidis A, Liapis V, Hay S, Panagopoulos V et al (2014) Pharmacologic inhibition of bone resorption prevents cancer-induced osteolysis but enhances soft tissue metastasis in a mouse model of osteolytic breast cancer. Int J Oncol 45:532–540. https://doi.org/10.3892/ijo.2014.2468
Lamoureux F, Picarda G, Garrigue-Antar L, Baudhuin M, Trichet V, Vidal A et al (2009) Glycosaminoglycans as potential regulators of osteoprotegerin therapeutic activity in osteosarcoma. Cancer Res 69:526–536. https://doi.org/10.1158/0008-5472.CAN-08-2648
Body JJ, Greipp P, Coleman RE, Facon T, Geur F, Fermand JP et al (2003) A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer 97:887–892. https://doi.org/10.1002/cncr.11138
Qiao B, Shui W, Cai L, Guo S, Jiang D (2015) Human mesenchymal stem cells as delivery of osteoprotegerin gene: homing and therapeutic effect for osteosarcoma. Drug Design Dev Ther 9:969–976. https://doi.org/10.2147/DDDT.S77116
Higgs JT, Jarboe JS, Lee JH, Chand D, Lee CM, Deivanayagam C et al (2015) Variants of osteoprotegerin lacking TRAIL binding for therapeutic bone remodeling in osteolytic malignancies. Mol Cancer Res 13:819–827. https://doi.org/10.1158/1541-7786.MCR-14-0492
Zinonos I, Labrinidis A, Lee M, Liapis V, Hay S, Ponomarev V et al (2011) Anticancer efficacy of Apo2L/TRAIL is retained in the presence of high and biologically active concentrations of osteoprotegerin in vivo. J Bone Miner Res 26:630–643. https://doi.org/10.1002/jbmr.244
Yu X, Kong W, Zheng K (2013) Expression of osteoprotegerin and osteoprotegerin ligand in giant cell tumor of bone and its clinical significance. Oncol Lett 5:1133–1139. https://doi.org/10.3892/ol.2013.1199
Yamagishi T, Kawashima H, Ogose A, Ariizumi T, Sasaki T, Hatano H et al (2016) Receptor-activator of nuclear KappaB ligand expression as a new therapeutic target in primary bone tumors. PLoS ONE 11:e0154680. https://doi.org/10.1371/journal.pone.0154680
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Deligiorgi, M.V., Panayiotidis, M.I., Griniatsos, J. et al. Harnessing the versatile role of OPG in bone oncology: counterbalancing RANKL and TRAIL signaling and beyond. Clin Exp Metastasis 37, 13–30 (2020). https://doi.org/10.1007/s10585-019-09997-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-019-09997-8