
Abstract
Bone metastasis involves tumor-induced osteoclast activation, resulting in skeletal tumor progression as well as skeletal disorders. Aberrant expression of receptor activator of NF-κB ligand (RANKL), an essential cytokine for osteoclast differentiation, induced by the metastatic tumor cells is responsible for the pathological bone resorption in bone metastasis. A fully human anti-RANKL neutralizing antibody has been developed to block osteoclast activation and is now used for the treatment of patients with bone metastasis and multiple myeloma. On the other hand, numerous studies have revealed that the RANKL/RANK system also contributes to primary tumorigenesis as well as metastasis through osteoclast-independent processes. Furthermore, emerging clinical and preclinical evidence has suggested anti-tumor immune effects of RANKL blockade when added to immune checkpoint inhibitor therapies. Study on the pleiotropic functions of RANKL in tumorigenesis and metastasis is now expanding beyond the bone field and has been established as one of the most important areas of “RANKL biology”.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The tumor necrosis factor (TNF) family cytokine RANKL is an essential cytokine for osteoclastogenesis [1]. RANKL binds to its receptor RANK expressed on osteoclast precursor cells to trigger osteoclast differentiation. The importance of the RANKL/RANK system is not limited to bone remodeling. RANKL exactly acts as a multifunctional cytokine that influences diverse physiological processes, including immune organ development and mammary gland maturation [1]. Pathologically, excess RANKL signal leads to abnormal osteoclast activation and bone loss in rheumatoid arthritis, osteoporosis, and bone metastasis. Bone metastasis critically involves osteoclastic bone resorption for tumor burden in bone, producing skeletal-related events (SREs), such as fracture, spinal cord compression, and the need for bone irradiation or surgery [2]. As demonstrated by the clinical benefits conferred by anti-RANKL treatment, RANKL is crucial for pathological osteoclast activation that causes SREs in bone metastasis. On the other hand, recent studies have revealed that the RANKL/RANK system also crucially contributes to tumorigenesis and metastasis through osteoclast-independent processes in various situations. This review presents a comprehensive overview of the pathological roles of RANKL in cancer development and metastasis.
Osteoclast-dependent roles of RANKL in bone metastasis and multiple myeloma
Osteolytic bone metastasis and RANKL
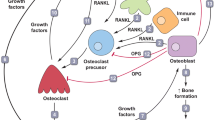
Bone is one of the most preferential metastatic target sites for certain cancers [3]. In particular, 65–75% of patients with advanced breast and prostate cancer, 30–40% of patients with advanced lung cancer, and 14–45% of patients with advanced melanoma develop bone metastases [4]. Bone stores large amounts of growth factors including transforming growth factor (TGF)-β and insulin-like growth factors (IGFs), which can promote the proliferation and survival of tumor cells. Thus, bone is an exclusively favorable environment for tumor metastasis, namely “seed and soil theory” proposed by Stephen Paget in 1889 [5]. Bone metastasis often causes serious skeletal complications including bone pain, pathologic fractures, hypercalcemia and spinal cord compression, which significantly worsen the quality of life in the patients [2]. Bone metastasis can be largely classified as osteolytic and osteoblastic [6]. Osteolytic bone metastasis is often seen in breast cancer and lung cancer. In bone, the tumor cells produce various cytokines or factors, such as parathyroid hormone-related peptide (PTHrP), prostaglandin E2, interleukin (IL)-6, IL-8, IL-11, and TNF-α, to induce RANKL expression in bone marrow stromal cells such as osteoblasts, resulting in enhanced osteoclast formation and bone resorption [7]. Tumor-induced osteoclastic bone resorption subsequently results in the release of calcium (Ca2+) and growth factors such as TGF- β and IGFs from the degraded bone matrices, which further promote tumor progression. The intercellular network by tumor cells, bone marrow stromal cells, and osteoclasts referred to as the "vicious cycle", is a central mechanism underlying the pathogenesis of the osteolytic metastasis (Fig. 1) [3]. Studies using the mouse model of bone metastasis achieved by injecting the human breast cancer cell line MDA-MB-231, which forms osteolytic metastasis, demonstrated that in vivo neutralization of RANKL by osteoprotegerin (OPG), which is a decoy receptor for RANKL, inhibited skeletal tumor burden and osteolysis via suppressing osteoclast activity [8, 9]. Oral administration of a small-molecule inhibitor against RANKL signaling also suppressed tumor-induced osteoclast differentiation and the tumor burden in a mouse model of bone metastasis using MDA-MB-231 cells [10]. RANKL-induced osteoclast activation plays a central role in the development of osteolytic lesions.

Roles of RANKL in bone metastasis. RANKL contributes to bone metastasis by not only activating osteoclast activation but also acting as a chemotactic factor for recruiting the tumor cells to bone. Tumor cells induce RANKL expression on bone marrow stromal cells such as osteoblasts via various cytokines and factors, leading to osteoclastic bone resorption. Increased bone resorption causes the release of growth factors from bone matrix, thereby further increasing tumor proliferation. Soluble RANKL is dispensable for osteoclast activation in bone metastasis, suggesting that membrane-bond RANKL (mRANKL) is mainly involved in this process. On the other hand, soluble RANKL promotes the homing of tumor cells to bone tissues, by directly acting on tumor cells expressing RANK. Chemokines derived from bone marrow stromal cell and bone marrow endothelial cell, such as CXCL12, CX3CL1, and CCL2, also contributes to the tumor migration to bone
Osteoblastic metastasis of prostate cancer and RANKL
Osteoblastic metastasis, which is commonly seen in prostate cancer, involves excessive activation of osteoblasts adjacent to metastatic tumor cells [11]. Factors locally produced by the tumor cells, including bone morphogenetic proteins (BMPs), fibroblast growth factors (FGFs), IGFs, TGF-β, and endothelin-1, promote osteoblastogenesis and bone formation [11]. Bone metastasis of prostate cancer most often presents as osteoblastic lesions but is also accompanied by mixed osteolytic features, thus indicating the effectiveness of RANKL inhibition. Production of PTHrP, IL-6 and RANKL is involved in the osteoclastic activity of prostate cancer [12]. Notably, administration of OPG inhibited the skeletal tumor progression in experimental bone metastasis models using human prostate cancer cell lines, LNCaP and LuCaP23.1 [13, 14]. Nevertheless, a study on human prostate tumor samples showed that not only RANKL and RANK but also OPG expression in the prostate tumor was positively correlated with the aggressiveness [15]. OPG by prostate cancer cells also has the capacity to facilitate tumor survival by inhibiting TRAIL-induced apoptosis [16]. The formation of mixed osteolytic/osteoblastic lesions in bone metastasis of prostate cancer would be influenced by the ratio of RANKL to OPG in the local microenvironment.
RANKL in multiple myeloma
Multiple myeloma is a malignant disorder caused by uncontrolled proliferation of monoclonal plasma cells in the bone marrow [17]. Osteolytic lesion formation is a hallmark of multiple myeloma, leading to SREs associated with morbidity and mortality. MIP-1α/β plays a critical role in multiple myeloma-induced osteolysis [18]. MIP-1α/β produced by multiple myeloma cells stimulates osteoclast progenitor cells and induces RANKL expression in bone marrow stromal cells. Interaction between VLA-4 on multiple myeloma cells and VCAM-1 on bone marrow stromal cells is implicated in bone marrow homing and progression of multiple myeloma cells. VLA-4–VCAM-1 adhesion upregulates MIP-1α/β production, promoting osteoclastogenesis [19, 20]. A primary t(4;14)(p16.3;q32.3) chromosomal translocation in multiple myeloma also leads to upregulation of MIP-1 via FGFR3 expression [17]. In addition, multiple myeloma cells exert osteoclastic activity by producing RANKL and soluble factors that induce RANKL on bone marrow stromal cells such as PTHrP [17]. It has been recently shown that RANKL-induced osteoclastic bone resorption activates dormant myeloma cells in the bone marrow [21]. Preclinical studies using the mouse models of multiple myeloma demonstrated that RANKL inhibition by OPG or RANK-Fc administration reduced tumor burden and the formation of osteolytic lesions [22, 23], indicating that RANKL-induced osteoclastogenesis largely contributes to the development of skeletal lesions in multiple myeloma. On the other hand, multiple myeloma inhibits osteoblastic bone formation by expressing the WNT pathway inhibitors such as Dickkopf-1, sclerostin, and secreted Frizzled-related proteins [17].
Overall, RANKL is evidently important for the expansion of tumor cells in bone through activating osteoclastic bone resorption. A human monoclonal anti-RANKL antibody denosumab is currently used for the prevention of SREs in patients with bone metastasis and in patients with multiple myeloma [24].
Osteoclast-independent roles of RANKL in bone metastasis and tumorigenesis
RANKL as an attractant for tumor metastasis to bone
RANKL contributes to the pathogenesis of bone metastasis in another way. Bone metastasis is a complex process including escaping from the primary tumor, invading into circulation, surviving in the circulation, homing to the bone and thriving in the bone microenvironment. Another role of RANKL is relevant to the step of “homing to bone”. RANKL acts directly on RANK-expressing tumor cells and promotes cell migration via the induction of actin polymerization, which is a hallmark of chemokine receptor signaling [25]. Chemotaxis assays revealed that the tumor cells expressing RANK migrated according to the concentration gradient of soluble RANKL in vitro [10, 25,26,27,28], indicating the chemoattractant activity of RANKL. Actually, it has been shown that RANK is highly expressed in many different tumor cells that exhibit a propensity to metastasize to bone [15, 25, 26, 29]. Among the RANK-expressing metastatic cell lines, it is known that the murine melanoma cell line B16F10 does not trigger osteoclast activation [10, 25, 30]. Treatment with bisphosphonate had no effect on bone metastasis of B16F10 cells in the mouse model of intracardiac injection [25]. However, administration of OPG could suppress bone metastasis of B16F10 cells by inhibiting the migration of tumor cells to bone [25]. We also showed that oral administration of a small molecular inhibitor against RANKL inhibited bone metastasis of B16F10 cells by suppressing RANKL-induced tumor cell migration [10]. RANKL-induced tumor migration has been demonstrated using various RANK-expressing tumor cells, such as MDA-MB-231 cells, human breast cancer cell MCF-7, human breast cancer cell BT-474, human liver carcinoma cell Huh7, and human kidney carcinoma cell Caki-1 [25, 26, 31,32,33]. Furthermore, human clinical studies reported that the level of RANK expression in primary breast cancer and renal cell carcinomas is positively correlated with the frequency of bone metastases [29, 32]. B16F10 cells also have the capacity to metastasize to non-skeletal tissues such as the adrenal glands and ovaries [10, 25]. Neither OPG nor a small-molecule inhibitor against RANKL blocked the metastasis of B16F10 cells to non-skeletal tissues [10, 25], supporting the hypothesis that the chemoattractant activity of RANKL is one of the crucial mechanisms underlying the preferential metastasis of RANK-expressing tumor cells to bone.
RANKL is expressed in two types: membrane-bound form on the cell surface and soluble form. Like other TNF family molecules, RANKL is initially synthesized as the membrane-bound form, and the soluble form is produced by proteolytic ectodomain shedding mediated by metalloproteinases such as matrix metalloproteinase (MMP) 14, a disintegrin and metalloproteinase (ADAM) 10 and ADAM17/Tumor necrosis factor-α-converting enzyme (TACE) [34, 35]. Accumulating evidence from in vitro studies has suggested that membrane-bound RANKL exerts more effective osteoclastogenic action than soluble RANKL [35, 36]. However, the functional difference between the membrane-bound and soluble forms of RANKL had been poorly understood in vivo. Recently, we have elucidated the physiological and pathological significances of soluble RANKL by generating mice that selectively lack soluble RANKL [30]. Mice lacking the Tnfsf11 gene (encoding RANKL) exhibited severe osteopetrosis accompanied by a tooth eruption defect due to a complete lack of osteoclasts [37]. On the other hand, soluble RANKL-deficient mice had normal tooth eruption and did not show any discernible osteopetrotic phenotype, indicating that soluble RANKL is dispensable for physiological bone remodeling [30]. Furthermore, soluble RANKL made no contribution to ovariectomy-induced osteoporosis or periodontitis-induced bone loss. As suggested by the in vitro studies, it is likely that membrane-bound RANKL mainly functions in osteoclastogenesis. Furthermore, unlike RANKL-null mice [37], soluble RANKL-deficient mice had normal development of the immune organs, including lymph node development, medullary thymic epithelial cell differentiation, and microfold (M) cell differentiation in gut, as well as normal mammary gland maturation during pregnancy [30, 38]. Thus, direct cell–cell interaction through membrane-bound RANKL would be important.
Soluble RANKL is physiologically dispensable but plays an essential role as the chemoattractant factor for RANK-expressing tumor cells in bone metastasis. Soluble RANKL deficiency in mice markedly suppressed the metastasis of B16F10 cells to bone [30]. Intracardiac injection of B16F10 cells into wild-type mice resulted in rapid metastasis into bones. On the other hand, even at the early time point, tumor progression in bone was reduced in soluble RANKL-deficient mice compared with control littermates. In contrast, in mice subcutaneously transplanted with B16F10 cells, the primary tumor growth was not affected by the absence of soluble RANKL. As mentioned above, bone metastasis of B16F10 cells is dependent on RANKL-mediated tumor migration in vivo, but not on osteoclast differentiation [25]. Consistent with this, soluble RANKL deficiency had no effect on osteoclasts at the metastasis site [30]. Furthermore, by using RANK-negative B16F10 cells generated by the CRISPR/Cas9 system, we demonstrated that a deficiency of RANK in B16F10 cells markedly reduced the frequency of bone metastasis, indicating that RANK expressed on the surfaces of tumor cells is important for bone metastasis [30]. Unlike the parental RANK-positive B16F10 cells, there was no significant difference between wild-type and soluble RANKL-deficient mice in bone metastasis of RANK-negative B16F10 cells, indicating that soluble RANKL promotes bone metastasis by directly acting on RANK on tumor cells. In addition, we confirmed that soluble RANKL deficiency potently inhibited bone metastasis of the breast cancer cell line EO771 [30]. Even though EO771 cells have the capacity to trigger osteoclast activation, soluble RANKL deficiency did not affect osteoclast number at the tumor–bone interface, suggesting that direct cell–cell interaction through membrane-bound RANKL is mainly involved in tumor-induced osteoclast activation. Collectively, membrane-bound RANKL is sufficient for most of the physiological RANKL function, but soluble RANKL specifically contributes to bone metastasis by exerting a chemotactic activity in tumor cells expressing RANK (Fig. 1).
Other than soluble RANKL, several chemokines are also involved in tumor cell migration to bone (Fig. 1). The CXCL12/CXCR4 axis is involved in homing of breast cancer, prostate cancer, multiple myeloma and lung cancer cells to bone [39]. CXCL12 is highly expressed on the bone marrow stromal cells, especially CXCL12-abundant reticular (CAR) cells, which are scattered throughout the bone marrow and function as a hematopoietic stem cell niche [1]. CX3CR1 signaling also mediates metastasis of breast cancer cells specifically to bone [40, 41]. CX3CL1, a ligand for CX3CR1, is expressed on endothelial cells and stromal cells within the bone marrow. CCL2 produced by bone marrow endothelial cells promotes the migration of CCR2-expressing prostate cancer cells to the bone [42]. Chemokine receptor-expressing cells move through the gradient towards the area of the higher concentration of chemokine. Like chemokines, we consider that the physiological concentration gradient of soluble RANKL from bone marrow to serum is important for exerting a chemotactic activity in tumor cells. In the bone, several types of mesenchymal lineage cells including osteocytes and osteoblasts express RANKL for bone metabolism. Although the previous reports indicate that the osteocytes express a much higher amount of RANKL than osteoblasts and bone marrow stromal cells [1], further analyses will be required to understand the source of soluble RANKL in the case of bone metastasis. Soluble RANKL deficiency had no effect on the metastasis of B16F10 and EO771 cells into non-skeletal tissues including ovaries and adrenal glands [30], suggesting that RANKL-mediated tumor migration is not associated with the metastasis to organs that express low levels of RANKL. Taken together, bone tissue possesses chemotactic factors that can promote tissue-specific homing of cancer cells.
Despite many studies of human cancers, it remains controversial whether there is a correlation between serum RANKL concentration and incidence of bone metastasis from solid tumors, such as breast cancer, lung cancer, prostate cancer, and renal cancer [43, 44]. The incidence of bone metastasis is evaluated after the tumors develop into the clinically detectable size. In this context, it is necessary to consider the possibility that serum RANKL levels can be affected in various ways after the metastasis is established. Interestingly, a recent report showed that high serum levels of soluble RANKL and high RANKL/OPG ratio are associated with the presence of disseminated tumor cells in the bone marrow of breast cancer patients, which is the initial phase of the metastasis [45]. Furthermore, patients with high RANKL serum levels had a significantly increased risk of developing bone metastases [45]. Therefore, soluble RANKL may be effective as a serum biomarker for estimating the risk of developing bone metastases, but further investigation is needed to validate the potential value of soluble RANKL in various situations.
RANKL-mediated control of the metastasis phenotype of tumor cells
RANKL was originally identified as a T cell-derived cytokine. Several preclinical studies highlight the importance of RANKL on T cells within the tumor microenvironment as a driver of tumor metastasis. RANKL on tumor-infiltrating inflammatory cells including T cells and macrophages suppresses the expression of a metastasis suppressor Maspin via IKKα activation in prostate cancer cells, thereby promoting cancer metastasis [46]. RANKL on tumor-infiltrating regulatory T (Treg) cells is involved in pulmonary metastasis of mammary tumor cells [47]. Cancer-associated myofibroblasts recruit Treg cells into primary mammary tumors by releasing T-cell attracting chemokines such as CCL5. Tumoral Treg cells highly express RANKL, which enhances the pulmonary metastasis of RANK+ mammary tumor cells via the IKKα-Maspin pathway. Administration of RANK-Fc markedly inhibited the pulmonary metastasis of mammary carcinoma cells. Therefore, RANKL/RANK-mediated interplay between tumor-infiltrating immune cells and tumor modulates metastatic potential of tumor cells.
RANKL in the mammary gland and breast cancer
The RANKL/RANK system is indispensable for the formation of the lactating mammary gland during pregnancy [48]. Mammary glands of RANKL- and RANK-deficient mice develop normally during puberty but fail to form lobuloalveolar structures for milk production during lactation, owing to increased apoptosis and impaired proliferation of mammary alveolar epithelial cells. RANKL induces the proliferation of mammary alveolar epithelial cells through NF-κB–cyclin D1 activation and inhibitor of DNA binding protein 2 (Id2) activation [49, 50]. The sex steroid hormone progesterone stimulates progesterone receptor-expressing luminal epithelial cells to induce the expression of RANKL, which subsequently acts on RANK on luminal epithelial and basal/myoepithelial cells to promote their proliferation. Mammary stem cells (MaSC), which reside in the basal/myoepithelial compartment, give rise to all lineages of the mammary gland including luminal epithelial and basal/myoepithelial cells [48]. RANKL on luminal epithelial cells also directly stimulates MaSC, increasing the MaSC pool during pregnancy.
Preclinical studies have revealed the intrinsic role of RANKL signaling in mammary tumorigenesis [48]. Hormone replacement therapy (HRT), estrogen alone or the combination of estrogen and progestins, is used for the prevention and treatment of osteoporosis in postmenopausal women, but the combination HRT is known to increase the risk of developing breast cancer compared to estrogen-alone therapy. RANKL signaling is crucial for progestin-driven breast cancer [51, 52]. Progestins such as medroxyprogesterone acetate (MPA) trigger RANKL expression on luminal epithelial cells, leading to increased proliferation of mammary epithelial cells, expansion of MaSC, and inhibition of DNA damage-induced apoptosis in mammary epithelial cells. Pharmacological inhibition of RANKL suppressed MPA-induced mammary tumor in mice, and spontaneous mammary tumor development in the mouse mammary tumor virus (MMTV)-neu transgenic mice model. Deletion of RANK in the mammary epithelium also delayed the onset of MPA-induced mammary cancer. In addition, RANKL signal was reported to induce epithelial-mesenchymal transition in human mammary epithelial cells [53]. Importantly, RANKL signaling is involved in the proliferation of breast cancer susceptibility gene 1 (BRCA1) mutant mammary progenitor cells [54, 55]. Genetic inactivation of RANK in the mammary gland or pharmacologic RANKL-blockade abrogated the occurrence of Brca1 mutation-driven breast cancer in mice. Interestingly, six SNPs in the TNFRSF11A locus (encoding RANK) were found to be significantly associated with breast cancer risk in women with BRCA1 mutations [54]. Moreover, in the pilot window study “BRCA-D”, cell proliferation in breast tissues from BRCA1-mutation carriers was decreased after denosumab treatment [55], indicating the therapeutic potential value of RANKL inhibition for the prevention of breast cancer.
RANKL and lung cancer
RANKL signaling is involved in primary lung cancer. High RANKL, high RANK, and low OPG are found to be associated with a worse prognosis for lung cancer patients [56]. In a mouse model of KrasG12D mutation-induced lung cancer, selective deletion of RANK in pneumocytes inhibited tumor progression. RANKL directly drives the expansion of lung cancer stem-like cells by regulating mitochondrial respiration. Moreover, female sex hormones enhance KrasG12D-driven lung cancer via the RANKL/RANK pathway. These findings might explain the sex-biased tendencies in the progression of lung cancer.
RANKL for anti-tumor immune response
Cancer immunotherapy, especially immune checkpoint inhibitors (ICI) and CAR-T cells has recently attracted considerable interest in the file of oncology as well as immunology. ICI such as anti-CTLA4 antibody, anti-PD-1 antibody, and anti-PD-L1 antibody can restore and augment anti-tumor immune responses by blocking the immune-suppressive molecules. ICI has opened up a new direction of cancer treatment, but a substantial proportion of patients still do not respond to ICI, indicating the need to develop additional therapeutic options. Recently, human clinical studies have demonstrated the significant efficacy of the combination therapy with denosumab and anti-CTLA-4, anti-PD-1 or anti-PD-L1 antibodies in patients with metastatic melanoma and lung cancer [57, 58]. Interestingly, preclinical studies using melanoma, prostate cancer, and colon adenocarcinoma cell lines showed that addition of anti-RANKL antibody to ICI enhanced the anti-tumor efficacy by increasing effector T cell function in mouse subcutaneous tumor models or natural killer cell function in lung metastasis models [59, 60]. Furthermore, the efficacy of a recently developed bispecific antibody co-targeting RANKL and PD-1 has been validated in mouse models of cancer [61]. Given various immune cells expressing RANKL and RANK such as macrophages, dendritic cells, myeloid-derived suppressor cells, and T cells within the tumor microenvironment, it can be speculated that RANKL modulates local immunosuppressive circumstance advantage for tumor. A recent study has shown that excessive amounts of TGF-β induced by osteoclastic bone resorption affect anti-tumor T cell function in bone metastasis of prostate cancer [62], suggesting that RANKL can affect anti-tumor immune response in an osteoclast-dependent manner under certain conditions. Taken together, these findings warrant further investigation on the molecular mechanism of how RANKL/RANK signaling regulates anti-tumor immunity.
Clinical studies evaluating osteoclast-independent effects of RANKL blockade
RANKL contributes to the pathogenesis of bone metastasis by driving the vicious cycle as an essential cytokine for osteoclastogenesis. Denosumab has demonstrated remarkable efficacy as a blockade of SREs in patients with bone metastasis and multiple myeloma. On the other hand, a number of preclinical mouse studies unveiled the osteoclast-independent functions of RANKL in primary tumorigenesis and metastasis, implicating the potential of RANKL blockade for treatment and prevention of tumor development and metastasis. However, it is notable that the osteoclast-independent effects of RANKL blockade need to be precisely and carefully validated under the various tumor conditions in human. In a randomized double-blind phase III trial study, denosumab was reported to be non-inferior to the bisphosphonate zoledronic acid in preventing SREs, but to significantly improve overall survival (OS) of patients with non-small-cell lung cancer (NSCLC) compared to the bisphosphonate, implying an osteoclast-independent effect of denosumab [63, 64]. A randomized phase III trial of patients with multiple myeloma also showed that denosumab has almost the same effect as zoledronic acid in terms of SRE prevention, but is superior in terms of progression-free survival [65]. The placebo-controlled phase III trial (ABCSG-18) provides convincing evidence that denosumab improves disease-free survival of hormone receptor-positive postmenopausal breast cancer patients receiving aromatase inhibitor therapy, whereas bone metastasis-free survival has not been evaluated yet [66]. In contrast, a randomized phase III trial (SPLENDOUR) in a large cohort of patients with advanced-stage NSCLC has recently concluded no significant difference in OS between standard chemotherapy alone and the combination with denosumab [67]. Regarding the prevention of bone metastasis, a randomized placebo-controlled study showed that RANKL inhibition with denosumab delayed bone metastasis in men with prostate cancer [68]. On the other hand, the most recent phase III trial D-CARE for women with high-risk early-stage breast cancer has reported that denosumab did not delay bone metastasis compared with placebo. In a window study, D-BEYOND focusing on denosumab treatment for pre-menopausal breast cancer patients, a short time treatment of denosumab did not suppress tumor proliferation, but significantly increased the number of tumor-infiltrating lymphocytes accompanied by decreased Treg, thus suggesting an immunomodulatory effect of denosumab. Ongoing clinical trials would further address the osteoclast-independent anti-tumor effects of denosumab (Table 1).
Concluding remarks
RANKL has multiple functions in our bodies, and emerging evidence from preclinical studies has revealed that RANKL contributes to cancer development and metastasis in multiple ways, not only through the osteoclast-dependent pathway but also the osteoclast-independent pathway. With the progression of cancer immunotherapy, the potential activity of RANKL to regulate anti-tumor immunity has attracted much attention in the oncology field. Actually, clinical trials for the efficacy of the combination of denosumab with ICI are also now underway (Table 1). Accumulating clinical data as well as state-of-the-art techniques such as mutiomics and single-cell analysis actively utilized in the oncology field would help to provide a more comprehensive picture of the benefits of RANKL blockade in cancer treatment, and new insights to guide the development of more effective combined cancer therapies from the viewpoint of RANKL inhibition.
References
Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, Sawa S, Nitta T, Takayanagi H (2017) Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev 97:1295–1349
Wilkinson AN, Viola R, Brundage MD (2008) Managing skeletal related events resulting from bone metastases. BMJ 337:a2041
Mundy GR (2002) Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2:584–593
Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, Goncalves F (2017) Bone metastases: an overview. Oncol Rev 11:321
Paget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 1:571–573
Roodman GD (2004) Mechanisms of bone metastasis. N Engl J Med 350:1655–1664
Weilbaecher KN, Guise TA, McCauley LK (2011) Cancer to bone: a fatal attraction. Nat Rev Cancer 11:411–425
Morony S, Capparelli C, Sarosi I, Lacey DL, Dunstan CR, Kostenuik PJ (2001) Osteoprotegerin inhibits osteolysis and decreases skeletal tumor burden in syngeneic and nude mouse models of experimental bone metastasis. Cancer Res 61:4432–4436
Canon JR, Roudier M, Bryant R, Morony S, Stolina M, Kostenuik PJ, Dougall WC (2008) Inhibition of RANKL blocks skeletal tumor progression and improves survival in a mouse model of breast cancer bone metastasis. Clin Exp Metastasis 25:119–129
Nakai Y, Okamoto K, Terashima A, Ehata S, Nishida J, Imamura T, Ono T (2019) Takayanagi H Efficacy of an orally active small-molecule inhibitor of RANKL in bone metastasis. Bone Res 7:1
Logothetis CJ, Lin SH (2005) Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer 5:21–28
Wong SK, Mohamad NV, Giaze TR, Chin KY, Mohamed N, Ima-Nirwana S (2019) Prostate cancer and bone metastases: the underlying mechanisms. Int J Mol Sci 20:2587
Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, Mizokami A, Fu Z, Westman J, Keller ET (2001) Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest 107:1235–1244
Kiefer JA, Vessella RL, Quinn JE, Odman AM, Zhang J, Keller ET, Kostenuik PJ, Dunstan CR, Corey E (2004) The effect of osteoprotegerin administration on the intra-tibial growth of the osteoblastic LuCaP 23.1 prostate cancer xenograft. Clin Exp Metastasis 21:381–387
Li X, Liu Y, Wu B, Dong Z, Wang Y, Lu J, Shi P, Bai W, Wang Z (2014) Potential role of the OPG/RANK/RANKL axis in prostate cancer invasion and bone metastasis. Oncol Rep 32:2605–2611
Holen I, Croucher PI, Hamdy FC (2002) Eaton CL osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res 62:1619–1623
Terpos E, Ntanasis-Stathopoulos I, Gavriatopoulou M, Dimopoulos MA (2018) Pathogenesis of bone disease in multiple myeloma: from bench to bedside. Blood Cancer J 8:7
Abe M, Hiura K, Wilde J, Moriyama K, Hashimoto T, Ozaki S, Wakatsuki S, Kosaka M, Kido S, Inoue D, Matsumoto T (2002) Role for macrophage inflammatory protein (MIP)-1α and MIP-1β in the development of osteolytic lesions in multiple myeloma. Blood 100:2195–2202
Abe M, Hiura K, Ozaki S, Kido S, Matsumoto T (2009) Vicious cycle between myeloma cell binding to bone marrow stromal cells via VLA-4-VCAM-1 adhesion and macrophage inflammatory protein-1α and MIP-1β production. J Bone Miner Metab 27:16–23
Mori Y, Shimizu N, Dallas M, Niewolna M, Story B, Williams PJ, Mundy GR, Yoneda T (2004) Anti-α4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 104:2149–2154
Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL et al (2015) Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun 6:8983
Croucher PI, Shipman CM, Lippitt J, Perry M, Asosingh K, Hijzen A, Brabbs AC, van Beek EJ, Holen I, Skerry TM, Dunstan CR, Russell GR, Van Camp B, Vanderkerken K (2001) Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood 98:3534–3540
Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, Michaeli J, Epstein J, Choi Y (2001) Multiple myeloma disrupts the TRANCE/ osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci U S A 98:11581–11586
Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, Lichinitser M, Fujiwara Y, Yardley DA, Viniegra M, Fan M, Jiang Q, Dansey R, Jun S, Braun A (2010) Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol 28:5132–5139
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla CV, Komnenovic V, Kong YY, Schreiber M, Dixon SJ, Sims SM, Khokha R, Wada T, Penninger JM (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440:692–696
Zhang L, Teng Y, Zhang Y, Liu J, Xu L, Qu J, Hou K, Yang X, Liu Y, Qu X (2012) C-Src-mediated RANKL-induced breast cancer cell migration by activation of the ERK and Akt pathway. Oncol Lett 3:395–400
Zhang L, Teng Y, Zhang Y, Liu J, Xu L, Qu J, Hou K, Liu Y, Qu X (2012) Proteasome inhibitor bortezomib (PS-341) enhances RANKL-induced MDA-MB-231 breast cancer cell migration. Mol Med Rep 5:580–584
Tang ZN, Zhang F, Tang P, Qi XW, Jiang J (2011) RANKL-induced migration of MDA-MB-231 human breast cancer cells via Src and MAPK activation. Oncol Rep 26:1243–1250
Santini D, Schiavon G, Vincenzi B, Gaeta L, Pantano F, Russo A, Ortega C, Porta C, Galluzzo S, Armento G, La Verde N, Caroti C, Treilleux I, Ruggiero A, Perrone G, Addeo R, Clezardin P, Muda AO, Tonini G (2011) Receptor activator of NF-kB (RANK) expression in primary tumors associates with bone metastasis occurrence in breast cancer patients. PLoS ONE 6:e19234
Asano T, Okamoto K, Nakai Y, Tsutsumi M, Muro R, Suematsu A, Hashimoto K, Okamura T, Ehata S, Nitta T, Takayanagi H (2019) Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat Metab 1:868–875
Song FN, Duan M, Liu LZ, Wang ZC, Shi JY, Yang LX, Zhou J, Fan J, Gao Q, Wang XY (2014) RANKL promotes migration and invasion of hepatocellular carcinoma cells via NF-κB-mediated epithelial-mesenchymal transition. PLoS ONE 9:e108507
Mikami S, Katsube K, Oya M, Ishida M, Kosaka T, Mizuno R, Mochizuki S, Ikeda T, Mukai M, Okada Y (2009) Increased RANKL expression is related to tumour migration and metastasis of renal cell carcinomas. J Pathol 218:530–539
Armstrong AP, Miller RE, Jones JC, Zhang J, Keller ET, Dougall WC (2008) RANKL acts directly on RANK-expressing prostate tumor cells and mediates migration and expression of tumor metastasis genes. Prostate 68:92–104
Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, Nakamura K, Seiki M, Tanaka S (2006) Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-κB ligand. J Biol Chem 281:36846–36855
Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H (2000) Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-κB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun 275:768–775
Miyamoto T, Arai F, Ohneda O, Takagi K, Anderson DM, Suda T (2000) An adherent condition is required for formation of multinuclear osteoclasts in the presence of macrophage colony-stimulating factor and receptor activator of nuclear factor κB ligand. Blood 96:4335–4343
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323
Xiong J, Cawley K, Piemontese M, Fujiwara Y, Zhao H, Goellner JJ, O’Brien CA (2018) Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun 9:2909
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56
Jamieson-Gladney WL, Zhang Y, Fong AM, Meucci O, Fatatis A (2011) The chemokine receptor CX3CR1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Res 13:R91
Shen F, Zhang Y, Jernigan DL, Feng X, Yan J, Garcia FU, Meucci O, Salvino JM, Fatatis A (2016) Novel small-molecule CX3CR1 antagonist impairs metastatic seeding and colonization of breast cancer cells. Mol Cancer Res 14:518–527
Loberg RD, Day LL, Harwood J, Ying C, St John LN, Giles R, Neeley CK, Pienta KJ (2006) CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia 8:578–586
Jung K, Lein M (1846) Bone turnover markers in serum and urine as diagnostic, prognostic and monitoring biomarkers of bone metastasis. Biochim Biophys Acta 425–38:2014
Kraj M, Owczarska K, Sokołowska U, Centkowski P, Pogłód R, Kruk B (2005) Correlation of osteoprotegerin and sRANKL concentrations in serum and bone marrow of multiple myeloma patients. Arch Immunol Ther Exp (Warsz) 53:454–464
Rachner TD, Kasimir-Bauer S, Göbel A, Erdmann K, Hoffmann O, Browne A, Wimberger P, Rauner M, Hofbauer LC, Kimmig R, Bittner AK (2019) Prognostic value of RANKL/OPG serum levels and disseminated tumor cells in nonmetastatic breast cancer. Clin Cancer Res 25:1369–1378
Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, Cheresh DA, Karin M (2007) Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin. Nature 446:690–694
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M (2011) Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 470:548–553
Schramek D, Sigl V, Penninger JM (2011) RANKL and RANK in sex hormone-induced breast cancer and breast cancer metastasis. Trends Endocrinol Metab 22:188–194
Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL, Boyle WJ, Khokha R, Penninger JM (2000) The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 103:41–50
Kim NS, Kim HJ, Koo BK, Kwon MC, Kim YW, Cho Y, Yokota Y, Penninger JM, Kong YY (2006) Receptor activator of NF-kappaB ligand regulates the proliferation of mammary epithelial cells via Id2. Mol Cell Biol 26:1002–1013
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A, Glimcher L, Pasparakis M, Khokha R, Ormandy CJ, Widschwendter M, Schett G, Penninger JM (2010) Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 468:98–102
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, Pinkas J, Branstetter D, Dougall WC (2010) RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468:103–107
Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A, Climent F, Soler MT, Muñoz P, Viñals F, Tometsko M, Branstetter D, Dougall WC, González-Suárez E (2012) RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res 72:2879–2888
Sigl V, Owusu-Boaitey K, Joshi PA, Kavirayani A, Wirnsberger G et al (2016) RANKL/RANK control Brca1 mutation. Cell Res 26:761–774
Nolan E, Vaillant F, Branstetter D, Pal B, Giner G, Whitehead L, Lok SW, Mann GB, Rohrbach K, Huang LY, Soriano R, Smyth GK, Dougall WC, Visvader JE, Lindeman GJ (2016) RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med 22:933–939
Rao S, Sigl V, Wimmer RA, Novatchkova M, Jais A et al (2017) RANK rewires energy homeostasis in lung cancer cells and drives primary lung cancer. Genes Dev 31:2099–2112
Smyth MJ, Yagita H, McArthur GA (2016) Combination anti-CTLA-4 and anti-RANKL in metastatic melanoma. J Clin Oncol 34:e104–e106
Liede A, Hernandez RK, Wade SW, Bo R, Nussbaum NC, Ahern E, Dougall WC, Smyth MJ (2018) An observational study of concomitant immunotherapies and denosumab in patients with advanced melanoma or lung cancer. Oncoimmunology 7:e1480301
Ahern E, Harjunpää H, Barkauskas D, Allen S, Takeda K, Yagita H, Wyld D, Dougall WC, Teng MWL, Smyth MJ (2017) Co-administration of RANKL and CTLA4 antibodies enhances lymphocyte-mediated antitumor immunity in mice. Clin Cancer Res 23:5789–5801
Ahern E, Harjunpää H, O’Donnell JS, Allen S, Dougall WC, Teng MWL, Smyth MJ (2018) RANKL blockade improves efficacy of PD1-PD-L1 blockade or dual PD1-PD-L1 and CTLA4 blockade in mouse models of cancer. Oncoimmunology 7:e1431088
Dougall WC, Roman Aguilera A, Smyth MJ (2019) Dual targeting of RANKL and PD-1 with a bispecific antibody improves anti-tumor immunity. Clin Transl Immunol 8:e01081
Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, Sharma P (2019) Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell 179:1177–90.e13
Henry DH, Costa L, Goldwasser F, Hirsh V, Hungria V, Prausova J, Scagliotti GV, Sleeboom H, Spencer A, Vadhan-Raj S, von Moos R, Willenbacher W, Woll PJ, Wang J, Jiang Q, Jun S, Dansey R, Yeh H (2011) Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J Clin Oncol 29:1125–1132
Scagliotti GV, Hirsh V, Siena S, Henry DH, Woll PJ, Manegold C, Solal-Celigny P, Rodriguez G, Krzakowski M, Mehta ND, Lipton L, García-Sáenz JA, Pereira JR, Prabhash K, Ciuleanu TE, Kanarev V, Wang H, Balakumaran A, Jacobs I (2012) Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: subgroup analysis from a randomized phase 3 study. J Thorac Oncol 7:1823–1829
Raje N, Terpos E, Willenbacher W, Shimizu K, García-Sanz R, Durie B, Legieć W, Krejčí M, Laribi K, Zhu L, Cheng P, Warner D, Roodman GD (2018) Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol 19:370–381
Gnant M, Pfeiler G, Steger GG, Egle D, Greil R, Fitzal F, Wette V, Balic M, Haslbauer F, Melbinger-Zeinitzer E, Bjelic-Radisic V, Jakesz R, Marth C, Sevelda P, Mlineritsch B, Exner R, Fesl C, Frantal S, Singer CF (2019) Adjuvant denosumab in postmenopausal patients with hormone receptor-positive breast cancer (ABCSG-18): disease-free survival results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 20:339–351
Peters S, Danson S, Hasan B, Dafni U, Reinmuth N et al (2020) A randomized open-label phase III trial evaluating the addition of denosumab to standard first-line treatment in advanced NSCLC: The European Thoracic Oncology Platform (ETOP) and European Organisation for Research and Treatment of Cancer (EORTC) SPLENDOUR Trial. J Thorac Oncol 15:1647–1656
Smith MR, Saad F, Coleman R, Shore N, Fizazi K et al (2012) Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet 379:39–46
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research B (18H02919) and Challenging Research (Pioneering) (17K19582) from the Japan Society for the Promotion of Science (JSPS); The Japanese Society for Bone and Mineral Research Rising Stars Grant; and grants from Astellas Foundation for Research on Metabolic Disorders, The Tokyo Society of Medical Sciences, Taiju Life Social Welfare Foundation, MSD K.K., Astellas Research Support, Mitsubishi Tanabe Pharma Corporation Research Support and Kobayashi Foundation for Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The Department of Osteoimmunology is an endowment department, supported with an unrestricted grant from AYUMI Pharmaceutical Corporation, Chugai Pharmaceutical, MIKI HOUSE and Noevir.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Okamoto, K. Role of RANKL in cancer development and metastasis. J Bone Miner Metab 39, 71–81 (2021). https://doi.org/10.1007/s00774-020-01182-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-020-01182-2