
Abstract
Almost a quarter century has passed since discovery of receptor activator of NF-κB ligand (RANKL). This discovery had a major impact on identification of mechanisms regulating osteoclast differentiation and function, establishment of a research field bridging bone and the immune system (osteoimmunology), and development of a fully human anti-RANKL neutralizing antibody (denosumab). Denosumab is now clinically available for treatment of osteoporosis and cancer-induced bone diseases in the US, Europe and many other countries, including Japan. Denosumab is a so-called blockbuster drug, with sales of 5.0 billion US dollars in 2019. This is a real success story from bench to bedside. In this review, the pivotal roles of the RANKL/RANK/OPG system in osteoclast differentiation and function are shown. RANKL is a ligand required for osteoclast generation, RANK is the receptor for RANKL, and osteoprotegerin (OPG) is a decoy receptor for RANKL. The review covers recent results showing the importance of RANKL on osteoblasts in regulation of osteogenesis and the role of RANKL-RANK dual signaling in coupling of bone resorption and formation, including demonstration of RANKL reverse signaling that we had previously hypothesized. Possible applications of anti-RANKL antibody in treatment of cancer are also discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bones are dynamic tissues in which bone resorption and formation are continuously repeated in a homeostatic mechanism of “coupling” between these processes. Osteoclasts are cells that resorb bone, while osteoblasts form bone. The bone volume resorbed by osteoclasts is strictly controlled to be equal to that formed by osteoblasts under normal conditions. If conditions such as menopause and aging cause bone resorption to increase relative to bone formation, metabolic bone diseases such as osteoporosis develop. Thus, there are precise mechanisms that control coupling between bone resorption and formation.
In their pioneering work, Rodan and Martin [1] hypothesized a role of osteoblasts in hormonal control of bone resorption. Osteoblasts are somehow involved in regulation of osteoclastogenesis. Suda et al. [2] advanced this hypothesis and proposed the presence of a hypothetical factor, osteoclast differentiation factor (ODF), that is produced on osteoblasts by bone resorbing factors such as 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] and parathyroid hormone (PTH). There were many attempts to identify ODF, but all resulted in failure until the late 1990s, until discovery of the RANKL/RANK/OPG system. In this review, an overview of this system is given.
Discovery of three key factors in the molecular mechanism of osteoclastogenesis
Discovery of OCIF/OPG
Mouse coculture system
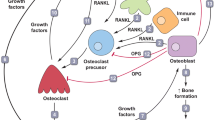
Until the late 1980s, it was difficult to prepare osteoclasts in in vitro culture, and as an alternative, these cells were directly prepared from bones. Takahashi et al. [3] established a mouse coculture system for osteoclasts, in which osteoblasts isolated from mouse calvaria were cocultured with spleen cells containing osteoclast progenitors. Osteoclasts formed only when osteoblasts and spleen cells were cocultured in the presence of 1,25(OH)2D3. Suda et al. [2] hypothesized that direct contact between osteoblasts and osteoclast progenitors was essential for osteoclast differentiation (Fig. 1a). In this hypothesis, osteoclast progenitors are “seeds” and osteoblasts are “soil” that provide a suitable microenvironment for osteoclast formation in bone. Chambers [4] reported a similar hypothesis at almost the same time.

Mechanisms of osteoclast development. a Hypothesis of osteoclast development (before RANKL identification). b Osteoclast formation from mouse spleen cells with sRANKL and M-CSF in the absence of osteoblasts (Yasuda et al. Proc Natl Acad Sci USA 1998;95:3597–3602 [14] Copyright (1998) National Academy of Sciences). c A model illustrating a mechanism through which osteoblasts/stromal cells regulate osteoclast differentiation and activation (after RANKL identification). 1,25(OH)2D3 1,25-dihydroxyvitamin D3, PTH parathyroid hormone, PGE2 prostaglandin E2, IL-11 interleukin-11, ODF osteoclast differentiation factor, M-CSF macrophage colony-stimulating factor, RANK receptor activator of NF-κB, RANKL receptor activator of NF-κB ligand, sRANKL soluble RANKL, OPG osteoprotegerin
Hypothesis for osteoclast development
In the hypothesis proposed by Suda et al. [2], a membrane-bound factor (ODF) is induced on osteoblasts or stromal cells in response to bone-resorbing factors such as 1,25(OH)2D3, PTH, prostaglandin E2 (PGE2), and interleukin (IL)-11 (Fig. 1a). Osteoclast progenitors with ODF receptor recognize ODF by cell-to-cell contact and differentiate to osteoclasts. Macrophage colony-stimulating factor (M-CSF) produced by osteoblasts/stromal cells is also required for osteoclast differentiation [5]. ODF is a hypothetical factor that is proposed to be commonly induced on membranes of osteoblasts/stromal cells by bone-resorbing factors such as 1,25(OH)2D3, PTH and IL-11, and to differentiate osteoclast progenitors to osteoclasts with M-CSF through binding to ODF receptor on the progenitors [2].
Isolation and molecular cloning of OCIF/OPG
Tsuda et al. [6] at Snow Brand Milk Products purified and identified a novel factor called osteoclastogenesis inhibitory factor (OCIF). OCIF was purified from conditioned medium of human primary fibroblasts (IMR-90 cells) with a combination of column chromatography and a bioassay using bone marrow cells treated with 1,25(OH)2D3. The main points of the strategy were the suggestion of an inhibitor(s) of ODF in Suda’s hypothesis [2]; and the expected presence of the inhibitor in conditioned medium of fibroblasts. Since ODF was a hypothetical factor, there was no inhibitor of the hypothetical factor at that time. Tsuda et al. proposed a new hypothesis, based on the Suda hypothesis. Thus, since fibroblasts are present in the whole body, it seemed unlikely that these cells would produce an inhibitor of osteoclast formation. We now know that IMR-90 cells produce a number of growth factors and cytokines [7].
We subsequently cloned human OCIF cDNA using the amino-acid sequences of the protein and identified it as a novel member of the tumor necrosis factor (TNF) receptor family [8]. OCIF is a secreted protein without a transmembrane domain that inhibits all in vitro osteoclast formation elicited through three distinct signaling pathways stimulated by 1,25(OH)2D3, PTH and IL-11. Administration of OCIF to rats increased bone volume, accompanied by a decrease of active osteoclast number [8]. During preparation of the manuscript (Ref. [8]), Simonet et al. [9] at Amgen independently cloned the same molecule in the expressed sequence tag (EST) cDNA project. The Amgen group found a novel member of the TNF receptor family without functional information and produced transgenic (TG) mice overexpressing the protein to identify its function. The TG mice showed osteopetrosis with inhibition of osteoclast differentiation. The protein was named OPG.
A difference in the strategies at Snow Brand Milk Products and Amgen is that we aimed to identify a hypothetical inhibitor of osteoclastogenesis by purification and bioassay, while Amgen discovered a novel protein in EST cDNA sequencing by chance and identified it as an inhibitor of osteoclastogenesis. It is of note that the same molecule was independently identified at almost the same time using two different strategies. The American Society for Bone and Mineral Research (ASBMR) President Committee on Nomenclature proposed OPG as the name of choice [10]. The physiological role of OPG is apparent through the finding that OPG-knockout (KO) mice exhibit severe osteoporosis due to enhanced osteoclast formation [11, 12]. Thus, OPG is a potent osteoclastogenesis inhibitory factor.
Discovery of RANKL
Evidence for OPG-binding protein
During characterization of OPG, we found evidence for an OPG-binding protein. The mouse stromal ST2 cell line supports osteoclastogenesis from mouse spleen cells with 1,25(OH)2D3 and dexamethasone (Dex) [13]. OPG bound to a single class of high affinity binding sites induced by 1,25(OH)2D3 and Dex in ST2 cells [8]. If the binding sites on treated ST2 cells were occupied by OPG, the cells failed to support osteoclastogenesis from spleen cells. The time course of the increase in binding sites coincided with that of osteoclast formation. These results strongly suggested that the sites were involved in cell-to-cell signaling between stromal cells and osteoclast progenitors, and that OPG inhibits osteoclastogenesis by interrupting this signaling through its binding sites [8]. Cross-linking using radioactive OPG revealed that OPG bound to a 40-kDa protein induced on the treated cells. Taken together, these results raised the possibility that the 40-kDa OPG-binding protein is a ligand for OPG, and is identical to ODF [8]. Since members of the TNF receptor family bind to ligands of the TNF family, we assumed that ODF was a novel member of the TNF ligand family.
Molecular cloning of ODF
To identify the 40-kDa OPG-binding protein, we screened a cDNA expression library of ST2 cells treated with 1,25(OH)2D3 and Dex using radioactive OPG. A cDNA clone encoding 316 amino acids (MW 36 kDa) was isolated [14]. The OPG-binding protein was a novel member of the TNF ligand family, as expected, with a type II transmembrane domain and extracellular C-terminal region. Expression of the gene was independently induced by 1,25(OH)2D3, PTH and IL-11, and the protein was produced on osteoblasts. Coimmunoprecipitation of the protein and OPG complex with anti-OPG Ab showed that the size of the protein was 40 kDa. The soluble form of the protein together with M-CSF induced osteoclast formation from spleen cells in the absence of osteoblasts (Fig. 1b). This was the first time that osteoclasts had been generated from their progenitors without coculture with osteoblastic cells, the common method for in vitro generation of osteoclasts. The osteoclasts formed numerous resorption pits on dentine slices. Fixed COS7 cells expressing the recombinant protein induced osteoclast formation from spleen cells, suggesting that the protein mediates cell-to-cell signaling essential for osteoclastogenesis. Taken together, the protein satisfied the major criteria for ODF in terms of its biological activity and regulation of its expression by bone-resorbing factors. Therefore, we concluded that the protein was ODF, a long-sought after ligand mediating an essential signal on osteoclast progenitors for their differentiation into osteoclasts [14] (Fig. 1c).
Lacey et al. [15] at Amgen cloned the same molecule, and called it OPG ligand (OPGL), after screening a cDNA expression library of the mouse myelomonocytic cell line, 32D, using fluorescence-activated cell sorting analysis with a fusion protein of OPG and an Fc-fragment (OPG-Fc). Soluble OPGL administered to mice induced rapid and severe hypercalcemia, which was inhibited by OPG treatment. We and others used a similar approach to identify ODF/OPGL as an OPG-binding protein [14, 15]. Contemporaneously, Anderson et al. [16] at Immunex and Wong et al. [17] in academia identified the same molecule regulating T-cell and dendritic cell functions, and called it RANKL and TNF-related activation-induced cytokine (TRANCE), respectively. Anderson et al. [16] reported that RANKL stimulated the activation of NF-κB in dendritic cells and it augmented the ability of the cells to stimulate T cells. In contrast, Wong et al. [17] reported that RANKL (TRANCE) stimulated c-Jun N-terminal kinase (JNK) specifically in cells of the T cell lineage and they suggested a role for RANKL in the regulation of the T cell-dependent immune response. As standard nomenclature for ODF/OPGL/RANKL/TRANCE, RANKL was proposed by the ASBMR President’s Committee on Nomenclature [10].
RANKL-KO mice exhibit osteopetrosis with no osteoclasts, marrow spaces, or tooth eruption, indicating that RANKL is essential for osteoclast development [18]. These mice also have profound growth retardation, defects in early differentiation of T- and B-cells, and lack all lymph nodes. These results showed that RANKL is essential for osteoclast development in vivo.
Discovery of RANK
RANKL directly binds to osteoclast progenitors, suggesting that a membrane-bound receptor may be present on the cells [14, 15]. RANK, a novel member of the TNF receptor family, was known to be a receptor for RANKL in T-cell and dendritic cell interaction [16], but the receptor responsible for RANKL-mediated osteoclastogenesis had not been identified. Some ligands of the TNF family bind to several receptors of the TNF receptor family, and it was suspected that RANKL might bind to another member of the TNF receptor family, but not to RANK. We cloned the RANKL receptor from mouse osteoclast progenitors by panning and identified it as RANK [19]. A polyclonal Ab against soluble RANK (sRANK) mimicked RANKL function by clustering of RANK. In contrast, sRANK and a Fab fragment of anti-RANK polyclonal Ab completely inhibited RANKL-mediated osteoclastogenesis by binding to RANKL and RANK, respectively. While OPG inhibited RANKL-mediated osteoclastogenesis by interrupting the binding of RANKL to RANK, it had no effect on anti-RANK Ab-mediated osteoclastogenesis. Taken together, these results provided the first evidence that RANK is the sole signaling receptor essential for in vitro RANKL-mediated osteoclastogenesis and that OPG acts as a decoy receptor for RANKL to compete against RANK [19].
Hsu et al. [20] at Amgen made TG mice overexpressing sRANK and showed that the mice exhibited osteopetrosis, similar to OPG-TG mice, based on which RANK was predicted to be a receptor for RANKL in vivo. Dougall et al. [21] at Immunex found evidence that RANK was the receptor for RANKL in vivo by showing that RANK-KO mice had an almost identical phenotype to that of RANKL-KO mice. A summary of these results is illustrated in a model of osteoclast differentiation and activation (Fig. 1c). RANKL is important for differentiation, fusion, survival and activation of osteoclasts. We know that RANKL on osteocytes and osteoblasts regulates osteoclastogenesis in bone remodeling and modeling, respectively [22, 23].
Genetic and pharmacological models related to RANKL
Genetic models
To investigate the effects and functions of RANKL in vivo, we made TG mice overexpressing soluble RANKL (sRANKL) [24]. The sRANKL TG mice exhibited severe osteoporosis with an increase of osteoclasts. As mentioned above, RANKL-KO mice exhibit osteopetrosis with no osteoclasts [18]. X-ray images of the genetic models including wild-type (WT), sRANKL TG, and RANKL-KO showed a normal status, osteoporosis, and osteopetrosis, respectively (Fig. 2a). Recent studies using mice that specifically lack sRANKL showed that sRANKL is dispensable for physiological bone remodeling [25, 26].

Bone images in RANKL-related models. a X-ray images of genetic models including WT, TG, and KO mice. b Micro-CT images of pharmacological models including control, sRANKL-, Ad-sRANKL-, and Anti-RANKL-Ab administration to normal mice. RANKL receptor activator of NF-κB ligand, sRANKL soluble RANKL, WT wild type mice, TG sRANKL transgenic mice, KO RANKL-knockout mice, Ad-sRANKL adenovirus harboring sRANKL
Pharmacological models
Genetic animal models are useful, but several months are needed to interbreed the mice with other TG mice or KO-mice. As an alternative, we established three pharmacological animal models: (1) a novel rapid bone loss model by administration of glutathione-S transferase-RANKL fusion protein to mice (sRANKL) [27]; (2) a novel mouse model of hypercalcemia with anorexia by overexpression of sRANKL using an adenovirus vector (Ad-sRANKL) [28]; and (3) a novel mouse model of osteopetrosis by administration of a denosumab-like anti-mouse RANKL neutralizing monoclonal Ab (clone OYC1, Anti-RANKL) [29].
The sRANKL-mediated bone loss model was rapidly established within 24–50 h. Two or three intraperitoneal injections of sRANKL are sufficient to induce osteoporotic bone loss with an increase of osteoclasts. This model is useful for evaluation of pharmaceuticals and/or candidates for treatment of osteoporosis [27]. The Ad-sRANKL injection model showed very severe osteoporosis and hypercalcemia with anorexia. The serum sRANKL level in this model was about 1.5 μg/ml, while that in WT mice was 0.1 ng/ml [28]. In the anti-RANKL Ab-treated osteopetrosis model, a single subcutaneous injection of anti-RANKL Ab increased bone mass with marked decreases in osteoclast surface and number, as well as decreases in osteoblast surface, mineral apposition rate (MAR), and bone formation rate (BFR) after 2 weeks [29]. Micro-CT images of the pharmacological models including control, sRANKL-, Ad-sRANKL-, and anti-RANKL-Ab administration to normal mice showed a normal status, osteoporosis, severe osteoporosis, and osteopetrosis, respectively (Fig. 2b). Two to 14 days are required to make these animal models. These inducible models of osteoporosis and osteopetrosis in normal mice exhibit exact mirror images in terms of the change in bone mass and are useful for research on osteoclast biology and bone metabolism in vivo.
RANKL reverse signaling
WP9QY (W9) is a peptide designed to be structurally similar to one of the cysteine-rich domains in TNF receptor type I, and is known to bind to TNFα and block its activity [30]. W9 also binds to RANKL and inhibits RANKL-induced osteoclastogenesis in vitro and in vivo [31]. We provided the first evidence that W9 enhances osteoblastic differentiation/mineralization in in vitro and increases bone mass in vivo [32]. Histomorphometrical analysis of mice treated with W9 showed that the peptide had a weak inhibitory effect on osteoclast number and surface in distal femoral metaphysis, but markedly increased MAR and BFR in femoral diaphysis. As a RANKL antagonist, it is surprising that W9 has a bone anabolic effect in vivo. However, knockdown of RANKL expression in treated osteoblastic cells reduces the effect of W9, and W9 has a weak effect on RANKL-KO osteoblasts in vitro. These results show that RANKL is involved in W9-mediated osteoblastogenesis [32]. We have hypothesized that W9 exerts its bone anabolic activity through RANKL reverse signaling [32,33,34] (Fig. 3a).

Hypotheses of regulation of coupling between bone resorption and formation. a Bidirectional signaling through RANKL/RANK may control bone remodeling. b Activated osteoclasts stimulate osteoblasts to produce GFs and GFRs through RANKL reverse signaling. GFs and GFRs produced by osteoblasts may work in an autocrine/paracrine manner. W9 WP9QY, RANKL receptor activator of NF-κB ligand, RANK receptor activator of NF-κB, GF growth factor, GFR growth factor receptor, OB osteoblast, OC osteoclast
It is well known that RANKL transmits the osteoclast differentiation signal through RANK. Since W9 is an artificial synthetic peptide that can bind to RANKL, we further hypothesized that the endogenous ligand for RANKL is RANK. Binding of RANKL and RANK may transmit a bidirectional signal to activate osteoclasts and osteoblasts through the RANKL forward and reverse signals, respectively (Fig. 3a). Reverse signaling occurs among members of the TNF family, including TNFα, CD40L, and FasL, and a membrane-bound ligand transmitting its signal as a receptor [35]. OPG and RANK both bind to RANKL, and the greater osteoclastogenesis and osteoblastogenesis found in OPG-KO mice [11, 12] suggest that bidirectional signaling is enhanced without OPG, because OPG is a decoy receptor for RANKL. The observation of similar phenotypes of RANKL- and RANK-KO mice also supports this hypothesis [18, 21].
Once signaling through RANKL is activated in osteoblasts, the cells produce many growth factors (GFs) and their receptors (GFRs) to activate differentiation in an autocrine manner. These GFs and GFRs produced by RANKL reverse signaling may subsequently and sequentially affect neighboring cells to further activate differentiation in a paracrine manner (Fig. 3b). The reverse signal from RANK on osteoclasts to RANKL on osteoblasts, and the forward signal from RANKL to RANK may play important roles in coupling of bone formation and resorption.
Evidence for our hypothesis was recently found by Ikebuchi et al. [36] in vivo, with the further suggestion of the importance of extracellular vesicles with RANK secreted from osteoclasts regulating osteoblastogenesis. Modulators of RANKL reverse signaling such as W9 and RANKL agonist Abs may be promising drug candidates for treatment of metabolic bone diseases. W9 accelerated BMP-2-induced calvarial bone regeneration and stimulated osteoblast differentiation in mice [37], and recovered alveolar bone loss by suppressing osteoclastogenesis and enhancing osteoblastogenesis in OPG-KO mice [38]. Local administration of W9 also promoted bone formation in a rat femur delayed-union model [39]. An artificial RANKL bifunctional Ab inhibiting osteoclastogenesis and activating RANKL reverse signaling prevented decreased bone formation in ovariectomized mice [36].
Therapeutic potential of anti-RANKL Ab in immuno-oncology
Denosumab was developed by Amgen and is now widely used in treatment of osteoporosis and cancer-induced bone diseases. Clinical trials of denosumab in patients with prostate and lung cancers have shown significantly improved overall survival [40, 41]. A retrospective analysis of denosumab treatment of patients with non-small-cell lung cancer (NSCLC) and bone metastases also showed good overall survival [42]. RANKL antagonists have anticancer effects through (1) direct action; (2) osteoclast-dependent indirect action; and (3) T-cell dependent indirect action (Table 1).
Direct action of RANKL on RANK-positive cancer cells
RANKL is involved in metastasis of melanoma cells to bones [43]. Recent study has reported that sRANKL triggers bone metastasis by exerting chemotactic activity in tumor cells such as melanoma and breast cancer [26]. Involvement of RANKL in metastasis of prostate cancer cells is also known [44]. It is reported that RANK drives lung cancer even though the source of RANKL is not identified [45]. Systemic sRANKL and/or local RANKL production by cells in the tumor microenvironment would be the source. RANKL/RANK signaling plays a pivotal role in proliferation of mammary cancer cells [46, 47]. OPG-Fc or RANK-Fc inhibits metastasis and proliferation by binding to RANK-positive cancer cells [43, 46, 47]. Tumor-infiltrating regulatory T (Treg) cells produce RANKL and stimulate metastasis of RANK-positive mammary cancer cells [48]. RANKL on Treg cells acts directly on RANK on the mammary cancer cells.
Osteoclast-dependent indirect action of RANKL on cancer cells
Osteoclasts generated by stimulation with RANKL on osteoblasts and osteocytes resorb bones. GFs such as transforming growth factor (TGF)-β and insulin-like growth factor (IGF)-1 are released from bones, and stimulate cancer cells to proliferate and release bone-resorbing factors such as PTH-related protein (PTHrP) and IL-6. These factors stimulate osteoblasts and osteocytes to produce RANKL. Repetition of these processes forms a so-called vicious cycle, through which cancer cells in bones proliferate. This cycle can be interrupted by anti-RANKL Ab [49].
T-cell dependent indirect action of RANKL on cancer cells
Akiyama et al. [50] showed that RANKL-RANK signaling plays crucial roles in development of medullary thymic epithelial cells (mTECs) that express tissue-specific antigens (TSAs) during embryogenesis. Development of mTECs is stimulated in OPG-KO mice and abrogated in RANKL-KO mice. Thymic central tolerance is a critical process that not only prevents autoimmunity, but also presents a challenge to generation of antitumor immune responses. The mTECs eliminate self-reactive T cells by displaying a diverse repertoire of TSAs that are also shared by tumors. Therefore, while protecting against autoimmunity, mTECs simultaneously limit generation of tumor-specific effector T cells by expressing tumor self-antigens [50]. We and others have shown that anti-RANKL Ab inhibits development of mTECs, which suppresses growth of tumors transplanted in nude mice and prolongs survival of mice transplanted with cancer cells by producing tumor-reactive T cells with tumor-specific antigens [51, 52]. Inhibition of RANKL rescues these T cells from thymic deletion. Transplantation of splenic lymphocytes of mice treated with anti-RANKL Ab reduces the growth of tumors transplanted in nude mice, which suggests that anti-RANKL Ab enhances cancer immunity via T-cells.
Mechanisms through which RANKL inhibition can exert therapeutic effects
As mentioned above, there are at least three mechanisms that can explain the anticancer effects of RANKL antagonists (Table 1). (1) Inhibition of the direct action of RANKL on RANK-positive cancer cells, such as mammary and prostate cancer cells; thus, the targets are limited to RANK-positive cancer cells. (2) Inhibition of the osteoclast-dependent indirect action of RANKL on cancer cells; this approach for interrupting a vicious cycle is only applicable to primary or secondary (metastatic) cancer cells in bones. (3) Inhibition of T-cell dependent indirect action of RANKL on cancer cells, using RANKL antagonists that enhance cancer immunity by generating tumor-reactive T cells with tumor-specific antigens through inhibiting development of mTECs. This approach is applicable to various cancer cells, but may be limited to younger patients due to thymic involution with age. The approach may also be a double-edged sword that cuts two ways: generation of tumor-reactive T cells, but also of self-reactive T cells that show autoimmune side effects. Denosumab has been widely used for treatment of osteoporosis and cancer-induced bone diseases, and thus, the risk may be low in elderly patients. However, it will be important to ensure patient safety in clinical trials, especially for younger patients.
Application of anti-RANKL Ab to immuno-oncology
Immune checkpoint inhibitors (ICIs) such as anti-programmed cell death protein 1 (PD-1) Ab and anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) Ab are used clinically for treatment of many cancers. Binding of programmed cell death 1 ligand 1 (PD-L1) on cancer cells to PD-1 on T cells prevents the T cells from killing the cancer cells. Inhibition of the interaction of PD-L1 and PD-1 with an ICI (anti-PD-L1 Ab or anti-PD-1 Ab) allows the T cells to kill the cancer cells. In contrast, binding of B7 on dendritic cells to CTLA-4 on T cells prevents T cells from killing the cancer cells (Table 2).
Smyth et al. [53] reported a case of rapidly advancing metastatic melanoma with aggressive and symptomatic bone metastases treated with a combination of ipilimumab (anti-CTLA-4 Ab) and denosumab for palliation. This was the first report showing a synergistic effect of an ICI with denosumab in a patient. Similar observations have been reported in mice, showing the remarkable synergistic effects of anti-PD-1, anti-PD-L1, anti-CTLA-4 and anti-RANKL Abs on cancer immunity [53, 54]. Since restriction of cancer immunity takes place in the thymus and periphery, a combination of immunotherapies targeting central (i.e. thymic) and peripheral tolerance should work synergistically. This mechanism strongly supports the above findings. Several clinical trials are currently in progress to study the efficacy of denosumab in combination with ICIs for melanoma (CHARLI), renal cell carcinoma (KEYPAD), and NSCLC (POPCORN). It is likely that we will witness a historic moment in immune-oncology in the near future.
Conclusion
The discovery of the RANKL/RANK/OPG system at the bench has resulted in the bedside translation of denosumab within 12 years. This is a tremendous success in both basic and translational research in bone biology in the last decade. Translation of basic research to clinical applications is achieved by addressing the biological significance of basic findings, and this approach to science can save the lives of patients. Establishment of the mechanisms of osteoclastogenesis has created the new field of osteoimmunology, while denosumab has improved the quality of life for patients with osteoporosis and cancer-induced bone diseases. Further successes are likely in determining the mechanisms of RANKL/RANK dual signaling in regulation of the bone and immune system, and in improving cancer immunity using combinations of denosumab and ICIs.
Change history
13 January 2021
In the original publication of the article, reference 12 was published incorrectly as follows.
References
Rodan GA, Martin TJ (1981) Role of osteoblasts in hormonal control of bone resorption–a hypothesis. Calcif Tissue Int 33:349–351
Suda T, Takahashi N, Martin TJ (1992) Modulation of osteoclast differentiation. Endocr Rev 13:66–80
Takahashi N, Akatsu T, Uadagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ, Suda T (1988) Osteoblastic cells are involved in osteoclast formation. Endocrinology 123:2600–2602
Chambers TJ (1992) Regulation of osteoclast development and function. In: Rifkin BR, Gay CV (eds) Biology and physiology of the osteoclast. CRC Press, Boca Raton, pp 105–128
Takahashi N, Uadagawa N, Akatsu T, Tanaka H, Isogai Y, Suda T (1991) Deficiency of osteoclasts in osteopetrotic mice is due to a defect in the local microenvironment provided by osteoblastic cells. Endocrinology 128:1792–1796
Tsuda E, Goto M, Mochizuki S, Yano K, Kobayashi F, Morinaga T, Higashio K (1997) Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun 234:137–142
Higashio K, Shima N, Goto M, Itagaki Y, Nagao M, Yasuda H, Morinaga T (1990) Identity of a tumor cytotoxic factor from human fibroblasts and hepatocyte growth factor. Biochem Biophys Res Commun 170:397–404
Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K (1998) Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 139:1329–1337
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang M-S et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
The American Society for Bone and Mineral Research President’s Committee on Nomenclature (2000) Proposed standard nomenclature for new tumor necrosis factor family members involved in the regulation of bone resorption. J Bone Miner Res 15:2293–2296
Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268
Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Gomibuchi T, Yano K, Shima N, Washida N, Tsuda E, MorinagaT HK, Ozawa H (1998) Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun 247:610–615
Udagawa N, Takahashi N, Akatsu T, Sasaki T, Yamaguchi A, Kodama H, Martin TJ, Suda T (1989) The bone marrow-derived stromal cell lines MC3T3–G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen cells. Endocrinology 125:1805–1813
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 95:3597–3602
Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR et al (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176
Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390:175–179
Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS, Frankel WN, Lee SY, Choi Y (1997) TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem 272:25190–25194
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323
Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K, Morinaga T, Higashio K (1998) RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun 253:395–400
Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A et al (1999) Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA 96:3540–3545
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17:1235–1241
Mizuno A, Kanno T, Hoshi M, Shibata O, Yano K, Fujise N, Kinosaki M, Yamaguchi K, Tsuda E, Murakami A, Yasuda H, Higashio K (2002) Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J Bone Miner Metab 20:337–344
Xiong J, Cawley K, Piemontese M, Fujiwara Y, Zhao H, Goellner JJ, O’Brien CA (2018) Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun 9:2909
Asano T, Okamoto K, Nakai Y, Tsutsumi M, Muro R, Suematsu A, Hashimoto K, Okamura T, Ehata S, Nitta T, Takayanagi H (2019) Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat Metab 1:868–875
Tomimori Y, Mori K, Koide M, Nakamichi Y, Ninomiya T, Udagawa N, Yasuda H (2009) Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J Bone Miner Res 24:1194–1205
Enomoto T, Furuya Y, Tomimori Y, Mori K, Miyazaki J, Yasuda H (2011) Establishment of a new murine model of hypercalcemia with anorexia by overexpression of soluble receptor activator of NF-κB ligand using an adenovirus vector. J Bone Miner Metab 29:414–421
Furuya Y, Mori K, Ninomiya T, Tomimori Y, Tanaka S, Takahashi N, Udagawa N, Uchida K, Yasuda H (2011) Increased bone mass in mice after single injection of anti-receptor activator of nuclear factor-κB ligand-neutralizing antibody: evidence for bone anabolic effect of parathyroid hormone in mice with few osteoclasts. J Biol Chem 286:37023–37031
Takasaki W, Kajino Y, Kajino K, Murali R, Greene MI (1997) Structure-based design and characterization of exocyclic peptidomimetics that inhibit TNFα binding to its receptor. Nat Biotechnol 15:1266–1270
Aoki K, Saito H, Itzstein C, Ishiguro M, Shibata T, Blanque R, Mian AH, Takahashi M, Suzuki Y, Yoshimatsu M, Yamaguchi A, Deprez P, Mollat P, Murali R, Ohya K, Horne WC, Baron R (2006) A TNF receptor loop peptide mimic blocks RANK ligand-induced signaling, bone resorption, and bone loss. J Clin Invest 116:1525–1534
Furuya Y, Inagaki A, Khan M, Mori K, Penninger JM, Nakamura M, Udagawa N, Aoki K, Ohya K, Uchida K, Yasuda H (2013) Stimulation of bone formation in cortical bone of mice treated with a receptor activator of nuclear factor-kB ligand (RANKL)-binding peptide that possesses osteoclastogenesis inhibitory activity. J Biol Chem 288:5562–5571
Khan A, Alles N, Soysa NS, Mamun M, Nagano K, Mikami R, Furuya Y, Yasuda H, Ohya K, Aoki K (2013) The local administration of TNF-α and RANKL antagonist peptide promotes BMP-2-inducedboneformation. J Oral Biosci 55:47–54
Nakamura M, Nakamichi Y, Koide M, Yamashita T, Ara T, Nakamura H, Penninger JM, Furuya Y, Yasuda H, Udagawa N (2017) The W9 peptide directly stimulates osteoblast differentiation via RANKL signaling. J Oral Biosci 59:146–151
Eissner G, Kolch W, Scheurich P (2004) Ligands working as receptors. Reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev 15:353–366
Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, Kariya Y, Kato G, Tabata Y, Penninger JM, Udagawa N, Aoki K, Suzuki H (2018) Coupling of bone resorption and formation by RANKL reverse signalling. Nature 561:195–200
Sugamori Y, Mise-Omata S, Maeda C, Aoki S, Tabata Y, Murali R, Yasuda H, Udagawa N, Suzuki H, Honma M, Aoki K (2016) Peptide drugs accelerate BMP-2-induced calvarial bone regeneration and stimulate osteoblast differentiation through mTORC1 signaling. BioEssays 38:717–725
Ozaki Y, Koide M, Furuya Y, Ninomiya T, Yasuda H, Nakamura M, Kobayashi Y, Takahashi N, Yoshinari N, Udagawa N (2017) Treatment of OPG-deficient mice with WP9QY, a RANKL-binding peptide, recovers alveolar bone loss by suppressing osteoclastogenesis and enhancing osteoblastogenesis. PLoS ONE 12:e0184904
Sawa M, Wakitani S, Kamei N, Kotaka S, Adachi N, Ochi M (2018) Local Administration of WP9QY (W9) peptide promotes bone formation in a rat femur delayed-union model. J Bone Miner Metab 36:383–391
Smith MR, Saad F, Coleman R, Shore N, Fizazi K et al (2012) Denosumab and bone-metastasis free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet 379:39–46
Scagliotti GV, Hirsh V, Siena S, Henry DH, Woll PJ, Manegold C, Solal-Celigny P, Rodriguez G, Krzakowski M, Mehta ND, Lipton L, García-Sáenz JA, Pereira JR, Prabhash K, Ciuleanu TE, Kanarev V, Wang H, Balakumaran A, Jacobs I (2012) Overall survival improvement in patients with lung cancer and bone metastases treated with denosumab versus zoledronic acid: subgroup analysis from a randomized phase 3 study. J Thorac Oncol 7:1823–1829
Udagawa H, Niho S, Kirita K, Umemura S, Matsumoto S, Yoh K, Goto K (2017) Impact of denosumab use on the survival of untreated non-squamous non-small cell lung cancer patients with bone metastases. J Cancer Res Clin Oncol 143:1075–1082
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla CV, Komnenovic V, Kong Y-Y, Schreiber M, Dixon SJ, Sims SM, Khokha R, Wada T, Penninger JM (2006) Regulation of cancer cell migration and bone metastasis by RANKL. Nature 440:692–696
Luo JL, Tan W, Ricono JM, Korchynskyi O, Zhang M, Gonias SL, Cheresh DA, Karin M (2007) Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 446:690–694
Rao S, Sigl V, Wimmer RA, Novatchkova M, Jais A et al (2017) RANK rewires energy homeostasis in lung cancer cells and drives primary lung cancer. Genes Dev 31:2099–2112
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ, Hanada R, Joshi PA, Aliprantis A, Glimcher L, Pasparakis M, Khokha R, Ormandy CJ, Widschwendter M, Schett G, Penninger JM (2010) Osteoclast differentiation factor RANKL controls development of progestin driven mammary cancer. Nature 468:98–102
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R, Pinkas J, Branstetter D, Dougall WC (2010) RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 468:103–107
Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M (2011) Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 470:548–553
Mundy G (2002) Metastasis to bone: causes, consequences and therapeutic opportunities. Nature Rev Cancer 2:584–593
Akiyama T, Shimo Y, Yanai H, Qin J, Ohshima D, Maruyama Y, Asaumi Y, Kitazawa J, Takayanagi H, Penninger JM, Matsumoto M, Nitta T, Takahama Y, Inoue J (2008) The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity 29:423–437
Akiyama T, Yanai H, Akiyama N, Yasuda H (2012) Potentiator of cancer immunity containing RANKL antagonist. Japan Patent Kokai WO 2012/133914. 4 Oct 2012
Khan IS, Mouchess ML, Zhu ML, Conley B, Fasano KJ, Hou Y, Fong L, Su MA, Anderson MS (2014) Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J Exp Med 211:761–768
Smyth MJ, Yagita H, McArthur GA (2016) Combination anti-CTLA-4 and anti-RANKL in metastatic melanoma. J Clin Oncol 34:e104–e106
Ahern E, Smyth MJ, Dougall WC, Teng MWL (2018) Roles of the RANKL-RANK axis in antitumour immunity—implications for therapy. Nat Rev Clin Oncol 15:676–693
Acknowledgements
I thank all collaborators, especially Yoshiya Tomimori, Tetsuro Enomoto, and Yuriko Furuya, for their help in the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Hisataka Yasuda is an employee of Oriental Yeast Co., Ltd.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Yasuda, H. Discovery of the RANKL/RANK/OPG system. J Bone Miner Metab 39, 2–11 (2021). https://doi.org/10.1007/s00774-020-01175-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-020-01175-1