
Abstract
Genetic variation is an important attribute of forest tree populations enabling them to adapt to spatial and temporal variations in environmental conditions. In particular, Scots pine (Pinus sylvestris L.) has an exceptionally broad area of dispersal covering different climates and soil conditions, but the genetic variability in extreme conditions has not been studied. We hypothesized that the genetic variability of P. sylvestris is enhanced at its soil fertility limit in bogs where stunted trees form a sparse canopy with reduced light competition, but in highly turbulent conditions generating ideal conditions for distant pollen dispersal. A total of 180 individuals were studied from three bog populations using five nuclear (nSSR) and five chloroplastic (cpSSR) polymorphic microsatellite loci (simple sequence repeats, SSR). According to both marker systems, high and similar level of genetic diversity (ca. 99 % within the populations and 1 % among the populations) was observed for nuclear (F ST = 0.007 and R ST = 0.022) and chloroplast (F ST = 0.015 and R ST = 0.023) genomes. Despite the low genetic differentiation among the populations, there was evidence of geographic genetic differentiation in the chloroplast genomes, suggesting that isolation by distance might be a possible mechanism shaping the present distribution of genetic variability. In addition, significant but low spatial genetic structure along habitat wetness gradient was found in nuclear genomes in one site. Significant cyto-nuclear linkage disequilibrium was shown between one of the cpSSR loci to all nSSR loci. The results demonstrate a huge within-population genetic variability in these sites and underscore the importance of pollen gene flow in homogenizing populations on these geographic scales.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Presence of genetic variation in forest tree populations is the key factor determining adaptability of populations to environmental heterogeneity. Thus, understanding population genetic variability and its underlying individual and sub-population components constitutes an important means for conservation and management of genetic resources as well as for practical forestry breeding and silviculture applications, especially under conditions of global change (Brown 1978; Gregorius 1994; Hattemer 1987; Holliday et al. 2011; Ziehe et al. 1989). Recent developments in molecular methods for analysis of population genetic structure have vastly enhanced the body of knowledge of genetic variation in many important forestry species (Dzialuk et al. 2009; Newton et al. 1999; Pazouki et al. 2010; Petit et al. 1997; Scalfi et al. 2009; Shanjani et al. 2004, 2010, 2011). Molecular markers based on noncoding DNA regions typically have high variability, and thus, they provide appropriate means to gaining high-resolution information on within-species variability within populations and populations genetic structure (Bouzat 2001; Grassi et al. 2003; Pautasso 2009; Ribeiro et al. 2002) and for biodiversity protection (Hansen et al. 2005; Kadereit et al. 2004; Karp et al. 1998; Sgorbati et al. 2004). Molecular marker-based analyses have also allowed for gaining an insight into the historical species migration and population differentiation processes responsible for the species present distribution range (Petit et al. 2003). Although data archives on molecular variability have dramatically expanded in recent years, there is still limited information of genetic variability on many major forestry species, especially at their range margins.
Scots pine (Pinus sylvestris L., Pinaceae), like the majority of Pinus species, is an outbreeding wind-pollinated species that has a diploid genome with a chromosome number of 2n = 24 (Saylor 1972). It is a long-lived conifer with extremely broad distribution range that extends from latitudes of 70°N in northern Fennoscandia to 40°S in Spain and from longitudes of 5°W in Spain and Scotland to 135°E in Siberia (Cheddadi et al. 2006; Critchfield and Little 1966; Naydenov et al. 2007; Pyhäjärvi et al. 2007, 2008; Sinclair et al. 1999; Soranzo et al. 2000). Pinus sylvestris survived the last glaciations in four main refugia (Iberian Peninsula, north Apennine Peninsula, south of the Carpathian and Sudeten Mountains, and the Balkans) where it today is present as isolated populations at high 1000–2500 m. In addition to these refugia, P. sylvestris also survived the glaciation in the Russian plains, migrating into the central and western parts of Europe and to Fennoscandia once the glaciers had retreated (Bennett et al. 1991; Huntley and Birks 1983; Robledo-Arnuncio et al. 2004; Soranzo et al. 2000). This large population bottleneck has been considered as one of the key factors shaping the genetic diversity of P. sylvestris in its current habitats (Naydenov et al. 2007; Pyhäjärvi et al. 2007; Sinclair et al. 1999).
Despite the recent population bottleneck, this species covers not only a large climatic range, but also a wide range of soil habitats from wet peaty soils to excessively drained sand plains, and from fertile deep clay soils to infertile thin rocky soils and deep peat soils. In fact, in these infertile habitats, P. sylvestris is often the main dominant, suggesting a large adaptability of this species (Finér 1992; Gunnarsson and Rydin 1998; Niinemets et al. 2001). Due to importance for forestry, studies on P. sylvestris genetic diversity have mainly focused on stands with high productivity in fertile habitats (Kuznetsova and Mandre 2006; Maaten and Kurm 2007; Naydenov et al. 2005), and much less is known of population genetic structure in infertile habitats. Studies along fertility gradients over short spatial distance from mineral soils that support highly productive tall trees toward organic wet peaty soils that support stunted small trees have demonstrated major variations in tree growth rates and structure of leaves and shoots (Niinemets et al. 2001, 2002), but it is unclear to which extent the species adaptability to infertile conditions is affected by genetic and plastic components of variability.
Pinus sylvestris is the only pine species native to northern Europe, and it is the most common tree species in the Baltic states. In Estonia, P. sylvestris covers almost 38 % of Estonian forests (Pärt and Villard 2006) and is the key dominant species in extremely infertile raised bogs (Läänelaid 1981; Niinemets et al. 2001). In this study, genetic variability and population structure of P. sylvestris were analyzed in three distant naturally seeded bog populations that can be considered as the fertility limit of this species. In each site, the samples were collected along a soil fertility gradient from drier bog margins toward wetter bog interiors to characterize the small-scale variability in sub-population genetic structure. We hypothesized that there is high level of outbreeding and consequently high genetic variability at the nutrient limit of the species, and further that there is a spatial gradient in genetic relatedness indicative of the genetic component of phenotypic plasticity in the organic–mineral soil continuum. Like many other tree species (Hamrick et al. 1992), P. sylvestris reveals a high level of genetic variation at microsatellite loci (Belletti et al. 2012; Maaten and Kurm 2007; Nowakowska et al. 2014; Robledo-Arnuncio et al. 2005; Scalfi et al. 2009). Both nuclear and chloroplast microsatellite markers were used to characterize the genetic variability. Given that in the wind-pollinated species, pollen can arrive from relatively large distances, larger than the extent of seed dispersal, we further hypothesized that such a possible small-scale variation in genetic diversity is primarily reflected in nSSR markers rather than in paternally inherited cpSSR markers that reflect variability of pollen. We argue that these data provide important insight into overall genetic variability and sub-population differentiation in nutrient-limited sites. This information is a crucial value in understanding species adaptability to environmental heterogeneity and has important practical applications in selecting plant sources suitable for reforestation especially in nutrient-limited habitats, e.g., on destroyed habitats where tree growth is particularly strongly curbed by limited soil fertility.
Materials and methods
Study sites and sampling
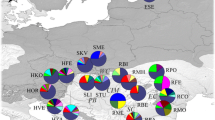
Pinus sylvestris was studied at three naturally regenerated low-elevation bog ecosystems in Estonia (Fig. 1): Laeva (58°27′N, 26°21′E, elevation 44 m, area ca. 7050 ha), Parika (58°30′N, 25°49′E, elevation 46 m, area ca. 2200 ha) and Kakerdaja (59°11′N, 25°31′E, elevation 80 m, area ca. 2400 ha). All sites sampled are characterized by a thick layer of acidic organic soil (peat, pH in distilled water 4.1–4.6) and are dominated or co-dominated by stunted (0.2–3 m) tall trees of P. sylvestris. In addition, all sites are characterized by vigorous, largely continuous moss cover, primarily dominated by Sphagnum species that fill the gaps in dwarf-shrub and grass layer. The sites differ in thickness of peat layer, water regime (bogs relaying on precipitation and transitional mire to bog areas relying on both precipitation and ground water or/and surface runoff water) and composition of herb, shrub and dwarf-shrub layer. In all sites, there are further characteristic gradients of water availability from drier and more fertile (exterior parts or central raised bog “islands”) toward wetter and less fertile parts (central or intermediate parts, Fig. 1) (Kännaste et al. 2013).

Location of sampling sites for three natural bog populations of Pinus sylvestris in Estonia and demonstration of tree sampling along the trails from drier (1) to wetter (60) habitats. Pie graphs refer to cpDNA haplotypes frequencies. For each population, they indicate the frequencies of the most common haplotypes (H12, H24, H36, H44, H47, H49, H63, H66, H67 and H78). The blue portion of the pie represents the shared haplotype frequencies, and the red portion indicates the frequency of private haplotypes. According to sample location along the trails, the populations at each site were divided among dry (KakerdajaD-[KD], ParikaD-[PD] and LaevaD-[LD]) and wet (KakerdajaW-[KW], ParikaW-[PW] and LaevaW-[LW]) sub-populations, 30 individuals in each sub-population
Kakerdaja site is a raised bog with a drier central bog “island” and extremely wet surrounding areas with numerous open-water hollows and a lake (6.7 ha). The shrub and dwarf-shrub layer is prevailed by Empetrum nigrum L., Andromeda polifolia L. and Ledum palustre L., and the herbaceous layer is dominated by Rhynchospora alba (L.) Vahl, Scheutzeria palustris L., Rubus chamaemorus L. and Trichophorum cespitosum L.
Laeva site supports a transitional bog with patches of raised bog in the central part with ca. 5-m-thick peat layer. In addition to P. sylvestris, the tree layer is co-dominated by Betula pubescens Ehrh., while the shrub and dwarf-shrub layer is dominated by Salix lapponum L. and Vaccinium oxycoccus L. in the transitional bog and by Chamaedaphne calyculata (L.) Moench, L. palustre and Vaccinium uliginosum L. in the raised bog. The herbaceous layer is dominated by Carex spp., Phragmites australis (Cav.) Trin. ex Steud. and Eriophorum vaginatum L.
Parika site supports a raised bog with transitional bog areas along the shores of two lakes (114 and 5 ha) present at the site. Additional co-dominants in tree and shrub layer are B. pubescens, Betula nana L. and Salix spp. The dwarf-shrub layer is dominated by V. oxycoccus, E. nigrum and A. polifolia, while the herbaceous layer by Eriophorum spp. and Carex spp. More details on the sites are provided by Kännaste et al. (2013).
Mature healthy current-year needles were collected from 60 individual trees at each site. Wooden path was present in the three bog sites, and the sampling started from the drier edges toward the wetter interior of the bogs in equidistant intervals. In Kakerdaja, the trail passed a drier ‘bog island’ (Fig. 1 for the mapped paths of foliage sampling). After collection, the needles were sealed in closed vials and stored at −80 °C until DNA extraction. The number of trees studied here from each population constitutes representative to characterize the genetic variation at the level of polymorphic loci with a great confidence (Nei 1978; Nybom 2004).
Molecular analysis
The frozen leaf samples were pulverized with a mortar and a pestle in liquid nitrogen, and genomic DNA was extracted from 100 mg of fresh needles using the DNasy Plant Mini Kit (Qiagen, Hilden, Germany). Initially, 12 nuclear (Auckland et al. 2002; Elsik et al. 2000; Soranzo et al. 1998; Zhou et al. 2002) and 7 chloroplastic (Vendramin et al. 1996) single sequence repeat (SSR, microsatellite) markers were tested. Ultimately, samples were genotyped using five nuclear microsatellite markers (nSSR, Table 1): SPAC11.8, SPAC12.5 (Soranzo et al. 1998), TX2123 (Elsik et al. 2000), TX3116 (Auckland et al. 2002) and TX4001 (Zhou et al. 2002), and five chloroplastic microsatellites (cpSSR, Table 2): Pt15169, Pt30204, Pt36480, Pt71936 and Pt87268 (Vendramin et al. 1996). Previous studies on the genetic variation of Estonian P. sylvestris populations (Kuznetsova and Mandre 2006; Maaten and Kurm 2007) have used much fewer markers than used here.
PCR amplifications were performed in a AB 2720 thermal cycler (Applied Biosystems, MA, USA). nSSR amplification was carried out in a total volume of 15 µl, containing 2.5 mM MgCl2, 1 × reaction buffer, 1 % DMSO (dimethyl sulfoxide), 200 mM of each dNTP, 2.5 pmol Cy5- or IRD700-labeled forward primer, 5 pmol reverse primer, 1 U Taq polymerase (Bio-Rad, CA, USA) and 20 ng of genomic DNA. The PCR profile for SPAC11.8, Tx4001 and Tx3116 was as follows: initial denaturation at 94 °C for 5 min, followed by 5 cycles of 30 s at 94 °C, 30 s at 60 °C and 30 s at 72 °C (decreasing the annealing temperature by 1 °C per cycle), followed by 30 cycles of 30 s at 94 °C, 30 s at 55 °C and 30 s at 72 °C, and a final extension step of 10 min at 72 °C. A similar touch-down profile was used for SPAC12.5 and Tx2123, but with annealing temperatures of 60–50 °C and 10 touchdown cycles. cpSSR amplifications were performed in 15 µl reactions using the same reagents as for nSSR markers. cpSSR amplification was performed with the following profile: 5 min denaturation at 95 °C, 1 min denaturation at 94 °C, 1 min annealing at 55 °C, 1 min extension at 72 °C with final extension step at 72 °C for 8 min for 25 cycles (Vendramin et al. 1996).
0.4 μl fluorescently labeled PCR product and the internal size standard, 0.4 μl of ROX-1000 (MapMarker, BioVenture Inc., TN, USA), were added to 10 μl of Hi-Di formamide (Applied Biosystems). The samples were denatured for 5 min at 95 °C, placed on ice, and then separated by capillary electrophoresis using a Prism 3130 Genetic Analyzer (Applied Biosystems).
Data analysis
For nSSR data, diploid genotypes for each individual were scored and following parameters were computed as averages over loci using GenAlEx version 6 (Peakall and Smouse 2006) according to Nei (1978): mean number of alleles per locus (N a), number of unique alleles (N p), effective number of alleles (N e), average observed heterozygosity (H o), average expected heterozygosity (H e) and the fixation index (inbreeding coefficient, F IS = (H e − H o)/H e).
Polymorphisms of cpSSRs were scored and used to define chloroplast haplotypes according to different combinations of length variants. Observed number of haplotypes (N h,a), number of private haplotypes (N h,p), unbiased effective number of haplotypes (N h,e) (Nielsen et al. 2003), unbiased haplotype diversity (uH) (Nei 1978) as well as the mean genetic distance among individual haplotypes following a stepwise mutation model (SMM) approach (D 2sh ) as described in Vendramin et al. (1998) were calculated using HAPLOTYPE ANALYSIS version 1.05 (Eliades and Eliades 2009).
Spatial genetic structure (SGS) was characterized using the analysis of kinship coefficients (F ij) between all pairs of individuals sampled across the trails through the bogs (Fig. 1) estimated as in Loiselle et al. (1995). Average pairwise F ij estimates were plotted against the pairwise spatial distances for individuals of each populations (Fig. 6a). All the analyses were carried out using SPAGeDi version 1.2 computer program (Hardy and Vekemans 2002).
The extent of population subdivision was evaluated by calculating Wright’s F ST (the inbreeding coefficients within populations relative to total) (Wright 1951) and the related R ST (Slatkin 1995). R ST is based on a stepwise mutation model and takes explicit account of likely mutational processes in microsatellite loci. These two indices were estimated across populations and between pairs of populations according to Weir and Cockerham (1984) and Michalakis and Excoffier (1996) by SPAGEDI version 1.2. The migration coefficient (N m) that characterizes historical gene flow was further calculated as [(1/F ST) − 1]/4).
F ST may be overestimated in the case of null alleles (Chapuis and Estoup 2007). We controlled for the potential effect of null alleles on genetic differentiation by calculating F ST(FREENA) values using the excluding null allele (ENA) method by Chapuis and Estoup (2007) in FREENA.
A principal coordinate analysis (PCoA) based on a pairwise distance matrix with data standardization was conducted by GenAlEx version 6 (Peakall and Smouse 2006). To test for a pattern of isolation by distance (Rousset 1997), a Mantel test with 1000 random permutations was performed with the matrix of pairwise genetic differentiation between populations, using F ST/(1 − F ST), and a matrix of geographic distance using GenAlEx v6 (Peakall and Smouse 2006). The rate of gene flow was estimated indirectly from the proportion of total diversity found among populations (Wright 1931, 1951). Based on pairwise genetic distance (Nei 1987), a neighbor joining dendrogram of the three studied P. sylvestris populations was constructed. Node consistency was evaluated through 1000 bootstrap replicates using MEGA 5 software (Tamura et al. 2011).
To gain further insight into the population structure, this analysis was repeated twice with differing grouping of samples. First, each population was divided according to sample location along the trails (Fig. 1) among two sub-parts, drier (KakerdajaD, LaevaD, ParikaD) versus wetter (KakerdajaW, LaevaW, ParikaW). Second, each population was divided among 6 groups, each including 10 genotypes along the trails from drier to wetter parts.
Cyto-nuclear linkage disequilibrium (LD) was estimated between each nSSR and cpSSR. The analytical framework of Basten and Asmussen (1997) was applied using the program CNDm to estimate a standardized estimate of an allelic D′ between each nSSR and cpSSR locus. CNDm applies a Monte Carlo approach to approximate a Fisher’s exact test for RxC contingency tables and tests for significant deviations from the null hypothesis of no allelic association (Basten and Asmussen 1997). We followed the approach of Latta et al. (2001) and Fields et al. (2014), wherein all nSSR and cpSSR were treated as bi-allelic by pooling all alleles other than the most common allele in the full sample into a single composite allele. Briefly, this approach is preferable for the present analysis, providing a single value for each locus–locus pair, while also generating the most intermediate allele frequencies, and maximizing the bounds on D′. Importantly, given finite sample sizes and the resultant large class of rare, private alleles generated by high mutation rates in microsatellite loci, which will rarely be in linkage equilibrium, the applied binning procedure should provide statistical robustness. In addition to quantifying cyto-nuclear LD across the nuclear and chloroplast genomes, we applied tests to determine evidence for significant LD within each genome. Specifically, we determined the statistical significance of nuclear LD between pairs of loci, under the null hypothesis of random allelic assortment, using a Monte Carlo approximation of Fisher’s exact test implemented in the software Arlequin (Excoffier and Lischer 2010). Arlequin uses a Markov chain extension of Fisher’s exact test for RxC contingency tables (Li and Merila 2010; Slatkin 1994). A total of 100,000 alternative tables were explored by the Markov chain (Li and Merila 2010; Slate and Pemberton 2007). We determined the statistical significance of chloroplast LD using LIAN (Haubold and Hudson 2000). Specifically, LIAN tests for independent assortment of alleles by computing the number of loci at which an each pair of haplotypes differs, generating a mismatch distribution V D, which can then be compared to the variance expected for linkage equilibrium V e. The null hypothesis of V D = V e is then tested using Monte Carlo simulation (Haubold and Hudson 2000).
Results
Overall allele frequencies and genetic diversity according to nuclear SSR (nSSR) markers
All five nSSR loci were highly polymorphic with the number of detected alleles per locus ranging from 5 (Tx2123) to 43 (SPAC12.5). The total number of alleles scored in 180 individuals over all loci was 138. High polymorphism was found within all three populations; on average 18.4 alleles per locus were found across the sites (Table 3), and the average expected heterozygosity over all investigated loci (H e) was also high (H e = 0.787, Table 3). The average observed heterozygosity (H o = 0.947) was significantly higher than the expected heterozygosity, resulting in a high negative mean inbreeding coefficient (F IS = −0.239, Table 3).
These statistics were similar for all three populations, i.e., all of them exhibited high H e and H o values and high negative F IS, indicating excess of heterozygotes and significant and negative departure from Hardy–Weinberg equilibrium (Table 3). Values of mean H o, H e and F IS were not significantly different among populations (P > 0.05).
Overall genetic diversity according to chloroplast SSR (cpSSR) markers
All five analyzed cpSSRs were polymorphic, giving a total of 32 size variants (5–8 per locus) (Tables 2, 4), although there were important variations in allele frequency for different loci (Fig. 2). The distribution of size variants for each region was unimodal (Fig. 3), with variants differing by 1 bp from each other, consistent with a stepwise mutation model (SMM). Two exceptions were found: one in the Pt71936 region where a 3-bp gap was observed and the other in the Pt36480 region where a 9-bp gap was detected. Different size variants resulted in altogether 86 different haplotypes among the 180 individuals surveyed from three populations (Fig. 1). Many of these haplotypes (62 %) were detected only once, 12 % were detected in two individuals, and the rest were found in 3–11 trees. A high proportion (67 %) of haplotypes found in this study were unique to a particular population, but most of them were observed only in one individual from a single population. None of the detected haplotypes was common to all populations, and the most frequent haplotype was found in three populations with an overall frequency of 6 %.


Frequency distribution of size variants of five Pinus sylvestris chloroplast microsatellite loci in each population (n = 60 for each population). Table 2 provides details of all the five markers used, and Fig. 2 provides the frequency distribution of all individual markers through all the data pooled
As the result of low frequency of specific size variants, the mean number of haplotypes across populations was very high (N h,a = 40, range 36–43 for different populations, Table 4). Analogously, the effective number of haplotypes 23.7–36.7 in different populations was also high (Table 4). The average haplotypic diversity or unbiased effective haplotypes (uH) was 0.981 (Table 4), further indicating a very high within-population diversity. The average mean distance among individuals within populations according to SMM (D 2sh ) had a moderately high value of 4.3, whereas the lowest within-population mean pairwise distance was observed for Laeva population that also showed the lowest haplotype diversity (Table 4).
Genetic differentiation among three bogs
The nSSR and cpSSR datasets were significantly, although not very strongly, correlated (r = 0.498, P < 0.01; Mantel test with pairwise genetic distance matrices generated by the different marker systems), and thus, both marker systems provided broadly similar conclusions about the genetic variability within and among populations with several differences. A low level of genetic differentiation among the three populations was observed according to both nSSR and cpSSR markers. According to nSSR, the coefficient of genetic differentiation among populations according to the infinite allele mutation (IAM model; F ST) was 0.007; after correction using FREENA, the overall F ST was only slightly lower (F ST(FREENA)) = 0.006), but the stepwise mutation model (SMM) that more strongly weights the differences between allele lengths suggested a somewhat higher, but still very low, differentiation among the populations (R ST = 0.022). The high value of the migration coefficient (N m = 35.6) indicated a high gene flow between the studied populations. Analogously, cpSSR markers indicated low but significant difference between populations (values of F ST of 0.015 and R ST of 0.023).
Analysis of molecular variance, AMOVA, using nuclear SSRs further revealed that the variation among populations accounted for about 1 % of the total variance, and within populations for 99 % of the total variance (Table 5). Chloroplast SSRs confirmed the presence of a similar degree of molecular variation among populations (based on AMOVA, differences among populations accounted for about 1 % of the total cpSSR variance) (Table 5).
To gain an insight into the genetic relationships among populations, principal coordinate analysis (PCoA) was carried out using both genetic distance datasets (Fig. 4a). The first principal coordinate explained 58 % of the total variation for both nSSR and cpSSR markers. This first principal coordinate clearly separated the Kakerdaja from the other more closely located populations (Laeva and Parika populations). A clear separation of these populations was revealed by high bootstrap values in the neighbor joining dendrogram calculated from genetic distance matrix (Fig. 4).

Relationships between the ordination scores of the principal coordinate analysis (PCoA, Nei 1978) based on both nSSR and cpSSR markers, and corresponding dendrograms (above each PCoA panel) showing the genetic relatedness of Pinus sylvestris populations. The analysis was conducted with all individuals sampled in the given site pooled (n = 60 for each site, left panels) and with equal sub-populations from drier and wetter parts of the site (n = 30 for each sub-population, Fig. 1 for the site codes and sampling strategy)
Within- and across-population genetic variability as driven by geographic distance and site water availability
Three types of analyses were carried out to gain an insight into within- and across-population differentiation in space and in dependence on within-site water availability by dividing the populations within the sites among two sub-populations, drier (KakerdajaD, LaevaD, ParikaD) and wetter (KakerdajaW, LaevaW, ParikaW). Second, simultaneous effects of smaller- and larger-scale spatial distances were analyzed for dry and wet sub-populations using the Mantel test. Finally, the populations within the given site were divided into six sub-populations from drier to wetter habitats along the trails, and variation among these sub-populations was analyzed. This analysis was complemented by the kinship analysis that allows for evaluation of the genetic variation over short spatial distances along the sampling trails (Fig. 1).
Compared with the analyses considering each site as a single population (Tables 3, 4), consideration of drier and wetter sub-populations within each site separately (Table 6) did not result in qualitatively different conclusions on the overall genetic variability. This analysis based on nSSR markers indicated that the overall genetic variability, degree of heterozygosity and inbreeding coefficient were similar among the sub-populations (Table 6). Only for LaevaD (drier part of Laeva population), the Shannon’s information index was less than in the other sub-populations, and also F IS in this population was more negative than in the others, indicating even higher outbreeding (Table 6a). Haplotype diversity and distribution of haplotypes in six bog sub-populations was almost the same (Table 6b).
According to phylogenetic analyses (neighbor joining) and PCoA analyses based on nSSR markers, LaevaD was indeed separated from other populations, and there was also certain evidence of wetter sub-populations, LaevaW and ParikaW, and drier sub-populations, LaevaD and ParikaD, to cluster together (Fig. 4b). However, in the case of cpSSR markers, phylogenetic analyses rather suggested closer relatedness of wet and dry pairs of Laeva and Parika sub-populations, and PCoA suggested only higher relatedness of wetter sub-populations, KakerdajaW and ParikaW.
At a larger scale, Mantel test according to nSSR markers did not show a significant correlation between geographic and genetic distance of sub-populations (Fig. 5a), whereas there was a significant correlation between geographic and genetic distance according to cpSSR markers (Fig. 5b). At a smaller scale, the kinship analysis demonstrated that there was a significant, although weak, trend of decreasing kinship with increasing spatial distance between individuals of Laeva population (Fig. 6a). Kakerdaja and Parika populations did not show a significant spatial trend in population genetic structure (Fig. 6a). Complementary to this analysis, dividing each population among six sub-populations, from the drier edge toward the wetter center, confirmed a certain spatial, wet–dry, gradient in sub-populations in Laeva site (Fig. 6c), but no such gradient was evident for Kakerdaja (Fig. 6b) and Parika site (Fig. 6d).

Relationships among the geographic and genetic distance according to nSSR and cpSSR markers of Pinus sylvestris populations separated according to water availability within the three sites. Diamond symbols represent the comparisons LaevaD versus ParikaD, LaevaD versus KakerdajaD and ParikaD versus KakerdajaD; circles the comparisons LaevaD versus LaevaW, ParikaD versus ParikaW and KakerdajaD versus KakerdajaW, triangles the comparisons LaevaD versus ParikaW, LaevaD versus KakerdajaW, ParikaD versus LaevaW, ParikaD versus KakerdajaW, KakerdajaD versus LaevaW and KakerdajaD versus ParikaW, and squares the comparisons LaevaW versus ParikaW, LaevaW versus KakerdajaW and ParikaW versus KakerdajaW (Fig. 1 for location of sample sites and definition of site codes)

Correlograms of kinship coefficients among P. sylvestris individuals sampled along the trails from dry to wet habitats in three sites (a), and relationships among ordination scores of principal component analyses (PCoA) for sub-populations positioned from dry to wet parts within each of the three sites (b–d). Figure 1 demonstrates the sampling strategy and site location. In b–d, the population sampled at each site (n = 60) was divided among 10 equal sub-populations from drier (driest = 1) toward wetter (wettest = 6) habitats, and PCoA was conducted according to Nei (1978) using unbiased genetic distances based on nSSRs (b–d)
Evidence for linkage disequilibrium across genomes
Table 7 presents the observed distribution of D′ between each of the nSSR and cpSSR loci. Of the genotyped loci, only Pt15169 shows evidence of significant cyto-nuclear LD, for all nSSR. Of the four remaining cpSSR loci, the average D′ was 0.13137, with a range of 0.093–0.17. On average, each nSSR showed evidence for significant LD to 2.8 other nSSR loci. For the cpSSR loci, we found no evidence of significant LD (P = 0.49), with an observed V D = 1.0725 and V e = 1.0727 (Monte Carlo resampled variance in V D = 0.0023). When Pt15169 was removed and the LIAN analysis rerun, V D = 0.9129 and V e = 0.8828 (Monte Carlo resampled variance in V D = 0.0018), with a P = 0.234.
Discussion
Genetic variation of Pinus sylvestris at its nutrient availability limit vis-à-vis overall variation in species range
Pinus sylvestris, a major fast-growing economically important forest tree species in fertile sites, is also extremely successful in colonizing sites with poor nutrient availability where it is typically the only trees species or a co-dominant species. The aim of this study was to assess the level of genetic variation of P. sylvestris at its lower nutrient limit in the bogs. The nuclear microsatellite markers used in this study detected high levels of genetic variation, with an average expected heterozygosity of 0.79 and observed heterozygosity of 0.95 (Table 3). This high genetic diversity matches high phenotypic variability observed for terpenoid contents and composition in these sites (Kännaste et al. 2013).
The high genetic diversity observed in our study is only partly surprising since a very large genetic variability has been recognized for a long time as a peculiar characteristic of woody plants (Hamrick et al. 1992). This high diversity in trees likely reflects their specific life history characteristics such as late reproduction, high longevity and presence of multiple overlapping generations in woody plants. These life history characteristics can buffer against the decrease in genetic variation due to population contractions (Austerlitz et al. 2000). Furthermore, species such as P. sylvestris, which do not have strong habitat specificity and are almost continually distributed, are expected to have greater within-population diversity than those with strong habitat preference and fragmented distribution. High genetic variability of P. sylvestris observed here is comparable with previous observations in other European sites: H e = 0.81 for three microsatellite loci (Scalfi et al. 2009) and H e = 0.847 for nine microsatellite loci (Belletti et al. 2012) in Italy; H e = 0.853 for three microsatellite loci in Poland (Nowakowska et al. 2014); and H e = 0.923 for three microsatellite loci in isolated Iberian mountain populations (Robledo-Arnuncio et al. 2005).
Considering the significant variation observed for quantitative phenotypic traits such as terpenoid content and composition (Kännaste et al. 2013), the Estonian P. sylvestris populations do not fit the expectation that increasing environmental heterogeneity in combination with local adaptations leads to reduced genetic diversity within populations (Hertel and Schneck 1999; Oleksyn et al. 1998; Wegmann et al. 2006). High genetic diversity within populations together with high heritable phenotypic variation observed at several quantitative traits (Perks and McKay 1997) suggests that these three populations studied (Laeva, Parika and Kakerdaja) have a high potential to produce diverse adaptive responses to environmental variation.
Deviations from the expected Hardy–Weinberg proportions were observed for all three populations. In general, the observed heterozygosity was higher than expected resulting in negative F IS values, indicating that the studied populations of P. sylvestris were highly outbred (F IS = −0.239; Table 3). Important departures from the Hardy–Weinberg expectations with an excess of heterozygotes were also found in another study of the Estonian P. sylvestris populations (Maaten and Kurm 2007). The non-conformity with the Hardy–Weinberg equilibrium can result from both increased randomness in mating (primarily due to pollen source) leading to enhanced gene flow as well as from spatial (sub-)population structure (Wahlund effect). The most commonly reported causes of negative values of the inbreeding coefficient are effective gene flow among populations. With regard to phenology, it is well known that the flowering of P. sylvestris overlaps over large climatic gradients, even over 500–1000 km latitudinal clines with strong temperature differences, but also that earlier pollen is expected to have a competitive advantage (Lindgren et al. 1995; Pessi and Pulkkinen 1994; Pulkkinen and Rantiolehtimaki 1995; Sarvas 1962). This advantage could have favored effective pollen dispersal from southern populations to northern populations, which presumably starts to shed pollen later.
High genetic diversity estimate based on haplotype frequencies (0.981) was observed in Estonian P. sylvestris populations using five polymorphic chloroplast microsatellites (Table 4). The observed value is similar to the values previously found in P. sylvestris in a large survey of European populations: 0.99 in Scotland (Provan et al. 1998), 0.92 in the northern Apennines, Italy (Scalfi et al. 2009), and 0.978 in isolated Iberian mountain populations (Robledo-Arnuncio and Gil 2005). In Scotland, high diversity of mtDNA was linked to gene flow from a Siberian refugee population (Sinclair et al. 1998). It seems that the biology of P. sylvestris has ensured the maintenance of its neutral genetic diversity throughout glacial periods and across its range despite having undergone probably different demographic processes in different areas.
Genetic variability among populations
In general, large intrapopulation but small interpopulation variation is found in P. sylvestris (Kuznetsova and Mandre 2006; Maaten and Kurm 2007), indicative of extensive gene flow between populations. In fact, comparisons of genetic structure of reproducing forest stands and the produced offspring have demonstrated significant changes in population genetic structures during reproduction in this species (Hattemer et al. 1993; Müller-Starck and Ziehe 1991; Ziehe et al. 1999), suggesting that important changes in population structure can occur already within a few generations. Nevertheless, the overall level of genetic diversities arising from the differentiation between populations found in this study using nSSRs (F ST = 0.007) is lower than that previously observed in the same species in other European countries (Müller-Starck et al. 1992). In the Scandinavian region, for instance, values of F ST = 0.02 were found in populations of P. sylvestris studied with different markers (Karhu et al. 1996). Allozymes gave a F ST = 0.03 between Sweden and Siberian populations (Wang et al. 1991) and a FST = 0.021–0.046 among many European stands (Prus-Glowacki et al. 1993, 2003). Analogously, based on cpSSR markers, the variation among populations (FST = 0.015) in our study is lower than the value among northern P. sylvestris populations in Scotland (Provan et al. 1998).
As suggested above, the most plausible explanation for high genetic diversity and low genetic differentiation in these P. sylvestris populations is high gene flow between populations. In particular, wind pollination has a homogenizing effect on the distribution of genetic variation. In fact, it has been suggested that pollen-mediated gene flow over several hundred kilometers in a south–north direction is possible for Finnish P. sylvestris (Varis et al. 2009). Fertile pollen grains were detected in the air when local female strobili were receptive, but male strobili were immature (Varis et al. 2009). The occurrence of airborne pollen in northern and northernmost sites overlapped with male flowering in more southern locations. The amount of pollen flow from outside of the study site has been 21–76 % in seed orchards in Sweden and Finland (El-Kassaby et al. 1989; Harju and Muona 1989; Pakkanen et al. 1991; Wang et al. 1991). In this regard, greater genetic variability in nutrient-limited sites might be directly linked to the open sparsely located canopies and highly turbulent wind conditions in these sites. Such conditions facilitate access of low-concentration distant pollen to the female strobili. The situation is much different in closed productive stands where turbulent conditions rarely occur.
Sub-population variability
As P. sylvestris has a very high phenotypic variability over small spatial scales (s. Introduction), it is tempting to assume that part of this small-scale variability is due to genotypic differentiation along environmental gradients, and as such, reflects adaptability to site conditions. Despite a strong gene flow, Mantel test according to cpSSR markers did demonstrate that genetic distance between populations increased with their geographic distance (Fig. 5b), indicating that there was a certain evidence of isolation by distance (IBD) of haplotypes even over these relatively short distances. No such pattern across populations was found for nSSR markers (Fig. 5a). This lack of spatial correlation over a larger scale reflected the presence of small-scale spatial population structure, from dry toward wetter parts of the bog, in one out of the three studied sites (cf. Figs. 5a, 6a, d). Although weak, the observed spatial correlation suggests that there can be a certain adaptive selection to habitat conditions at least in some conditions. Such in situ genetic adaptations to environmental conditions have been observed in some species (Jump et al. 2006; Peñuelas et al. 2007), but not in others (Nishimura and Setoguchi 2011). Currently, we cannot explain the occurrence of within-population spatial structure in only one site. We argue that site-specific conditions (presence of surrounding taller vegetation) as well as site history could have played a role.
Linkage disequilibrium across chloroplast and nuclear loci
Recent research has suggested that polymorphism in organelles may be important for adaptation in plants (Bock et al. 2014). Previous research has also shown that neutral demographic processes such as extinction/recolonization events can generate significant cyto-nuclear LD. For example, Fields et al. (2014) showed in a metapopulation of Silene latifolia that overall levels of cyto-nuclear LD were stable over approximately seven generations of the species due to repeated founder effects consistently recreating cyto-nuclear LD even as gene flow erodes it. In the present study, we observed significant cyto-nuclear LD between one of the cpSSR loci to all nSSR loci. This significant association may indicate that this particular cpSSR locus may be linked to a locus under selection or that this locus belongs to an area of the chloroplast genome experiencing different mutation rates, which has been observed in other plant species (Sloan et al. 2008). The lack of significant LD within the sampled cpSSR loci, even though there was significant LD between nSSR loci, is likely the result of high mutation rates which results in homoplasy across the species range (Desplanque et al. 2000; Luo et al. 1995; Strauss et al. 1993).
Conclusions
According to the present results, high and similar levels of genetic diversity and little (but significant) genetic differentiation were detected for both chloroplast and nuclear genome. Despite low genetic differentiation by both markers, clear geographic patterns in the distribution of genetic variation across the populations were found for chloroplastic markers. Clustering of bog populations suggested that isolation by distance might be a possible mechanism shaping the present distribution of genetic variability among the populations. The results further underscore the importance of pollen gene flow in homogenizing populations on this geographic scale. Overall, the results of this study suggest that nSSRs and cpSSRs have high resolution for detecting neutral molecular variation on small scales; and that the biology of P. sylvestris provides for the long-term maintenance of high within population and low among-population diversity in neutral DNA regions. Existence of the small-scale spatial structure of P. sylvestris in one site further suggests that there could be adaptive selection operating under some conditions, and its importance seems to be generally low, suggesting that most of the phenotypic variability observed in this species along small-scale environmental gradients is plastic rather than genetic.
Data archiving statement
Primer sequences of five nuclear microsatellites SPAC11.8, SPAC12.5, TX2123, TX3116 and TX4001 which are previously developed by Soranzo et al. (1998), Elsik et al. (2000), Auckland et al. (2002) and Zhou et al. (2002) respectively, and five chloroplastic microsatellites Pt15169, Pt30204, Pt36480, Pt71936 and Pt87268 that are developed by Vendramin et al. (1996), were used in this study and have been deposited at GenBank: accession number AJ223770 for SPAC11.8, AJ223772 for SPAC12.5, AF143960 for TX2123, AY304034 for PtTX3116 and AF286619 for TX4001.
References
Auckland LD, Bui T, Zhou Y, Shepherd M, Williams CG (2002) Conifer microsatellite handbook. Corporate Press, Raleigh
Austerlitz F, Mariette S, Machon N, Gouyon PH, Godelle B (2000) Effects of colonization processes on genetic diversity: differences between annual plants and tree species. Genetics 154:1309–1321
Basten CJ, Asmussen MA (1997) The exact test for cytonuclear disequilibria. Genetics 146:1165–1171
Belletti P, Ferrazzini D, Piotti A, Monteleone I, Ducci F (2012) Genetic variation and divergence in Scots pine (Pinus sylvestris L.) within its natural range in Italy. Eur J For Res 131:1127–1138. doi:10.1007/s10342-011-0584-3
Bennett KD, Tzedakis PC, Willis KJ (1991) Quaternary refugia of north European trees. J Biogeogr 18:103–115
Bock DG, Andrew RL, Rieseberg LH (2014) On the adaptive value of cytoplasmic genomes in plants. Mol Ecol 23:4899–4911. doi:10.1111/mec.12920
Bouzat JL (2001) The population genetic structure of the Greater Rhea (Rhea americana) in an agricultural landscape. Biol Conserv 99:277–284. doi:10.1016/S0006-3207(00)00193-2
Brown AHD (1978) Isozymes, plant population genetic structure and genetic conservation. Theor Appl Genet 52:145–157
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. doi:10.1093/molbev/msl191
Cheddadi R et al (2006) Imprints of glacial refugia in the modern genetic diversity of Pinus sylvestris. Glob Ecol Biogeogr 15:271–282
Critchfield WB, Little EL (1966) Geographic distributions of the pines of the world. U.S.D.A. Serv Misc Public 991, Washington, DC
Desplanque B, Viard F, Bernard J, Forcioli D, Saumitou-Laprade P, Cuguen J, van Dijk H (2000) The linkage disequilibrium between chloroplast DNA and mitochondrial DNA haplotypes in Beta vulgaris ssp maritima (L.): the usefulness of both genomes for population genetic studies. Mol Ecol 9:141–154. doi:10.1046/j.1365-294x.2000.00843.x
Dzialuk A, Muchewicz E, Boratynski A, Montserrat JM, Boratynska K, Burczyk J (2009) Genetic variation of Pinus uncinata (Pinaceae) in the Pyrenees determined with cpSSR markers. Plant Syst Evol 277:197–205. doi:10.1007/s00606-008-0123-y
Eliades NG, Eliades DG (2009) HAPLOTYPE ANALYSIS: software for analysis of haplotypes data. Distributed by the authors. Forest genetics and forest tree breeding, Georg-Augst University Goettingen, Germany
El-Kassaby YA, Rudin D, Yazdani R (1989) Levels of outcrossing and contamination in two Pinus sylvestris L. seed orchards in northern Sweden. Scand J For Res 4:41–49
Elsik CG, Minihan VT, Hall SE, Scarpa AM, Williams CG (2000) Low-copy microsatellite markers for Pinus taeda L. Genome 43:550–555. doi:10.1139/gen-43-3-550
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Fields PD, McCauley DE, McAssey EV, Taylor DR (2014) Patterns of cyto-nuclear linkage disequilibrium in Silene latifolia: genomic heterogeneity and temporal stability. Heredity 112:99–104. doi:10.1038/hdy.2013.79
Finér L (1992) Nutrient concentrations in Pinus sylvestris growing on an ombrotrophic pine bog, and the effects of PK and NPK fertilization. Scand J For Res 7:205–218
Grassi F et al (2003) Genetic isolation and diffusion of wild grapevine Italian and Spanish populations as estimated by nuclear and chloroplast SSR analysis. Plant Biol 5:608–614. doi:10.1055/s-2003-44689
Gregorius HR (1994) Strategies for conserving genetic diversity in the face of global change. In: Boyle TJB, Boyle CEB (eds) Temperate ecosystems and global change. Springer, Berlin, pp 157–182
Gunnarsson U, Rydin H (1998) Demography and recruitment of Scots pine on raised bogs in eastern Sweden and relationships to microhabitat differentiation. Wetlands 18:133–141
Hamrick JL, Godt MJW, Shermanbroyles SL (1992) Factors influencing levels of genetic diversity in woody plant-species. In: Adams WT, Strauss SH, Copes DL, Griffin AR (eds) Population genetics of forest trees, vol 42, pp 95–124
Hansen AJ, Knight RL, Marzluff JM, Powell S, Brown K, Gude PH, Jones A (2005) Effects of exurban development on biodiversity: patterns, mechanisms, and research needs. Ecol Appl 15:1893–1905. doi:10.1890/05-5221
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Harju A, Muona O (1989) Background pollination in Pinus sylvestris seed orchards. Scand J For Res 4:513–520
Hattemer HH (1987) Are the EEC directives on forest reproductive material genetically adequate? Silvae Genet 36:94–102
Hattemer HH, Starke R, Ziehe M (1993) Changes of genetic structures in beech populations. In: Muhs H-J, VON Wühlisch G (eds) The scientific basis for the evaluation of the genetic resources of beech. Comm. Europ. Communities, Working Doc. F.II.3-SJ/0009, pp. 233–248
Haubold B, Hudson RR (2000) LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics 16:847–848. doi:10.1093/bioinformatics/16.9.847
Hertel H, Schneck V (1999) Genetic and phenotypical variation of scots pine (Pinus sylvestris L.) populations due to seed origin and environmental conditions at experimental sites. For Genet 6:65–72
Holliday JA, Suren H, Aitken SN (2011) Divergent selection and heterogeneous migration rates across the range of Sitka spruce (Picea sitchensis). Proc R Soc Lond B. doi:10.1098/rspb.2011.1805
Huntley B, Birks HJB (1983) An Atlas of past and present pollen maps for Europe: 0–13,000 years ago. Cambridge University Press, Cambridge
Jump AS, Hunt JM, Martínez-Izquierdo JA, Peñuelas J (2006) Natural selection and climate change: temperature-linked spatial and temporal trends in gene frequency in Fagus sylvatica. Mol Ecol 15:3469–3480
Kadereit JW, Griebeler EM, Comes HP (2004) Quaternary diversification in European alpine plants: pattern and process. Philos Trans R Soc Lond B-Biol Sci 359:265–274. doi:10.1098/rstb.2003.1389
Kännaste A, Copolovici L, Pazouki L, Suhhorutsenko M, Niinemets Ü (2013) Highly variable chemical signatures over short spatial distances among Scots pine (Pinus sylvestris) populations. Tree Physiol 33:374–387. doi:10.1093/treephys/tpt013
Karhu A, Hurme P, Karjalainen M, Karvonen P, Karkkainen K, Neale D, Savolainen O (1996) Do molecular markers reflect patterns of differentiation in adaptive traits of conifers? Theor Appl Genet 93:215–221. doi:10.1007/bf00225748
Karp A, Isaac PG, Ingram DS (1998) Molecular tools for screening biodiversity: plants and animals. Chapman & Hall, London
Kuznetsova T, Mandre M (2006) Chemical and morphological indication of the state of lodgepole pine and Scots pine in restored oil shale opencast mining areas in Estonia. Oil Shale 23:366–384
Läänelaid A (1981) Pines on bogs as indicators of fires in the past. In: Laasimer L, Trass H, Kask M (eds) Anthropogenous changes in the plant cover of Estonia. Academy of Sciences of the Estonian S.S.R. Institute of Zoology and Botany—“Man and Biosphere”. Republican Committee of the Estonian S.S.R., Tartu, pp 91–94
Latta RG, Linhart YB, Mitton JB (2001) Cytonuclear disequilibrium and genetic drift in a natural population of ponderosa pine. Genetics 158:843–850
Li MH, Merila J (2010) Extensive linkage disequilibrium in a wild bird population. Heredity 104:600–610. doi:10.1038/hdy.2009.150
Lindgren D, Paule L, Xihuan S, Yadzani R, Segerström U, Tallin JE, Lejdebro ML (1995) Can viable pollen carry Scots pine genes over long distances? Grana 34:64–69
Loiselle BA, Sork VL, Nason J, Graham CAJB (1995) Spatial genetic structure of a tropical understorey shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Luo H, Van Coppenolle B, Seguin M, Boutry M (1995) Mitochondrial DNA polymorphism and phylogenetic relationships in Hevea brasiliensis. Mol Breeding 1:51–63. doi:10.1007/bf01682089
Maaten T, Kurm M (2007) Analysis of genetic variation of Scots pine populations in Estonia based on DNA microsatellite markers. For Stud 46:45–56
Michalakis Y, Excoffier L (1996) A genetic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142:1061–1064
Müller-Starck G, Ziehe M (1991) Genetic variation in populations of Fagus sylvatica L., Quercus robur L. and Q. petraea Liebl. in Germany. In: Müller-Starck G, Ziehe M (eds) Genetic variation in european populations of forest trees. Sauerländer’s Verlag, Frankfurt-am Main, pp 125–140
Müller-Starck G, Baradat PH, Bergmann F (1992) Genetic variation in European tree species. New Forest 6:23–47
Naydenov KD, Tremblay FM, Alexandrov A, Fenton NJ (2005) Structure of Pinus sylvestris L. populations in Bulgaria revealed by chloroplast microsatellites and terpenes analysis: provenance tests. Biochem Syst Ecol 33:1226–1245
Naydenov K, Senneville S, Beaulieu J, Tremblay F, Bousquet J (2007) Glacial vicariance in Eurasia: mitochondrial DNA evidence from Scots pine for a complex heritage involving genetically distinct refugia at mid-northern latitudes and in Asia Minor. BMC Evol Biol 7:233
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Newton AC, Allnutt TR, Gillies ACM, Lowe AJ, Ennos RA (1999) Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol 14:140–145. doi:10.1016/s0169-5347(98)01555-9
Nielsen R, Tarpy DR, Reeve HK (2003) Estimating effective paternity number in social insects and the effective number of alleles in a population. Mol Ecol 12:3157–3164. doi:10.1046/j.1365-294X.2003.01994.x
Niinemets Ü, Ellsworth DS, Lukjanova A, Tobias M (2001) Site fertility and the morphological and photosynthetic acclimation of Pinus sylvestris needles to light. Tree Physiol 21:1231–1244
Niinemets Ü, Cescatti A, Lukjanova A, Tobias M, Truus L (2002) Modification of light-acclimation of Pinus sylvestris shoot architecture by site fertility. Agric For Meteorol 111:121–140
Nishimura M, Setoguchi H (2011) Homogeneous genetic structure and variation in tree architecture of Larix kaempferi along altitudinal gradients on Mt. Fuji. J Plant Res 124:253–263
Nowakowska JA, Zachara T, Konecka A (2014) Genetic variability of Scots pine (Pinus sylvestris L.) and Norway spruce (Picea abies L. Karst.) natural regeneration compared with their maternal stands. For Res Pap 75(1):47–54
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155. doi:10.1111/j.1365-294X.2004.02141.x
Oleksyn J, Tjoelker MG, Reich PB (1998) Adaptation to changing environment in Scots pine populations across a latitudinal gradient. Silva Fennica 32:129–140
Pakkanen A, Pulkkinen P, Vakkari P (1991) Pollen contamination in the years 1988–1989 in some old Scots pine seed orchards of northern Finnish origin Reports from the Foundation for Forest Tree Breeding 3 Helsinki, Finland, pp 3–8
Pärt T, Villard MA (2006) Ecological traps: avian and human perspectives. J Ornithol 147:23
Pautasso M (2009) Geographical genetics and the conservation of forest trees. Perspect Plant Ecol Evol Syst 11:157–189. doi:10.1016/j.ppees.2009.01.003
Pazouki L et al (2010) Genetic diversity and relationships among Pistacia species and cultivars. Conserv Genet 11:311–318. doi:10.1007/s10592-009-9812-5
Peakall R, Smouse PE (2006) GENALEX V. 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peñuelas J, Ogaya R, Boada M, Jump AS (2007) Migration, invasion and decline: changes in recruitment and forest structure in a warming-linked shift of European beech forest in Catalonia (NE Spain). Ecography 30:829–837
Perks MP, McKay HM (1997) Morphological and physiological differences in Scots pine seedlings of six seed origins. Forestry 70:223–232. doi:10.1093/forestry/70.3.223
Pessi AM, Pulkkinen P (1994) Temporal and spatial variation of airborne Scots pine pollen. Grana 3:151–157
Petit JR et al (1997) Four climate cycles in Vostok ice core. Nature 387:359–360
Petit RJ et al (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565. doi:10.1126/science.1083264
Provan J, Soranzo N, Wilson NJ, McNicol JW, Forrest GI, Cottrell J, Powell W (1998) Gene-pool variation in caledonian and European Scots pine (Pinus sylvestris L.) revealed by chloroplast simple-sequence repeats. Proc R Soc Lond B Biol Sci 265:1697–1705
Prus-Glowacki W, Urbaniak L, Zubrowska-Gil M (1993) Allozyme differentiation in Mid-Eropean and Scandinavian population of Scots pine (Pinus sylvestris). Genetica Polonica 34:159–176
Prus-Glowacki W, Stephan BR, Bujas E, Alia R, Marciniak A (2003) Genetic differentiation of autochthonous populations of Pinus sylvestris (Pinaceae) from the Iberian peninsula. Plant Syst Evol 239:55–66. doi:10.1007/s00606-002-0256-3
Pulkkinen P, Rantiolehtimaki A (1995) Viability and seasonal distribution patterns of Scots pine pollen in Finland. Tree Physiol 15:515–518
Pyhäjärvi T, Garcia-Gil MR, Knurr T, Mikkonen M, Wachowiak W, Savolainen O (2007) Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics 177:1713–1724. doi:10.1514/genetics.107.077099
Pyhäjärvi T, Salmela MJ, Savolainen O (2008) Colonization routes of Pinus sylvestris inferred from distribution of mitochondrial DNA variation. Tree Genet Genomes 4:247–254. doi:10.1007/s11295-007-0105-1
Ribeiro MM, Mariette S, Vendramin GG, Szmidt AE, Plomion C, Kremer A (2002) Comparison of genetic diversity estimates within and among populations of maritime pine using chloroplast simple-sequence repeat and amplified fragment length polymorphism data. Mol Ecol 11:869–877. doi:10.1046/j.1365-294X.2002.01490.x
Robledo-Arnuncio JJ, Gil L (2005) Patterns of pollen dispersal in a small population of Pinus sylvestris L. revealed by total-exclusion paternity analysis. Heredity 94:13–22. doi:10.1038/sj.hdy.6800542
Robledo-Arnuncio JJ, Alia R, Gil L (2004) Increased selfing and correlated paternity in a small population of a predominantly outcrossing conifer, Pinus sylvestris. Mol Ecol 13:2567–2577. doi:10.1111/j.1365-294X.2004.02251.x
Robledo-Arnuncio JJ, Collada C, Alia R, Gil L (2005) Genetic structure of montane isolates of Pinus sylvestris L. in a Mediterranean refugial area. J Biogeogr 32:595–605. doi:10.1111/j.1365-2699.2004.01196.x
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Sarvas R (1962) Investigations on the flowering and seed crop of Pinus sylvestris. Commun Inst For Fenn 53:1–198
Saylor LC (1972) Karyotype analysis of the genus Pinus—subgenus Pinus. Silvae Genetica 21:155–163
Scalfi M, Piotti A, Rossi M, Piovani P (2009) Genetic variability of Italian southern Scots pine (Pinus sylvestris L.) populations: the rear edge of the range. Eur J Forest Res 128:377–386. doi:10.1007/s10342-009-0273-7
Sgorbati S et al (2004) A survey of genetic diversity and reproductive biology of Puya raimondii (Bromeliaceae), the endangered queen of the Andes. Plant Biol 6:222–230. doi:10.1055/s-2004-817802
Shanjani PS, Vettori C, Giannini R, Khavari-Nejad RA (2004) Intraspecific variation and geographic patterns of Fagus orientalis Lipsky chloroplast DNA. Silvae Genetica 53:193–197
Shanjani PS, Vendramin GG, Calagari M (2010) Genetic diversity and differentiation of Fagus orientalis Lipsky in Hyrcanian forests revealed by nuclear and chloroplast microsatellite markers. Conserv Genet 11:2321–2331. doi:10.1007/s10592-010-0118-4
Shanjani PS, Vendramin GG, Calagari M (2011) Altitudinal genetic variations among the Fagus orientalis Lipsky populations in Iran. Iran J Biotechnol 9:11–20
Sinclair WT, Morman JD, Ennos RA (1998) Multiple origins for Scots pine (Pinus sylvestris L) in Scotland: evidence from mitochondrial DNA variation. Heredity 80:233–240. doi:10.1038/sj.hdy.6882870
Sinclair WT, Morman JD, Ennos RA (1999) The postglacial history of Scots pine (Pinus sylvestris L.) in western Europe: evidence from mitochondrial DNA variation. Mol Ecol 8:83–88. doi:10.1046/j.1365-294X.1999.00527.x
Slate J, Pemberton JM (2007) Admixture and patterns of linkage disequilibrium in a free-living vertebrate population. J Evol Biol 20:1415–1427. doi:10.1111/j.1420-9101.2007.01339.x
Slatkin M (1994) Linkage disequilibrium in growing and stable populations. Genetics 137:331–336
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Sloan DB, Barr CM, Olson MS, Keller SR, Taylor DR (2008) Evolutionary rate variation at multiple levels of biological organization in plant mitochondrial DNA. Mol Biol Evol 25:243–246. doi:10.1093/molbev/msm266
Soranzo N, Provan J, Powell W (1998) Characterization of microsatellite loci in Pinus sylvestris L. Mol Ecol 7:1260–1261
Soranzo N, Alia R, Provan J, Powell W (2000) Patterns of variation at a mitochondrial sequence-tagged-site locus provides new insights into the postglacial history of European Pinus sylvestris populations. Mol Ecol 9:1205–1211. doi:10.1046/j.1365-294x.2000.00994.x
Strauss SH, Hong YP, Hipkins VD (1993) High levels of population differentiation for mitochondrial DNA haplotypes in Pinus radiata, muricata, andattenuata TAG. Theor Appl Genet 86:605–611. doi:10.1007/bf00838716
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Varis S, Pakkanen A, Galofre A, Pulkkinen P (2009) The extent of south-north pollen transfer in Finnish scots pine. Silva Fennica 43:717–726
Vendramin GG, Lelli L, Rossi P, Morgante M (1996) A set of primers for the amplification of 20 chloroplast microsatellites in Pinaceae. Mol Ecol 5:595–598
Vendramin GG, Anzidei M, Madaghiele A, Bucci G (1998) Distribution of genetic diversity in Pinus pinaster Ait. as revealed by chloroplast microsatellites. Theor Appl Genet 97:456–463. doi:10.1007/s001220050917
Wang X-R, Szmidt AE, Lindgren DAG (1991) Allozyme differentiation among populations of Pinus sylvestris (L.) from Sweden and China. Hereditas 114:219–226. doi:10.1111/j.1601-5223.1991.tb00328.x
Wegmann D, Currat M, Excoffier L (2006) Molecular diversity after a range expansion in heterogeneous environments. Genetics 174:2009–2020. doi:10.1534/genetics.106.062851
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright S (1951) The genetical structure of populations. Ann Eugen 15:323–354
Zhou Y, Bui T, Auckland LD, Williams CG (2002) Undermethylated DNA as a source of microsatellites from a conifer genome. Genome 45:91–99. doi:10.1139/g01-119
Ziehe M, Gregorius HR, Glock H, Hattemer HH, Herzog S (1989) Gene resources and gene conservation in forest trees: general concepts. In: Scholz F, Gregorius HR, Rudin D (eds) Genetic effects of air populations in forest tree populations. Springer, Heidelberg, pp 173–186
Ziehe M, Hattemer HH, Müller-Starck R, Müller-Starck G (1999) Genetic structures as indicators for adaptation and adaptational potentials. In: Mátyás C (ed) Forest genetics and sustainability. Kluwer Academic Publishers, Dordrecht, pp 75–89
Acknowledgments
We thank Dr. Astrid Kännaste and Dr. Lucian Copolovici for the help in sampling Scots pine populations and Seyedeh Azimeh Motallebi Hasankola for the help to prepare some graphs. Financial support for the study has been provided by the Estonian Ministry of Science and Education (institutional Grant IUT-8-3), and the European Commission through the European Regional Fund (the Center of Excellence in Environmental Adaptation).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Jarmo Holopainen.
Rights and permissions
About this article

Cite this article
Pazouki, L., Shanjani, P.S., Fields, P.D. et al. Large within-population genetic diversity of the widespread conifer Pinus sylvestris at its soil fertility limit characterized by nuclear and chloroplast microsatellite markers. Eur J Forest Res 135, 161–177 (2016). https://doi.org/10.1007/s10342-015-0928-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-015-0928-5