
Abstract
Self-incompatible plants as well as common plant species are expected to be especially vulnerable to the deleterious effects of fragmentation on genetic diversity. Paypayrola blanchetiana (Violaceae) is a common, self-incompatible understory tree in the East Brazilian Atlantic forest. Its autochorous seed dispersal and occurrence in dense, well-separated clusters makes it an interesting model for studies of gene flow and genetic structuring on a small geographic scale. A previous study has found remarkably low fruit set and frequent seed abortions in this species in several populations situated in forest fragments. We tested the hypothesis that P. blanchetiana is affected by inbreeding and loss of genetic diversity in a fragmented landscape. Nine nuclear and three plastid microsatellite loci were genotyped for 285 individuals (149 adult trees and 136 saplings) across seven populations in five forest fragments in a 240-km2 sugarcane-rainforest matrix. We found a low to moderate genetic diversity in nuclear loci of P. blanchetiana, population structuring on a small geographical scale and high levels of inbreeding. Haplotype distributions confirmed that seed dispersal is very limited. There were, however, no signs for lower genetic diversity or higher inbreeding in populations situated in the smallest forest fragments. Furthermore, genetic diversity was not lower in the sapling cohort, which was created in post-fragmentation condition. Therefore, we may be witnessing the genetic consequences of this species’ biology, rather than immediate effects of fragmentation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Brazilian Atlantic coastal forest is home to approximately 20,000 species of vascular plants, of which 8000 (40%) are endemic (Myers et al. 2000). Today, this forest is greatly reduced in extent and highly fragmented, with only c. 7–8% remaining (Galindo-Leal and Camara 2003). Conservation of its biodiversity will depend, among various factors, on the ability of species to maintain genetic diversity within and connectivity among its populations (Krutovsky and Neale 2005). When studying plants of fragmented tropical forests, special attention should be paid to common, outcrossing species, because their genetic diversity is expected to suffer stronger declines under fragmentation conditions than naturally rare and/or frequently selfing species (Aguilar et al. 2008).
The scientific consensus today is that gene flow among tropical forest trees is quite extensive (Ward et al. 2005, Dick et al. 2008) and has generally been sufficient to prevent short- to medium-term losses of genetic diversity through genetic drift and inbreeding (Young et al. 1996, Kramer et al. 2008). Spatial genetic structuring on local scales should depend on seed dispersal modes and may occur when seed dispersal is limited by gravity (Dick et al. 2008 and references therein). There is also a trend for moderate genetic differentiation (mean Fst = 0.116) of tropical tree populations across larger geographical scales (> 50 km; Dick et al. 2008). Nevertheless, many of the studied species are tall canopy trees, which occur naturally at low densities and may be pre-adapted to long-range pollen- and (to a lesser degree) seed-mediated gene flow through wide-ranging dispersers such as birds, bats, and large bees (Chase et al. 1996, Ward et al. 2005).
On the other hand, many tropical trees have seeds that travel only short distances either due to autochory or because primary or secondary dispersers have small home ranges. Such species tend to occur only locally and to form dense aggregations of conspecifics. If seed dispersal is very limited, they should thus depend much more on pollen dispersal to prevent genetic drift and keep populations interconnected. Furthermore, local aggregations with many floral resources may discourage pollinators to fly long distances (Stacy et al. 1996). Such tree species, which are most typical of the forest understory, may therefore be interesting special cases with regard to genetic differentiation. The few previous studies suggest that genetic differentiation on local scales (< 50 km) can be considerable (Domínguez et al. 2005, Moraes-Filho et al. 2015) or even extremely high (Dutech et al. 2002, Lasso et al. 2011) in understory trees and shrubs with an aggregated distribution. On the other hand, an interesting example of ample pollen flow in an understory tree with a more evenly spaced distribution has been presented by Jha and Dick (2010). A tendency for reduced gene flow and local-scale genetic differentiation may also lead to stronger negative impacts of habitat fragmentation, because populations of short-range–dispersing plants could become more effectively isolated.
Paypayrola blanchetiana Tul. (Violaceae) is a small tree endemic to the Atlantic forest of Brazil (Martini et al. 2007). It is locally common in the Atlantic forest of the northeast Brazilian state of Pernambuco (M. Braun, pers. obs.). The species occurs in the understory, typically in dense aggregations that are well-separated from each other. Seed dispersal is autochorous, and we expect that trees growing next to each other tend to be closely related (half-sibs). Plants are self-incompatible, do not reproduce clonally, and consistently produced very few seeds, despite near-continuous flowering, during a 2-year monitoring of phenology (Braun et al. 2012, b). Ninety percent of developing fruits were aborted without noticeable insect damage or other visible causes. Inbreeding depression following near-neighbor breeding has been considered as a main cause for these abortions. Fruit/flower ratios declined as a function of high patch flowering, which suggests that pollination among putatively related neighboring plants may not function adequately (Braun et al. 2012). Furthermore, the restricted dispersal mode should predispose this species to strong genetic structuring (Hamrick et al. 1993). In highly fragmented landscapes, which are common in the Atlantic and other tropical forests, many populations will be lost, and distances between remaining populations increase considerably (Trindade et al. 2008). Fragmentation may thus accelerate insufficient gene flow and genetic drift and, consequently, the loss of genetic diversity and increasing genetic differentiation among populations.
In this study, we test these assumptions using nuclear as well as chloroplast microsatellite markers, targeting populations of P. blanchetiana in several fragments of forest separated by sugarcane cultivation. We are interested in the following aspects: (1) Is genetic diversity lower than mean values usually observed for outcrossing tropical trees? (2) Do we find indications for lower genetic diversity in saplings compared to adults, and in small fragments compared to large ones? (3) Is there significant inbreeding that could account for the observed low fruit set? (4) Are populations genetically different from each other, in response to reduced gene flow?
Materials and methods
Population sampling and DNA extraction
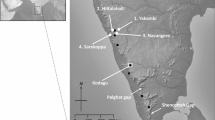
Sampling took place at the sugarcane field from the Usina São José S.A. (hereafter, USJ), situated in the Igarassú and Araçoiaba municipalities in the state of Pernambuco, Brazil. USJ has about 100 Atlantic forest fragments varying in size from less than 10 to ca. 500 ha, which are embedded in vast areas of sugarcane cultivation. Seven populations, situated in five different fragments, were sampled (Fig. 1). Fresh leaves of 149 adult trees (> 1.5 m tall) and 136 saplings (< 0.7 m), 285 individuals in total, were collected from individuals separated by several meters and immediately silica-dried until DNA extraction, which followed the cetyltrimethylammonium bromide (CTAB) method described by Doyle and Doyle (1987) and modified following Ferreira & Grattapaglia (1995).

a Landscape configuration at Usina São José, Igarassú, PE (Brazil). Forest fragments shown in dark green. Most of the remaining area consists of sugar cane plantations. Area covered is approx. 15 km north-south and 14 km west-east. Inlets show locations of studied populations. b Populations 1–3 (Piedade_1–3) all within a single forest fragment; c population 4 (Pezinho); d populations 5 (Vespas), 6 (Gota), and 7 (Palmeira). Edge length of inlets ca. 2.8 km
Nuclear microsatellite amplification
Nine nuclear microsatellite markers, which were previously developed for P. blanchetiana (Braun et al. 2019), were used for genotyping the 285 collected individuals. Individual DNA concentrations were quantified with a NanoDrop™ 2000 spectrophotometer and adjusted (ca. 20 ng/μl). Sequence-specific forward primers with M13 (− 21) tail at the 5’end were coupled with a universal fluorescent–labeled M13 (− 21) primer (6-FAM; VIC; NED or PET) (Schuelke 2000). Amplifications were carried out on a Veriti 96-Well Thermal Cycler (Applied Biosystems), with final volumes of 12.5 μl, containing 5–10 ng DNA template, 0.2 μM forward primer with M13 tail, 0.8 μM reverse primer, 0.8 μM fluorescent-labeled M13 primer, 0.2 mM of each dNTP, 1.5 mM MgCl2, × 1 PCR buffer (10 mM Tris–HCl buffer, 50 mM KCl), and 1.25 U Taq DNA Polymerase (Invitrogen). Amplification consisted of a first step at 94 °C for 5 min, 30 cycles of 30 s at 94 °C, 45 s at annealing temperatures 56–66 °C, 45 s extension at 72 °C, followed by 8 cycles of 30 s at 94 °C, 53 °C for 45 s, and 45 s extension at 72 °C, with a final extension of 72 °C for 10 min.
Chloroplast marker selection and amplification
Three cpDNA loci were analyzed. The first (cpPB-15) is a polymorphic SSR marker (primer sequence see Braun et al. 2019). This locus is also present in Viola seoulensis (Cheon et al. 2015). We then tested a set of seven universal primer pairs (ccmp 2, 3, 4, 5, 6, 7, 10) for chloroplast SSR that were first described for Nicotiana tabacum by Weising and Gardner (1999) and another chloroplast SSR marker first described for Eucalyptus (EMCR74; Steane et al. 2005). Two primers (ccmp2, ccmp10; both with a mononucleotide repeat) amplified polymorphic products and were also used to genotype 110 individuals (15–16 from each population). The cpPB-15 locus was amplified under the same conditions as the nuclear loci, while the following mixture was applied for the universal primers: final volumes of 10 μl, containing 20–40 ng DNA template, 0.5 μM fluorescent-labeled forward primer, 0.5 μM reverse primer, 0.2 mM of each dNTP, 2.5 mM MgCl2, 10 mM Tris–HCl buffer, 50 mM KCl, and 1.5 U Taq DNA Polymerase (Invitrogen). Amplification consisted of a first step at 94 °C for 5 min, 30 cycles of 60 s at 94 °C, 60 s at annealing temperatures 54 °C, 60 s extension at 72 °C, final extension of 72 °C for 10 min. A subset (30%) of individuals was genotyped twice to ensure consistent results.
Genotyping procedure
All nuclear and cpSSR samples were genotyped on an ABI 3500 sequencer (Applied Biosystems®) and sized against 600 LIZ®. Prior to genotyping, four loci (normally) of the same individual, each with a different fluorescent marker, were pooled. One microliter of the pooled sample was added to 9.75 μl formamide and 0.25 μl of GeneScan 600 LIZ® size standard. Allele scoring was performed with GeneMapper and GENEIOUS programs.
Data analysis of nuclear SSR loci
The following genetic diversity indices within populations were calculated using FSTAT 2.9.3.2 (Goudet 1995) and Genepop web version 4.2 (Raymond and Rousset 1995): (1) percentage of polymorphic loci (P), (2) alleles per locus (A), (3) allelic richness (Rs), (4) observed heterozygosity (HO), (5) expected heterozygosity (HE) under Hardy–Weinberg equilibrium (Nei, 1978), and (6) inbreeding coefficient (FIS) (Weir and Cockerham 1984). Deviations from HWE and possible linkage disequilibrium between pairs of loci were detected using the ARLEQUIN 3.5.2.2 software package (Excoffier and Lischer 2010). The Micro-Checker 2.2.3 software (van Oosterhout et al. 2004) was employed to detect scoring errors due to stuttering, short allele dominance and null alleles, and GIMLET 1.3.3 (Valière 2002) was used to detect individuals with identical genotypes (i.e., possible clones). The program FreeNa (available at www1.montpellier.inra.fr/CBGP/software/FreeNA/) was used to calculate null allele frequencies across loci and correct pairwise FST values by excluding null alleles, using the “ENA” method described by Chapuit and Estoup (2007). The occurrence of recent effective population size reductions in one or more of the studied populations was tested using the Wilcoxon sign-rank test described by Cornuet and Luikart (1997) and implemented in the software BOTTLENECK (Piry et al. 1999). We ran the analysis employing a two-phased mutation model with 95% single-step mutations and 5% multiple-step mutations (recommended for microsatellites) over 1000 iterations.
We tested the hypotheses that (a) saplings had a lower genetic diversity than adult trees and (b) individuals in small (< 50 ha) fragments were less diverse than those in larger fragments by comparing (locus × population) A and HE values of saplings and adults through Mann-Whitney U tests for independent non-parametric data.
F-statistics were employed to quantify genetic diversity within and among populations (Weir and Cockerham 1984). Pairwise FST values among populations were calculated with ARLEQUIN 3.5.2.2. The partitioning of genetic diversity within and between populations was calculated with analysis of molecular variance (AMOVA), within the Arlequin software.
To test the hypothesis that genetic structuring between populations is a function of isolation-by-distance (Wright 1965), relationship between geographical and genetic distances was analyzed with a standard Mantel Test (Sokal and Rohlf 1995) provided by the ARLEQUIN 3.5.2.2 software. The significance was assessed through a randomization test using 10,000 Monte Carlo simulations.
A Bayesian analysis was performed using the program STRUCTURE 2.3.4 (Pritchard et al. 2000) to assign individuals to genetic clusters (K) and to estimate admixture proportions (Q) for each individual. We employed an admixture model assuming correlated allele frequencies, K = 1–7 with ten replicates each, a burn-in of 100,000 and 500,000 iterations. The optimum K value was calculated based upon a measure of ΔK that evaluates the second-order rate of change of the likelihood function with respect to K (Evanno et al. 2005). ΔK was calculated using Structure Harvester web version 0.6.94 (Earl and von Holdt 2012).
Data analysis of cpSSR loci
The data of the taxon-specific locus clPB-15 and the two universal loci ccmp2 and ccmp10 were combined to determine the haplotype of 110 individuals, equally distributed among the seven populations (15–16 per pop.). Haplotype diversity was characterized for each population through the number of haplotypes, gene diversity (HE), and haplotype richness (R_h). These, as well as Nei’s Genetic Distances, were calculated with HAPLOTYPES ANALYSES 1.05 (Eliades and Eliades 2009). A median-joining haplotype network was constructed using the program NETWORK 5.0.0.1 (Bandelt et al. 1999). To obtain the partition of haplotype variance among populations, AMOVA was calculated with ARLEQUIN 3.5.2.2. Φ values obtained by AMOVA for nuclear and cpSSR data were then used to calculate the ratio of gene flow through seeds vs. gene flow through pollen using the model proposed by Ennos (1994):
where ΦSC(B) is the level of population differentiation obtained from biparental (nuclear) markers and ΦSC(M) the level of differentiation obtained from maternally inherited (cpSSR) markers, assuming that cpDNA in P. blanchetiana is, as in most angiosperms, maternally inherited.
Results
Nuclear microsatellites
All nine SSR loci were polymorphic, with allele numbers ranging from three to seven and an average of 4.4 alleles per locus (Table 1). Observed and expected heterozygosity, calculated over all populations, ranged from 0.197 to 0.53 and from 0.206 to 0.742 respectively. The mean inbreeding coefficient FIS was 0.169 over all populations. Six loci departed significantly from Hardy–Weinberg equilibrium (Table 1).
Micro-Checker detected an excess of homozygotes, which may be due to the presence of null alleles, at six loci in 1–6 populations (most notably locus PB-36 with homozygote excess in six populations). Two pairs of individuals with identical genotypes were encountered (adult08 and juv15 in Piedade_1; adult08 and adult15 in Piedade_2). FreeNa detected null allele frequencies larger than 0.1 in 14 out of 63 calculations (nine loci and seven populations), including at six out of seven populations at the locus PB-36 and three out of seven populations at PB-20.
Genetic diversity and bottlenecks
Allelic richness (Rs) in populations of P. blanchetiana at USJ ranged from 2.43 to 3.12 among adult trees and from 2.39 to 3.18 among saplings, averaging 2.71 and 2.82 respectively (Table 2). We found low to average allelic diversity with total number of alleles per population ranging from 24 to 31 in adults and 22 to 30 in saplings. Mean observed and expected heterozygosity per population was 0.389 and 0.459 in adults and 0.399 and 0.485 in saplings, respectively (Table 2). Inbreeding coefficients were moderately high (mean 0.154 adults; 0.18 saplings) and deviations from HWE were detected in all populations, except two in the adult cohort. We found no difference in genetic diversity between adult trees and saplings (alleles per locus: z = − 0.16, p (2-tailed) = 0.873; expected heterozygosity: z = − 1.05, p (2-tailed) = 0.294). We therefore pooled the data for all subsequent analyses. Adults and saplings combined, all loci were polymorphic in all populations. Likewise, we found no difference in genetic diversity between populations from large and small fragments (alleles per locus: z = 1.3, p (2-tailed) = 0.194; expected heterozygosity: z = − 0.67, p (2-tailed) = 0.503). Signs of a recent reduction in population size (genetic bottleneck) were found in two out of seven populations, Piedade_2 and Vespas (Wilcoxon test p < 0.05).
Genetic differentiation and structuring among populations
Overall genetic differentiation of the seven populations of P. blanchetiana was moderate (FST = 0.125; Table 1), and slightly lower using the ENA-correction (FST = 0.12). Genetic differentiation among pairs of populations ranged from small to moderately high (Table 3; nuclear SSR). FST values were low when comparing populations within the same fragment (Piedade_1–3) and generally highest among the Pezinho fragment and other populations, as well as among Palmeira and other populations (except Vespas). The Mantel test revealed significant isolation-by-distance, suggesting fine-scale genetic structuring, but the linear relationship was not very strong (R2 = 0.28, p = 0.014; Fig. 2). All pairwise FST values were statistically significant at the .001 level (with one exception; see Table 3).

Relationship between geographical and genetic (Fst) distances between all pairs of populations
AMOVA results revealed that most of the genetic diversity of the nuclear SSR data was present within populations (87.24%) of P. blanchetiana (Table 4).
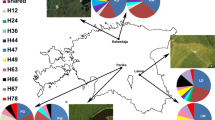
Bayesian analysis with STRUCTURE consistently identified K = 3 clusters. One cluster combined the three populations found in the Piedade fragment, another comprised the trees of the relatively isolated fragment Pezinho, while the remaining populations in the north of the study area formed the third cluster (Fig. 3).

Population structure in Paypayrola blanchetiana. a Bayesian assignment analysis for a K = 3 population model based on nine nuclear microsatellite loci (populations from north to south); b haplotype network and distribution across populations
Diversity of cpSSR and haplotype network
Analysis of three chloroplast markers across 110 individuals revealed four different haplotypes (Table 5). Haplotype diversity across populations was low to moderate (HE = 0–0.5; haplotype richness (R_h) = 0–1). No population contained more than two haplotypes. Nei’s genetic distance values among populations were generally high, but ranged from as low as 0.004 to 0.941 (Table 3). Most of the variation was distributed among populations (84.2%; Table 4), indicating strong genetic structuring. Haplotype H3 was fairly evenly distributed across the study area, while H1 was found mostly within the Piedade fragment in the south (Fig. 3b). H3 was also at the center of the median-joining network, being one mutational step from each of the other haplotypes. The two remaining haplotypes H2 and H4 were fixed (or nearly so) in a single population.
Pollen vs. seed flow
The genetic variance (AMOVA, Table 4) among populations for nuclear markers (ΦSC(B) = 0.128) and for cpSSR markers (ΦSC(M) = 0.84) translated into a ratio of pollen to seed flow of 33.8, indicating an approximately 34-fold gene flow excess through pollen.
Discussion
Genetic diversity and population inbreeding
We found comparatively low levels of genetic diversity in both nuclear and chloroplast markers of P. blanchetiana populations at Usina São José. There were only three or four alleles across all but three of the analyzed nuclear loci. Mean heterozygosity of the nuclear markers was moderate, but still low compared to levels found in many Neotropical canopy tree species (e.g., Collevatti et al. 2001a, 2009; Degen et al. 2004; André et al. 2008). The overall observed heterozygosity was even lower, creating a significant excess in homozygotes in six out of nine nuclear loci. This could be, in part, due to the presence of null alleles. However, null alleles were indicated for only two loci and did not significantly alter the results after correction.
Therefore, heterozygote deficiency is likely the consequence of non-random mating. The general magnitude of inbreeding following fragmentation has been assessed differently by previous comprehensive studies. Lowe et al. (2005) argue that inbreeding becomes evident shortly after impact, while subsequent reviews (Aguilar et al. 2008; Vranckx et al. 2012; Schlaepfer et al. 2018) did not find such an effect. Again, as in other parameters, general predictions appear to be difficult to make and individual plant species’ life history characters may play a more important role. Since P. blanchetiana is self-incompatible, we favor the explanation that seeds are frequently fathered by close relatives (e.g., half-sibs), thus creating significant biparental inbreeding. Biparental inbreeding has been observed in both obligate outcrossers and species with a mixed mating system, and levels of inbreeding could be quite high (Collevatti et al. 2001b; Degen et al. 2004). An important factor influencing these non-random mating processes in self-incompatible (SI) plants is a reduced mate availability due to reductions in S-allele diversity in small populations < 50–100 reproductive individuals (Byers & Meagher 1992; Young & Pickup 2010). In most SI plants, either one or both alleles at the S-locus have to be different for individuals to mate successfully. However, the large Piedade fragment contains hundreds of adult Paypayrola trees, thus likely maintaining a high diversity of S-alleles. Yet, it has similarly high Fis values, making this explanation less probable. Inbreeding in this case may in fact be best explained by the highly clustered spatial distribution pattern of P. blanchetiana, where pollination is often dominated by close-standing individuals (Garcia et al. 2005). Given the fact that pollinators tend to exploit aggregated floral resources very thoroughly, preferring to fly to the nearest plants (Nattero et al. 2011), pollen transfer among related plants should be very common.
Although the scale of inbreeding as indicated by average Fis values seems to be substantial, our data did not reveal any consistent tendencies among sites or age classes. Artifacts such as null alleles could have contributed to the high Fis to some degree, although such effects were tested for. Paternity analyses or direct analysis of aborted vs. non-aborted seeds, if feasible, would be appropriate to confirm the cause of inbreeding and its putative negative effect on fruit set in this species.
The present study identified no significant differences with regard to genetic diversity between saplings and adult trees, nor did we find such differences among plants in small vs. large fragments. Reviewing the mixed empirical signals concerning genetic declines following forest fragmentation, Kramer et al. (2008) predicted that population genetic effects of fragmentation events only become strongly evident after a fair number of generations. Likewise, Lowe et al. (2005) stressed that, while inbreeding and negative fitness effects are expressed shortly after disturbance, genetic diversity is lost much more slowly. However, the quantitative and systematic meta-analyses performed by Aguilar et al. (2008) and Vranckx et al. (2012) came to different conclusions, indicating that loss of genetic diversity following fragmentation was common among woody plants. The latter emphasized a strong effect of sample age (adult plants vs. progeny), while the former highlighted stronger responses in obligate outcrossers and common (vs. naturally rare) trees and significant losses of heterozygosity only after fragmentation events > 50 years ago. Schlaepfer et al. (2018) also demonstrated that loss of genetic diversity becomes more evident beyond 50 years of fragmentation time. Thus, although a reduction of genetic diversity could be expected in Paypayrola based on its biological characteristics (a common outcrosser), the time elapsed since fragmentation may have been too short.
Indeed, two of those fragments were part of a large forest block (> 5000 ha) until about 35–40 years ago. The third small fragment (Pezinho) was isolated, but in close proximity to this continuous forest (aerial photographs of USJ from 1975; Trindade et al. 2008), thus maintaining connectivity in the recent past. Factors unrelated to fragmentation may have been more important for shaping current allele distributions. One possibility is pre-fragmentation differences in historical tree distributions and densities across the landscape. For example, the highest numbers of alleles per population and age class (30–31) were found in the Piedade fragment, while in the populations outside Piedade a maximum of 28 alleles was found (sapling cohort in Gota). Extensive searches across USJ by the authors revealed that P. blanchetiana is quite common in Piedade, but generally much more sparsely distributed in other parts of USJ.
The fact that the sapling cohort maintained the same genetic diversity as the adult trees contradicts conclusions made by Vranckx et al. (2012), but agrees with predictions made in earlier reviews (Lowe et al. 2005, Kramer et al. 2008). Recently, Browne et al. (2015) obtained similar empirical results for the palm tree Oenocarpus bataua, with no apparent changes in genetic diversity parameters among tree generations and among fragments and continuous forest. We suggest that factors inherent to the individual mating system of a given species may play an important role in shaping the dynamics of allele distributions, especially on short-term time scales.
Genetic differentiation and gene flow
Overall genetic structuring
The degree of genetic structuring was unexpectedly high at the given geographical scale. Across distances of less than 15 km, we found pairwise differentiations similar to those typically recorded among populations separated by hundreds of kilometers. For example, Goetze et al. (2016) found similar pairwise Fst values among populations of a terrestrial bromeliad separated by 250–440 km. Genetic differentiation of Caryocar brasiliense populations across the Cerrado biome spanning 1500 km (Collevatti et al. 2001a) was of the same scale as P. blanchetiana in the present study (Fst 0.03–0.20). The terrestrial orchid Cypripedium calceolus showed the same magnitude of genetic differentiation, on a similar local scale, which has been attributed to a loss of connectivity due to habitat fragmentation (Minasiewicz et al. 2018). On the other hand, the wind-pollinated tree Castanopsis eyrei showed much weaker genetic differentiation among populations set apart by about 20 km (Mao et al. 2016). It is noteworthy that P. blanchetiana is self-incompatible and fully outcrossed, and therefore differentiation can be explained by limited gene flow and subsequent genetic drift in the relatively small and isolated stands, but not by frequent selfing events. Fst values are typically higher in plants with a mixed mating (i.e., frequently selfing) system than in obligate outcrossers (Duminil et al. 2009, Ballesteros-Mejia et al. 2016).
The Bayesian clustering method identified three separate genetic populations, which fall in line with their geographical distribution. The Pezinho population stands out as the purest stand with little admixture from the neighboring ones, despite its location between the other populations. The three populations found in the Piedade fragment are little differentiated (Fst ≤ 0.05), which indicates at least moderate gene flow among them. In the northern cluster, comprising the populations Vespas, Gota, and Palmeira (each in separate forest fragments), the pattern is more complex, but the fact that all three form a cluster can be explained by relatively recent fragmentation: Only in the 1970s or 1980s, when the last major forest clearing event took place at USJ, did Vespas, Gota, and Palmeira cease to belong to the same large forest block. In contrast, isolation of the Pezinho fragment took place before this time, which may be responsible for the large degree of genetic differentiation.
Haplotype structuring
We found a remarkably strong differentiation of haplotypes and among-population variation of chloroplast markers. Most populations were largely or entirely made up of a single haplotype, although there is evidence for occasional seed migration. Other studies that compared both nu-SSR and chloroplast markers likewise found higher Fst values for the latter, for example, about three times higher in an epiphytic forest bromeliad (Palma-Silva et al. 2009) and a terrestrial orchid (Minasiewicz et al. 2018). On the other hand, Chen et al. (2017) found the chloroplast-based genetic differentiation of the tree species Dipteronia dyeriana to be lower than the nu-SSR-based. Very high values of chloroplast differentiation, as in P. blanchetiana, were found in several bromeliads, for example, Aechmea calyculata. Populations of this rainforest epiphyte are widely spaced in the South Brazilian Atlantic forest and may have been historically separated by grasslands during colder periods in the pleistocene (Goetze et al. 2016). Another example of extremely high chloroplast marker divergence is the Alcantarea (Bromeliaceae) species complex from Southeast Brazilian inselbergs, which actually consists of several species with varying degrees of hybridization (Lexer et al. 2016). The among-population chloroplast diversity of 84% found in our study cannot be explained with natural barriers and points to an extremely restricted seed dispersal, which today is almost certain to be completely limited to within existing forest fragments. Very likely there was—at least in the past—some secondary seed dispersal by an unknown animal vector (possibly ants), since autochory alone could hardly account for the widespread occurrence of the species. Another factor hindering seed dispersal may be the extremely low seed output observed in this species (Braun et al. 2012).
Reasons for genetic structuring
Considering the isolation-by-distance relationship of Nei’s Genetic Distance and spatial distance, we conclude that limited gene flow is predisposing P. blanchetiana for genetic clustering at a small geographical scale. The species is likely pollinated by perching moths (Noctuidae, Geometridae) and to some degree by solitary bees and butterflies (Braun et al. 2012). The former pollinator group is capable of only limited pollen transfer distances mostly within forest blocks, as has been demonstrated for the moth-pollinated tree Cabralea canjerana (Melo & Franceschinelli 2016 and references therein). Despite a reputation as effective long-distance pollinators, resin-collecting female euglossine bees were not capable of carrying pollen across long distances or forest borders in the tropical vine Dalechampsia scandens (Opedal et al. 2017). These examples show that small forest-dwelling insects may not be able to mitigate the effects of genetic drift in a fragmented landscape with reduced plant populations.
Conclusions
The present study did not produce evidence that fragmentation has already had an effect on genetic diversity, although genetic distances seem to be influenced not only by geographical distances. However, we could show a strong tendency for reduced gene flow in P. blanchetiana, apparently driven by very limited seed dispersal as well as somewhat more wide-ranging, yet insufficient pollen flow. This biological condition may render this as well as other species with similar reproductive ecologies vulnerable for genetic erosion caused by the long-term effects of habitat fragmentation.
Our research is consistent with a trend in a yet small number of studies suggesting that genetic differentiation and reduced gene flow may actually be common in tree species which cluster locally and do not disperse seeds over large distances (see Domínguez et al. 2005; Lachenaud and Zhang 2008; Lasso et al. 2011). The view that gene flow in tropical trees is generally extensive may not uphold for a number of species, especially those growing in forest understory. More research is necessary on a variety of understory forest trees and shrubs with limited seed dispersal and spatial clustering.
References
Aguilar R, Quesada M, Ashworth L, Herrerias-Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188. https://doi.org/10.1111/j.1365-294X.2008.03971.x
André T, Lemes MR, Grogan J, Gribel R (2008) Post-logging loss of genetic diversity in a mahogany (Swietenia macrophylla King, Meliaceae) population in Brazilian Amazonia. Forest Ecol Manag 255:340–345. https://doi.org/10.1016/j.foreco.2007.09.055
Ballesteros-Mejia L, Lima NE, Lima-Ribeiro MS, Collevati RG (2016) Pollination mode and mating system explain patterns in genetic differentiation in Neotropical plants. PLoS One 11(7):e0158660
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Braun M, Dötterl S, Schlindwein C, Gottsberger G (2012) Can nectar be a disadvantage? Contrasting pollination natural histories of two woody Violaceae from the Neotropics. Int J Plant Sci 173:161–171. https://doi.org/10.1086/663167
Braun M, Esposito T, Huettel B, Pedrosa-Harand A (2019) Development and characterization of eleven microsatellite loci for the tropical understory tree Paypayrola blanchetiana Tul. (Violaceae). Mol Biol Rep 46:2529–2532. https://doi.org/10.1007/s11033-019-04622-z
Braun M, Piechowski D, Kazda M, Gottsberger G (2012) Fragment size and local flower density influence seed set of the understorey tree Paypayrola blanchetiana (Violaceae) in Brazilian Atlantic rain forest. J Trop Ecol 28:353–359. https://doi.org/10.1017/S0266467412000314
Browne L, Ottewell K, Karubian J (2015) Short-term genetic consequences of habitat loss and fragmentation for the neotropical palm Oenocarpus bataua. Heredity 115:389–395
Byers DL, Meagher TR (1992) Mate availability in small populations of plant species with homomorphic sporophytic self-incompatibility. Heredity 68:353–359
Chapuit M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chase MR, Moller C, Kesseli R, Bawa KS (1996) Distant gene flow in tropical trees. Nature 383:398–399. https://doi.org/10.1038/383398a0
Chen C, Lu RS, Zhu SS, Tamaki I, Qiu YX (2017) Population structure and historical demography of Dipteronia dyeriana (Sapindaceae), an extremely narrow palaeoendemic plant from China: implications for conservation in a biodiversity hot spot. Heredity 119:95–106. https://doi.org/10.5061/dryad.t8q1g
Cheon, K-S, Jang, S-K, Kim, K-A, Yang, J-C, Yoo, K-O (2015) The first complete chloroplast genome sequence from Violaceae (Viola seoulensis). GenBank: KP749924.1
Collevatti RG, Grattapaglia D, Hay JD (2001a) Population genetic structure of the endangered tropical tree species Caryocar brasiliense, based on variability at microsatellite loci. Mol Ecol 10:349–356. https://doi.org/10.1046/j.1365-294x.2001.01226.x
Collevatti RG, Grattapaglia D, Hay JD (2001b) High resolution microsatellite based analysis of the mating system allows the detection of significant biparental inbreeding in Caryocar brasiliense, an endangered tropical tree species. Heredity 86:60–67. https://doi.org/10.1046/j.1365-2540.2001.00801.x
Collevatti RG, Leoi LCT, Leite SA, Gribel R (2009) Contrasting patterns of genetic structure in Caryocar (Caryocaraceae) congeners from flooded and upland Amazonian forests. Biol J Linn Soc 98:278–290. https://doi.org/10.1111/j.1095-8312.2009.01287.x
Cornuet JM, Luikart G (1997) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Degen B, Bandou E, Caron H (2004) Limited pollen dispersal and biparental inbreeding in Symphonia globulifera in French Guiana. Heredity 93:585–591. https://doi.org/10.1038/sj.hdy.6800560
Dick CW, Hardy OJ, Jones FA, Petit RJ (2008) Spatial scales of pollen and seed-mediated gene flow in tropical rainforest trees. Trop Plant Biol 1:20–32. https://doi.org/10.1007/s12042-007-9006-6
Domínguez CA, Abarca CA, Eguiarte LE, Molina-Freaner F (2005) Local genetic differentiation among populations of the mass-flowering tropical shrub Erythroxylum havanense (Erythroxylaceae). New Phytol 166:663–672. https://doi.org/10.1111/j.1469-8137.2005.01359.x
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Duminil J, Hardy OJ, Petit RJ (2009) Plant traits correlated with generation time directly affect inbreeding depression and mating system and indirectly genetic structure. BMC Evol Biol 9:177. https://doi.org/10.1186/1471-2148-9-177
Dutech C, Seiter J, Petronelli P, Joly HI, Jarne P (2002) Evidence of low gene flow in a neotropical clustered tree species in two rainforest stands of French Guiana. Mol Ecol 11:725–738. https://doi.org/10.1046/j.1365-294x.2002.01475.x
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eliades N-G, Eliades DG (2009) HAPLOTYPE ANALYSIS: software for analysis of haplotypes data. Distributed by the authors. Forest Genetics and Forest Tree Breeding, Georg-Augst University Goettingen, Germany
Ennos RA (1994) Estimating the relative rates of pollen and seed migration among plant-populations. Heredity 72:250–259
Evanno G, Regnaut S, Goudet J (2005) Detecting the numbers of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Ferreira ME, Grattapaglia D (1995) Introdução ao uso de marcadores moleculares em análise genética. EMBRAPA-CENARGEN, Brasília. https://doi.org/10.13140/2.1.3757.8881
Galindo-Leal C, Camara IG (2003) Atlantic forest hotspot status: an overview. In: Galindo-Leal C, Camara IDG (eds) The Atlantic Forest of South America – biodiversity status, threats and outlook. Island Press, Washington, pp 3–11
Garcia C, Arroy JM, Godoy JA, Jordano P (2005) Mating patterns, pollen dispersal, and the ecological maternal neighbourhood in a Prunus mahaleb L. population. Mol Ecol 14:1821–1830. https://doi.org/10.1111/j.1365-294X.2005.02542.x
Goetze M, Palma-Silva C, Martini Zanella C, Bered F (2016) East-to-west genetic structure in populations of Aechmea calyculata (Bromeliaceae) from the southern Atlantic rainforest of Brazil. Bot J Linn Soc 181:477–490. https://doi.org/10.1111/boj.12416
Goudet J (1995) FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Hamrick JL, Murawski DA, Nason JD (1993) The influence of seed dispersal mechanisms on the genetic structure of tropical tree populations. Vegetatio 107:281–297
Jha S, Dick CW (2010) Native bees mediate long-distance pollen dispersal in a shade coffee landscape mosaic. PNAS 107:13760–13764. https://doi.org/10.1073/pnas.1002490107
Kramer AT, Ison JL, Ashley MV, Howe AF (2008) The paradox of forest fragmentation genetics. Conserv Biol 22:878–885. https://doi.org/10.1111/j.1523-1739.2008.00944.x
Krutovsky KV, Neale DB (2005) Forest genomics and new molecular genetic approaches to measuring and conserving adaptive genetic diversity in forest trees. In: Geburek T, Turok J (eds) Conservation and management of forest genetic resources in Europe. Arbora Publishers, Zvolen, pp 369–390
Lachenaud P, Zhang D (2008) Genetic diversity and population structure in wild stands of cacao trees (Theobroma cacao L.) in French Guiana. Ann For Sci 65:310. https://doi.org/10.1051/forest:2008011
Lasso E, Dalling JW, Bermingham E (2011) Strong spatial genetic structure in five tropical Piper species: should the Baker–Fedorov hypothesis be revived for tropical shrubs? Ecol Evol 1:502–516. https://doi.org/10.1002/ece3.40
Lexer C, Marthaler F, Humbert S, Barbará T, de la Harpe M, Bossolini E, Paris M, Martinelli G, Versieux LM (2016) Gene flow and diversification in a species complex of Alcantarea inselberg bromeliads. Bot J Linn Soc 181:505–520. https://doi.org/10.1111/boj.12372
Lowe AJ, Boshier D, Ward M, Bacles CFE, Navarro C (2005) Genetic resource impacts of habitat loss and degradation; reconciling empirical evidence and predicted theory for neotropical trees. Heredity 95:255–273. https://doi.org/10.1038/sj.hdy.6800725
Mao LH, Zhou XL, Fang YM (2016) Genetic diversity and population structure of Castanopsis eyrei based on simple sequence repeat markers. Genet Mol Res 15:1–8. https://doi.org/10.4238/gmr.15028256
Martini AM, Fiaschi P, Amorim AM, da Paixa OJL (2007) A hot-point within a hot-spot: a high diversity site within Brazil’s Atlantic Forest. Biodivers Conserv 16:3111–3128. https://doi.org/10.1007/s10531-007-9166-6
Melo A, Franceschinelli EV (2016) Gene flow and fine-scale spatial genetic structure in Cabralea canjerana (Meliaceae), a common tree species from the Brazilian Atlantic forest. J Trop Ecol 32:135–145. https://doi.org/10.1017/S0266467416000067
Minasiewicz J, Znaniecka JM, Górniak M, Kawiński A (2018) Spatial genetic structure of an endangered orchid Cypripedium calceolus (Orchidaceae) at a regional scale: limited gene flow in a fragmented landscape. Conserv Genet 19:1449–1460. https://doi.org/10.1007/s10592-018-1113-4
Moraes-Filho RM, Bonifácio-Anacleto F, Alzate-Marin AL (2015) Fragmentation effects and genetic diversity of the key semidecidual forest species Metrodorea nigra in Southwestern Brazil. Genet Mol Res 14:3509–3524. https://doi.org/10.4238/2015.April.15.15
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. https://doi.org/10.1038/35002501
Nattero J, Malerba R, Medel R, Coccucci A (2011) Factors affecting pollinator movement and plant fitness in a specialized pollination system. Plant Syst Evol 296:77–85. https://doi.org/10.1007/s00606-011-0477-4
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Opedal ØH, Falahati-Anbaran M, Albertsen E, Armbruster WS, Pérez-Barrales R, Stenøien HK, Pélabon C (2017) Euglossine bees mediate only limited long-distance gene flow in a tropical vine. New Phytol 213:1898–1908. https://doi.org/10.1111/nph.14380
Palma-Silva C, Lexer C, Paggi GM, Barbará T, Bered F, Bodanese-Zanettini MH (2009) Range-wide patterns of nuclear and chloroplast DNA diversity in Vriesea gigantea (Bromeliaceae), a neotropical forest species. Heredity 103:503–512. https://doi.org/10.1038/hdy.2009.116
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. Heredity 90:502–503. https://doi.org/10.1093/jhered/90.4.502
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Schlaepfer D, Braschler B, Rusterholz H-P, Bauer B (2018) Genetic effects of anthropogenic habitat fragmentation on remnant animal and plant populations: a meta-analysis. Ecosphere 9(10):e02488. https://doi.org/10.1002/ecs2.2488
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234. https://doi.org/10.1038/72708
Sokal RR, Rohlf FI (1995) Biometry: the principles and practice of statistics in biological research. Freeman and Company, NewYork
Stacy EA, Hamrick JL, Nason JD, Hubbell SP, Foster RB, Condit R (1996) Pollen dispersal in low-density populations of three Neotropical tree species. Am Nat 148:275–298. https://doi.org/10.1086/285925
Steane DA, Jones RC, Vaillancourt RE (2005) A set of chloroplast microsatellite primers for Eucalyptus (Myrtaceae). Mol Ecol Notes 5:538–541. https://doi.org/10.1111/j.1471-8286.2005.00981.x
Trindade MB, Lins-e-Silva ACB, da Silva HP, Figueira SB, Schessl M (2008) Fragmentation of the Atlantic Rainforest in the Northern Coastal Region of Pernambuco, Brazil: recent changes and implications for conservation. Bioremediat Biodivers Bioavail 2:5–13
Valière N (2002) GIMLET: a computer program for analysing genetic individual identification data. Mol Ecol Resour 2:377–379
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vranckx G, Jacquemyn H, Muys B, Honnay O (2012) Meta-analysis of susceptibility of woody plants to loss of genetic diversity through habitat fragmentation. Conserv Biol 26:228–237. https://doi.org/10.1111/j.1523-1739.2011.01778.x
Ward M, Dick CW, Gribel R, Lowe AJ (2005) To self or not to self…a review of outcrossing and pollen-mediated gene flow in neotropical trees. Heredity 95:246–254. https://doi.org/10.1038/sj.hdy.6800712
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.1111/j.1558-5646.1984.tb05657.x
Weising K, Gardner RC (1999) A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 42:9–19
Wright S (1965) The interpretation of population structure by F-statistics with special regards to system of mating. Evolution 19:395–420. https://doi.org/10.1111/j.1558-5646.1965.tb01731.x
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11:413–418. https://doi.org/10.1016/0169-5347(96)10045-8
Young AG, Pickup M (2010) Low S-allele numbers limit mate availability, reduce seed set and skew fitness in small populations of a self-incompatible plant. J Appl Ecol 47:541–548. https://doi.org/10.1111/j.1365-2664.2010.01798.x
Acknowledgments
The authors thank Usina São José S.A., municipality of Igarassú (Pernambuco) for facilitating collection of leaf material, and Dr. Theresa Liberal, Bruno Sampaio, and the Laboratory of Bioinformatics and Evolutionary Biology (Federal University of Pernambuco, Recife) for providing services of the ABI-3500 sequencing platform.
Data archiving statement
Genotype (SSR markers) and haplotype data have been deposited in the Tree Genes Database (acession-nr. TGDR 377; https://doi.org/10.5281/zenodo.4274480).
Funding
The study was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (grant no. 300532/2016-4) and Fundação de Amparo à Ciência e Tecnologia do Estado de Pernambuco (grant no. APQ-0207-2.03/13).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Additional information
Communicated by N. Tomaru
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Population structure
Rights and permissions
About this article

Cite this article
Braun, M., Dantas, L., Esposito, T. et al. Strong genetic differentiation on a small geographic scale in the Neotropical rainforest understory tree Paypayrola blanchetiana (Violaceae). Tree Genetics & Genomes 16, 85 (2020). https://doi.org/10.1007/s11295-020-01477-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-020-01477-5