
Abstract
Oriental beech (Fagus orientalis Lipsky) is a widespread monoecious and wind-pollinated tree species. It is one of the major components of the Hyrcanian forests of Iran and it is of both ecological and economical importance. Twelve beech stands were surveyed at 9 chloroplast (cp) and 6 nuclear (n) polymorphic microsatellite loci (simple sequence repeats, SSR) to provide information on distribution of genetic diversity within and among populations and on gene conservation and silvicultural management of this species. High levels of genetic differentiation were detected for the chloroplast genome (F ST = 0.80 and R ST = 0.95), in sharp contrast to the nuclear genome (F ST = 0.06, R ST = 0.05). The analysis of molecular variance (AMOVA) showed that 48% of the total cpSSR variation was attributable to differences among regions and 30% to differences among populations within regions, suggesting multiple origins of beech populations in Hyrcanian forests. Nuclear SSRs confirmed the presence of significant differentiation among populations and among geographic regions, even if, as expected, this was less pronounced than that found with cpSSRs (based on AMOVA, differences among regions and among populations within regions each contribute 5% to total nSSR variance). A highly significant correlation between genetic (nSSRs) and geographic distances (R 2 = 0.522) was estimated, thus showing an isolation by distance effect. The application of spatial analysis of molecular variance (SAMOVA) using both marker data allowed identification of genetically homogeneous groups of populations. Possible applications of these results for the certification of provenances and/or seed lots and for designing conservation programs are presented and discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Forest genetic resources, defined as the genetic variation present in the thousands of tree species on earth, constitute an inter-generational resource of vast social, economic and environmental importance. Forest genetic resources are threatened by several factors such as climate change, environmental pollution, habitat fragmentation and pest attacks. The capacity of forest tree species to survive these threats and to persist in spatially and temporally heterogeneous environments is dependent on their adaptive potential, which is mainly determined by the within-species genetic diversity. This is why conservation strategies for forest trees should be based on an evolutionary approach and focus on the maintenance of within-species genetic diversity (Geburek 1997).
Among the broad-leaved temperate forests of the northern hemisphere, Iran’s Hyrcanian forests are very particular. These forests, with a total area of 1.5 million hectares, are located along the southern coasts of the Caspian Sea and on the northern slopes of the Alborz mountains in the northern part of Iran. The Hyrcanian forest ecosystem is considered to be one of the last remnants of natural deciduous forest in the world. In this area beech survived intense climate and geological changes during the Quaternary, which occurred despite the fact that this area was not covered by ice and therefore was only indirectly influenced by the glaciations (Mobayen and Tregubov 1969). Hyrcanian commercial forests in Iran were nationalized in 1963. Since then, the area has declined significantly from 3.4 to less than 1.3 million ha in 2002. Mixed and pure stands of beech occupy less than 20% of the Hyrcanian forests and produce more than 35% of the total wood stock volume. A high proportion, about. 86%, of the trees are at least 100 years old and in some areas regeneration does not occur (Fishwick 1972; Anonymous 2000). Most of the beech forests in Iran are managed under the shelterwood silvicultural system and suffer from human interference. The rotation period for these forests is 120–125 years and complete natural regeneration is seldom achieved (Sagheb-Talebi and Schütz 2002). The two main technical reasons for the failure of this system in these forests are unsuitable harvesting methods during the last 40 years and lack of forest protection (Hosseini et al. 2000). There is an urgent need to improve conservation of this tree species through better forest management, with the aim of both genetic resource conservation and high quality timber production.
Information on the life-history traits of most forest tree species (e.g., mating system, pollen and seed dispersal mechanisms) and on their patterns of genetic variation are still lacking. In this context, the application of molecular markers in conservation programs is useful for generating data that can be used to extract relevant biological information, to identify hotspots of genetic diversity and to study geographic and phylogeographic structure (Newton et al. 1999).
The genetic diversity of oriental beech has been previously investigated using isoenzymes and restriction fragment length polymorphisms (RFLP) throughout the range of the species in Iran (Salehi Shanjani 2002). Allozyme analyses showed that the level of genetic diversity is high within populations and low among populations, with no evident geographic structure (F ST = 0.04, Salehi Shanjani et al. 2002). On the other hand, cpDNA studies (using PCR-RFLP markers, maternally inherited in beech) revealed that haplotype diversity within populations is low and population differentiation is high (F ST = 0.68, Salehi Shanjani et al. 2004). However, due to the relatively limited variation displayed by the markers used, little information was obtained from a practical point of view. Therefore in the present study genetic structure of F. orientalis was analyzed using chloroplast and nuclear microsatellite (SSR) markers, which are characterized by different modes of inheritance in beech (maternal and bi-parental, respectively) and are expected to display higher variation than the markers analysed previously in this species. The objectives of this study were (1) to provide basic information on gene conservation and management of beech forests, by studying the genetic diversity within and among populations/regions, (2) to shed light on the history of beech in Iran.
Materials and methods
Plant materials
Fagus orientalis(Lipsky) forests in Iran are located on the northern slopes of the Alborz Mountains, at an altitude ranging from about 600 to 2000 m above sea level. They form a forest strip 700 km long which covers the 3 provinces of Gilan, Mazandaran and Golestan. The sampled stands were chosen in order to provide maximum representation of the ecological conditions of the area. Dormant buds were collected from 12 natural oriental beech (Fagus orientalis) populations (each represented by at least 30 trees, see Table 1) in 5 different regions along the distribution range of the species from West to East. In each region 2–3 populations at the lowest, middle and highest altitude of the species distribution range were selected (Table 1). A subset of 168 individuals (14 samples per population) was also analysed using cpSSRs.
Molecular analysis
DNA was isolated from dormant buds (100 mg as starting material) using Nucleospin plant kit (Macherey-Nagel, Germany).
Nuclear microsatellites: Six nSSR primer pairs (Table 3) were amplified according to the following thermal profile: 5 min denaturation at 95°C followed by 30 cycles of 1 min denaturation at 95°C, 1 min annealing (Table 3), 1 min extension at 72°C, with a final extension step of 8 min at 72°C. PCR was carried out in a reaction volume of 25 μl, containing 10 ng of template DNA, 1× PCR buffer (GE Healthcare), MgCl2 as in Table 2, 0.2 mM dNTPs (GE Healthcare), 0.4 μM of each primer, 1 U of Taq DNA polymerase (GE Healthcare).
Chloroplast microsatellites: Nine cpSSR primer pairs (Table 2) were amplified according to the following thermal profile: 5 min 95°C, 30 cycles of 1 min denaturation at 95°C, 1 min annealing at 50°C (Table 2), 1 min extension at 72°C, with a final extension step of 8 min at 72°C. The PCR was performed in a final volume of 25 μl containing 10 ng of template DNA, 1× PCR buffer (GE Healthcare), 2.5 mM MgCl2, 0.2 mM dNTP, 0.2 μM of each primer, 1 U of Taq DNA polymerase (GE Healthcare).
The success of the amplification using cp- and n-SSRs was confirmed on 1.4% agarose gels. Amplified fragments were then multiplexed by size (mixed two by two). Standards (50, 100, 150, 200, 250 and 300 bp) were added to each mix before loading the samples onto Reprogel Long Read acrylamide gels (GE Healthcare), and run on an automated sequencer ALF Express (GE Healthcare) at 1500 V, 60 mA, 30 W, 55°C. The results of the run were then analysed using Fragment Manager 1.2 (GE Healthcare).
Data analysis
Polymorphisms of cpSSRs were scored and used to define chloroplast haplotypes according to different combinations of length variants (Table 4). For nSSR data, diploid genotypes for each individual were scored and following parameters were computed as averages over loci using GenAlEx v. 6 (Peakal and Smouse 2006): mean number of alleles per locus (A), number of unique alleles, number of locally common alleles, effective number of alleles (N e), average observed heterozygosity (H o), average expected heterozygosity (H e) computed according to Nei (1978) and fixation index (F IS).
The extent of population subdivision was evaluated by calculating Wright’s F ST (Wright 1951) and the related R ST (Slatkin 1995), with the latter taking explicit account of likely mutational processes at microsatellite loci. These two indices were estimated across populations and between pairs of populations, according to Weir and Cockerham (1984) and Michalakis and Excoffier (1996), by using the program SPAGEDI v1.1 (Hardy and Vekemans 2002).
FST may be overestimated in the case of null alleles (Chapuis and Estoup 2007). We controlled for the potential effect of null alleles on genetic differentiation by calculating FST(FREENA) values using the excluding null allele (ENA) method by Chapuis and Estoup (2007) in FREENA.
To take into consideration the high within-population variation, we estimate standardized F ST values using RecodeData v. 0.1 (Meirmans 2006) in conjunction with FSTAT. The data were recoded such that all alleles were population specific, maximizing differentiation. Standardized \( F^{\prime}_{\text{ST}} \) were then calculated following Hedrick (2005) as \( F^{\prime}_{\text{ST}} \) = F ST/F ST max. Mantel tests between pairwise F ST values, pairwise ENA corrected F ST values and pairwise \( F^{\prime}_{\text{ST}} \) was performed in ARLEQUIN 3.11 (Excoffier et al. 2005) using 1000 random permutations to evaluate if the different F ST calculations were significantly correlated.
A genetic distance matrix of pairwise F ST values was also used to perform a hierarchical analysis of molecular variance (AMOVA) (Excoffier et al. 1992) with Arlequin software version 3.11 (Excoffier et al. 2005). AMOVA was employed to estimate and partition the total molecular marker variances among regions, among populations within region, and within populations. Significance of partitioned variance components was estimated using a 1000 permutation procedure.
Principal coordinate analysis (PCoA) based on a pairwise distance matrix with data standardization was conducted by GenAlEx v.6 (Peakal and Smouse 2006). To test for a pattern of isolation by distance (Rousset 1997), a Mantel test with 1,000 random permutations was performed with the matrix of pairwise genetic differentiation between populations, using F ST/(1 − F ST), and a matrix of geographic distance using GenAlEx v.6 (Peakal and Smouse 2006).
Finally, a simulated annealing procedure implemented in the spatial analysis of molecular variance (SAMOVA) algorithm (Dupanloup et al. 2002) was used for both chloroplast and nuclear microsatellite data to define groups of populations that are geographically homogeneous and maximally differentiated from each other. The program repeatedly seeks the composition of a user defined number, K, of groups of geographically adjacent populations that maximizes F CT, the proportion of total genetic variance due to differences among groups of populations. The program was run for 10,000 iterations for K = {2,…,12}. The simulated annealing process was repeated 250 times to ensure that the final configuration of the K groups is not affected by a given initial configuration (Dupanloup et al. 2002). For each K, the configuration with the largest F CT values was retained as the best grouping of populations. SAMOVA was applied using cpSSR data too.
Results
The six nSSR loci were found to be highly polymorphic, with number of detected alleles per locus ranging from 6 (FS3-04) to 41 (FS4-46) (Table 3). The total number of alleles scored in 562 individuals over all loci was 141. Estimates of genetic diversity within populations are given as values averaged across loci (Table 5). High polymorphism was found within populations, on average more than 10 alleles per locus were observed (the mean number of alleles per locus varied from 7.67 ± 2.0 to 11.67 ± 3.2) and the probability that two randomly sampled alleles in a given populations are different was 66% (H e = 0.66). The mean expected heterozygosity (H e) over all investigated loci ranged from 0.50 ± 0.09 (Gorgan-1400) to 0.70 ± 0.11 (Sangdeh-900). No significant H e differences (P < 0.01) were observed among populations. The observed heterozygosity (H o = 0.604) was slightly lower than the expected heterozygosity (H e), causing a low but positive mean inbreeding coefficient (F IS = 0.045). However some populations showed high values of F IS, indicating deficiency of heterozygotes. Presence of null alleles at some loci cannot be excluded (FS1-25, FS4-46). Six out of the 12 populations showed an overall significant and positive departure from Hardy–Weinberg equilibrium (Table 5). Significantly less inbreeding (P < 0.05; one-tailed test) occurs on average in the unmanaged populations than in the stands with moderate and high management intensity, with exception of the Kheyrud region.
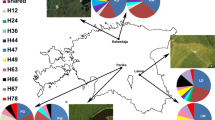
Two cpSSRs regions out of nine showed polymorphism (ccmp4 and ccmp7), with two and six length variants respectively, which combined into a total of 8 haplotypes (Table 2). Microsatellite haplotype distribution revealed that 7 out of the 12 populations had within population variation, while 5 populations displayed a fixed haplotype (Fig. 1). Almost all of the haplotypic variation resides among populations, with clear geographic distribution of haplotypic diversity. All haplotypes, but one, are geographically structured. The only exception is haplotype 3 which was found in two different regions (in 2 populations in the West and 1 population in the centre of the distribution range). The most frequent haplotype (haplotype 5, with a frequency of 44%) was detected in the populations distributed at longitudes ranging from 53° to 54°. The other haplotypes are represented in much lower proportions. Haplotypes 7 and 8 were found in two central populations (Kheirud region) and were highly divergent from all other haplotypes detected in eastern and western populations.

Frequency (right) and distribution (left) of haplotypes in F. orientalis based on the analysis of chloroplast microsatellite markers
The estimated coefficient of genetic differentiation among populations (F ST) was 0.058 for nSSRs, indicating a low but significant level of genetic differentiation. After the correction using FREENA, the overall F ST was only slightly lower (F ST(FREENA) = 0.057). The pairwise standardized Hedrick (2005) \( F^{\prime}_{\text{ST}} \) estimates were generally higher, but strongly correlated with the uncorrected pairwise F ST values (R 2 = 0.919, P < 0.00001) in the Mantel test. R ST (=0.048) gave slightly lower, but not significantly lower, differentiation values than F ST. Thus, low differences between allele lengths cause small variances and lower differentiation values between populations. On the other hand, using cpSSRs a marked and highly significant difference between populations was suggested by the F ST and R ST values, which were 0.80 and 0.95, respectively.
AMOVA using nuclear SSRs revealed that variation among regions accounted for 5% of the total variance, among populations within region and within populations for 5% and 90% of the total variation, respectively (Table 6). Chloroplast SSRs confirmed the presence of a much more pronounced and significant (P < 0.01) differentiation among regions and among populations within regions (based on AMOVA, differences among regions and among populations within regions account for 48% and 30% of the total cpSSR variance, respectively) (Table 6).
Pairwise F ST/(1 − F ST) was significantly correlated with geographical distance between populations for nSSRs (R = 0.552, P < 0.001, Fig. 2A), but not for cpSSRs (Fig. 2B). To better understand the relationships among populations, principal coordinate analysis (PCoA) was also carried out using both genetic distance datasets (Fig. 3). Using nSSR data, the first principal coordinate, which accounted for 50% of the total variation, clearly separated the Asalem populations (the western populations) and K-1200 (one of the central populations) from the eastern populations. The only exception among central populations was K-600 population, which clustered with eastern populations. The eastern populations were separated from the western populations along the second principal coordinate, which explained 21% of the total variation (Fig. 3). In fact, with the exception of K-600 population, all the western and eastern populations were clustered in different groups. Based on cpSSR data, the first principal coordinate, which accounted for 54% of the total variation, separated the two populations of Kheyrud (the central populations) from the rest. In contrast to the nSSR data, the cpSSR data did not classify populations according to their origin.

Isolation by distance analysis estimated using nSSRs (A) and cpSSRs (B): F ST/1 − F ST is plotted against the geographical distance among populations in kilometers

Two-dimensional graph based on the ordination scores of the principal coordinate analysis (PCoA) based on Nei′s unbiased genetic distances estimated using nSSRs (A) and cpSSRs (B): arrow shows the K-600 population (from central region) clustered with the eastern populations
We applied SAMOVA to both genotypic and haplotypic data using both F ST and R ST indices. The highest values of differentiation (F CT) were estimated at 3 and 6 groups using nuclear and chloroplast SSRs, respectively (Fig. 4).

Grouping of populations obtained following SAMOVA and using cpSSR (A) and nSSR (B) data
Discussion
Nuclear SSRs
The nuclear microsatellite markers used in this study detected high levels of genetic variation, with an average expected heterozygosity of 0.67. Mean number of alleles per locus was similar to that estimated in populations of European beech, however mean observed and expected heterozygosity was lower (Pastorelli et al. 2003; Buiteveld et al. 2007). In studies of other deciduous temperate tree species using microsatellite markers, higher polymorphism has been reported: e.g., H e = 0.87 at eight microsatellite loci in Magnolia obovata (Isagi et al. 2000); H e = 0.87 for six microsatellite loci in Quercus robur and Q. petraea (Streiff et al. 1998); H e = 0.87 for 16 microsatellite loci in Olea europaea (Štambuk et al. 2007).
Deviations from expected Hardy–Weinberg proportions were observed for some populations. In general, the observed heterozygosity was lower than expected generating positive, and in some cases significant, F IS values. Non-conformity with Hardy–Weinberg suggests nonrandom mating. Relative departures from Hardy–Weinberg expectations with a deficiency of heterozygotes were also found in allozyme studies of the Iranian beech populations (Salehi Shanjani et al. 2002). Overall, populations of F. orientalis are slightly, but significantly inbred (F IS = 0.060). The most commonly reported causes of positive values of the inbreeding coefficient are self-fertilization and bi-parental inbreeding, i.e. inbreeding originating by selfing or mating between related individuals (Hardy and Vekemans 2002).
Significantly less inbreeding was found in four of the five unmanaged populations than in their corresponding moderate and more intensively managed populations. The effects of forest management on genetic diversity of different tree species have been studied and in some cases the results contradict those of this study. For example, Buiteveld et al. (2007) recorded higher F IS values in stands with limited management than in intensively managed stands of Fagus sylvatica, suggesting that human interference or management does not increase inbreeding levels. Thomas et al. (1999) found that the breeding system of P. contorta was not affected by harvesting. However, enhanced inbreeding following harvest has been reported in Pinus strobus L. (Marquardt et al. 2007), Tsuga canadensis L. (Carr.) (Hawley et al. 2005), Scaphium macropodurn (Miq.) Beumee ex K. Heyne (Lee et al. 2002), Shorea megistophylla Dipterocarpaceae (Murawski et al. 1994), and P. sylvestris L. (Yazdani et al. 1985). On the other hand, we observed a low overall differentiation among populations at bi-parentally inherited nuclear markers (F ST = 0.058), which was also observed in F. sylvatica (Buiteveld et al. 2007).
Chloroplast SSRs
Beech species produce seeds with low dispersal potential: this is the main factor generating low within-population cpDNA diversity and high population differentiation, as seen in our results. Almost all haplotypes show a clear geographic structure, with higher diversity allocated to populations from western and central part of the natural range. It has been hypothesized that the formation of the Hyrcanian forests were strongly influenced by environmental conditions, especially the amount of precipitation, air humidity, temperature, type of humus and bed rock which significantly vary from East to West (Marvie-Mohadjer 1976; Parsapajouh 1976; Habibi 1985). The distribution pattern of F. orientalis cpDNA haplotypes may reflect migration history of beech during the Tertiary and suggest dispersal from Asalem (most polymorphic region, located in the West) to the East. Haplotype 3 is the only haplotype which was found in two different regions (in 2 populations in the West and 1 population in the Centre of the distribution range). This pattern could not be explained by gene flow, as these populations are 500 km apart and are distributed in different biogeographic zones. Thus, it seems most likely that existence of this haplotype (H3) at different regions is the results of mutation and subsequent fixation.
Most genetic surveys of chloroplast data have indicated a trend towards fixation (Dumolin-Lapègue et al. 1997; Marchelli et al. 1998; Hamilton 1999; Dutech et al. 2000) or have detected few cpDNA haplotypes within populations (Mohanty et al. 2000; Caron et al. 2000). In accordance with these results, a very limited number of haplotypes within populations (1–3 haplotypes) was also found in oriental beech. The higher haplotypic richness in the western populations could support the hypothesis of a possible migration of the Hyrcanian beech forests from West to East. Similar observation has been made for cpDNA PCR-RFLP in the same beech populations of Iran (Salehi Shanjani et al. 2004).
The cpSSR markers have been successfully used for large-scale population genetic studies in angiosperms, where inheritance is usually maternal (Lu et al. 2001; König et al. 2002; Lian et al. 2003; Aguinagalde et al. 2005). In angiosperms, cpSSR markers revealed a high level of genetic differentiation among populations. In a comparative study of cytoplasmic diversity (Duminil et al. 2006), the mean value of population subdivision measured for 97 plant species was G ST = 0.7. Species belonging to the family of Fagaceae and producing heavy seeds, like Fagus sylvatica, Quercus robur and Q. petraea, are characterized by even higher G ST values, ranging between 0.83 and 0.90 (Demesure et al. 1996; Dumolin-Lapègue et al. 1997).
Comparison of genetic and geographic structure at nSSRs and cpSSRs loci
It has been shown theoretically that, for many models of population structure, the level of genetic differentiation among populations is expected to be higher for maternally inherited cpDNA markers than for biparentally inherited nuclear genes (Birky et al. 1989; Petit et al. 1993; Ennos 1994; Hu and Ennos 1997). Several factors contribute to the higher genetic differentiation for chloroplast markers: (1) effective gene flow is limited to seed dispersal for maternally inherited DNA; (2) drift is expected to be twice as strong for a haploid genome compared to a diploid one; (3) it has been shown in some hermaphrodite tree species that the flowering, seed dispersal and recruitment patterns resulted in an effective number of trees contributing to the next generation as females that is much reduced compared to the effective number of trees acting as male parents (Dow and Ashley 1996). The latter factor, however, is not as ‘absolute’ as the first two. Indeed, differences in species’ pollination ecology or seed dispersal mechanisms might have important consequences on the balance between female and male reproductive success of individual trees.
As expected, the level of differentiation among populations of F. orientalis was higher for the chloroplast genome (F ST = 0.80 and R ST = 0.95) than for nuclear genome (F ST = 0.06, R ST = 0.05). Under the migration-drift equilibrium of the island model (Wright 1943) F ST for maternally inherited cpDNA exceeds F ST for biparentally and paternally inherited nuclear DNA (Ennos 1994). Assuming migration-drift equilibrium to estimate gene flow (Birky et al. 1989; Ennos 1994), the mean number of migrants per generation for the nuclear genome was Nm = 2.87 [Fstb = 1/(4Nmb + 1), Fstb = overall nuclear microsatellite loci] while for the maternally inherited chloroplast genome [Fstm = 1/(2Nmm + 1), Fstm = overall chloroplast microsatellite loci] was Nm = 0.125. The pollen to seed migration ratio estimated according to Ennos (1994), i.e. (pollen flow)/(seed flow) = {[(1/Fstb) − 1] − 2[(1/Fstm) − 1]}/[(1/Fstm) − 1] is equal to 22. These results show that pollen movement is significantly greater than seed movement. Therefore the low population differentiation for the nuclear genome is most likely because of pollen movement rather than seed dispersal. Beech is an anemophilous and mostly allogamous species, and its pollen can be dispersed over long distances (Comps et al. 1991). A previous study supports the hypothesis that the connection among populations of beech is mainly because of pollen flow (Salehi Shanjani et al. 2002).
An isolation-by-distance pattern was detected for nSSR, but not cpSSR, and the topology of the PCoA based on nSSR frequencies clearly reflects the geographic relationships among populations. These results indicate that the level of pollen flow between populations is negatively correlated with the geographical distance between populations, which may have caused the isolation-by-distance pattern for nDNA. An isolation-by-distance pattern has also been detected at nSSR in Fagus sylvatica (Buiteveld et al. 2007).
The AMOVA approach (Excoffier et al. 1992) has been widely used for the hierarchical analysis of the genetic diversity in a set of sampled populations. A physical or ecological criterion is often used to define a priori groups of populations on which a test of genetic structure is applied. Where no obvious criterion exists for the definition of groups of populations, the investigation of the genetic structure in a set of populations may be difficult. In such cases, algorithms such as SAMOVA can help to define the strongest structure of populations in genetic terms (Dupanloup et al. 2002). The ability of SAMOVA to define groups in which all the populations are not geographically adjacent (i.e. the samples in center, S-1900, is associated with samples from West of Hyrcanian zone, A-600) can thus allow identification of some groups of populations that are particularly important for conservation purposes.
The two clustering patterns, 6 groups using cpSSR and 3 groups using nSSR, show the genetic peculiarities of beech populations in the Hyrcanian zone, but lead to quite different groups of populations. The SAMOVA approach based on cpSSR data suggests the association of the populations from the eastern part of Hyrcanian zone (from regions Gorgan, Neka and Sangdeh). This result is in agreement with the hypothesis of a migration history of oriental beech from West (most polymorphic) to East of the Hyrcanian forests and a more recent expansion of beech in the eastern part of this range. The SAMOVA approach also groups close populations in different clusters, e.g., populations from different elevation of region Asalem, Kheirud and Neka. This may be due to the presence of very efficient barriers to seed flow that would have prevented short-range migrations between populations from these regions. Differences between the genetic structure identified by SAMOVA using nSSR and cpSSR markers is strongly associated with the amount of migrants exchanged between populations (seed and pollen) and confirms the important role played by natural barriers to gene flow (i.e. altitude).
Conclusion
Genetic diversity was high for all oriental beech populations. This study shows that a comparative analysis of the distribution of genetic variation using differently inherited markers allows a more in-depth understanding of genetic structure in relation to both ancient and more recent events. This information is typically scarce and very much needed for planning sound conservation programs. The higher inbreeding observed in the more intensively managed stands is most likely because of disturbance such as harvesting, which may have changed the long-term genetic composition in these populations. Only through the understanding of the changes that occur as a result of forest management (e.g., changes in breeding neighbourhood sizes as a result of the cutting of seed trees) is it possible to plan genetic diversity conservation in tree populations, a crucial step for sustainable resource management. Moreover, the definition of genetic zones based on molecular markers can be extremely useful for the certification of seed-lots and for the identification of suitable seed sources and/or autochthonous stands, greatly improving the processes controlling commercial uses of seeds, plants or wood. In general these data provide a scientific basis for formulation of programs of conservation of genetic resources of this species.
References
Aguinagalde I, Hampe A, Mohanty A, Martin JP, Duminil J, Petit RJ (2005) Effects of life-history traits and species distribution on genetic structure at maternally inherited markers in European trees and shrubs. J Biogeog 32(2):329–339
Anonymous (2000) Iran statistical yearbook. Statistical Center of Iran, Iran
Birky CW, Fuerst P, Maruyama T (1989) Organelle gene diversity under migration, and drift: equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to unclear genes. Genetics 121:63–627
Buiteveld J, Vendramin GG, Leonardi S, Kamer K, Geburek T (2007) Genetic diversity and differentiation in European beech (Fagus sylvatica L.) stands varying in management history. For Ecol Manage 247:98–106
Caron H, Dumas S, Marque G (2000) Spatial and temporal distribution of chloroplast DNA polymorphism in a tropical tree species. Mol Ecol 9:1089–1098
Chapuis M, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24(3):621–631
Comps B, Thiébaut B, Merzeau D (1991) Genetic variation in European beech stands. In: Müller-Starck G, Ziehe M (eds) Genetic variation in European populations of forest trees. Sauerländer's Verlag, Frankfurt am Main, pp 110–124
Demesure B, Comps B, Petit RJ (1996) Chloroplast DNA phylogeography of the European beech (Fagus sylvatica L.) in Europe. Evolution 50(6):2515–2520
Dow BD, Ashley MV (1996) Microsatellite analysis of seed dispersal and parentage of saplings in bur oaks, Quercus macrocarpa. Mol Ecol 5:615–627
Duminil J, Caron H, Scotti I, Cazal S, Petit RJ (2006) Blind population genetics survey of tropical rainforest trees. Mol Ecol 15(12):3505–3513
Dumolin-Lapègue S, Demesure B, Fineschi S, Le Corre V, Petit RJ (1997) Phylogenetic structure of white oaks throughout the European continent. Genetics 146:1475–1487
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define genetic structure of populations. Mol Ecol 11:2571–2581
Dutech C, Maggia L, Joly HI (2000) Chloroplast diversity in Vouacapoua americana (Caesalpiniaceae), a neotropical forest tree. Mol Ecol 9:1427–1432
Ennos RA (1994) Estimating the relative rates of pollen and seed migration among plant populations. Heredity 80:548–593
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fishwick RW (1972) The Caspian forests of Iran. Commonwealth Forestry Rev 51:295–306
Geburek T (1997) Isozymes and DNA markers in gene conservation of forest trees. Biodivers Conserv 6:1639–1654
Habibi H (1985) Investigation on soil of beech forests in Iran and its role on development of different type of beech forests. Iranian J Natu Res 39:6–18
Hamilton MB (1999) Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Mol Ecol 8:521–523
Hardy OJ, Vekemans X (2002) SPAGEDI: a versatile computer program to analyze spatial genetic structure at the individual or population level. Mol Ecol Notes 2:618–620
Hawley GJ, Schaberg PG, DeHayes DH, Brissette JC (2005) Silviculture alters the genetic structure of an eastern hemlock forest in Maine, USA. Can J For Res 35:143–150. doi:10.1.1391x04-148
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution 59:1633–1638
Hosseini S, Madjnonian B, Nieuwenhuis M (2000) Damage to natural regeneration in the Hyrcanian forests in Iran: a comparison of two typical timber extraction operations. J Forest Engineer 11:69–73
Hu XS, Ennos R (1997) On estimation of the ratio of pollen to seed flow among plant populations. Heredity 79:541–552
Isagi Y, Kanazashi T, Suzuki W, Tanaka H, Abe T (2000) Microsatellite analysis of the regeneration process of Magnolia obovata Thunb. Heredity 84:143–151
König AO, Ziegenhagen B, van Dam BC, Csaikl UM, Coart E, Degen B, Burg K, de Vries SMG, Petit RJ (2002) Chloroplast DNA variation of oaks in western central Europe and genetic consequences of human influences. For Ecol Manage 156:147–166
Lee CT, Wickneswari R, Mahani MC, Zakri AH (2002) Effect of selective logging on the genetic diversity of Scaphium macropodurn. Biol Conserv 104:107–118. doi:10.1016lS0006-3207(01)00159-8
Lian C, Oishi R, Miyashita N, Nara K, Nakaya H, Wu B, Zhou Z, Hogetsu T (2003) Genetic structure and reproduction dynamics of Salix reinii during primary succession on Mount Fuji, as revealed by nuclear and chloroplast microsatellite analysis. Mol Ecol 12:609–618
Lu SY, Peng CI, Cheng YP, Hong KH, Chiang TY (2001) Chloroplast DNA phylogeography of Cunninghamia konishii (Cupressaceae), an endemic conifer of Taiwan. Genome 44:797–807
Marchelli P, Gallo L, Scholz F, Ziegenhagen B (1998) Chloroplast DNA markers reveal a geographical divide across Argentinean southern beech Nothofagus nervosa (Phil.) Dim. et Mil. distribution area. Theor Appl Genet 97:642–646
Marquardt PE, Echt CS, Epperson BK, Pubanz DM (2007) Genetic structure, diversity, and inbreeding of eastern white pine under different management conditions. Can J For Res 37:2652–2662
Marvie-Mohadjer MR (1976) Some quantitative characteristics of Iranian beech forests. Iranian J Natu Res 34:77–97
Meirmans PG (2006) Using the AMOVA framework to estimate a standardised genetic differentiation measure. Evolution 60:2399–2402
Michalakis Y, Excoffier L (1996) A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142:1061–1064
Mobayen S, Tregubov V (1969) The vegetative map of Iran, No 14. Tehran University, Tehran, p 50
Mohanty A, Martin JP, Aguinagalde I (2000) Chloroplast DNA diversity within and among populations of the allotetraploid Prunus spinosa L. Theor Appl Genet 100:1304–1310
Murawski DA, Gunatilleke IAUN, Bawa KS (1994) The effects of selective logging on inbreeding in Shorea megistophylla (Dipterocarpaceae) from Sri Lanka. Conserv Biol 8:997–1002
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590
Newton AC, Allnutt TR, Gillies ACM, Lowe AJ, Ennos RA (1999) Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol 14:140–145
Parsapajouh D (1976) Research on physical characteristics of Iranian beech timbers in different growing stations. Iranian J Natu Res 34:21–34
Pastorelli R, Smulders MJM, Van’T Westender WPC, Vosman B, Giannini R, Vettori C, Vendramin GG (2003) Characterization of microsatellite markers in Fagua sylvatica L. and Fagus orientalis Lipsky. Mol Ecol Notes 3:76–78
Peakal R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Petit RJ, Kremer A, Wagner DB (1993) Finite island model for organelle and nuclear genes in plants. Heredity 71:630–641
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Sagheb-Talebi K, Schütz JP (2002) The structure of natural oriental beech (Fagus orientalis) in the Caspian region of Iran and potential for the application of the group selection system. Forestry 75(4):465–472
Salehi Shanjani P (2002) Genetic diversity of Oriental beech (Fagus orientalis Lipsky) and its relations with some of physiology, biochemistry and morphology characteristics in Iran. Dissertation, Teacher Training University
Salehi Shanjani P, Paule L, Khavari-Nejad RA, Gömöry D, Sagheb-Talebi K (2002) Allozymic variability in beech (Fagus orientalis Lipsky) forests over Hyrcanian zone. For Genet 9(4):297
Salehi Shanjani P, Vettori C, Giannini R, Khavari-nejad RA (2004) Intraspecific variation and geographic patterns of Fagus orientalis Lipsky chloroplast DNA. J Silveae Genet 53:193–197
Sebastiani F, Carnevale S, Vendramin GG (2004) A new set of mono- and di-nucleotide chloroplast microsatellites in Fagaceae. Mol Ecol Notes 4:259–261
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139:457–462
Štambuk S, Sutlović D, Bakarić P, Petricevic S, Andelinovic S (2007) Potential usefulness of microsatellite-based genotyping of Croatian olive (Olea europaea L.) in forensic casework. Croat Med J 48:434–441
Streiff R, Labbe T, Bacillieri R, Steinkellner H, Glössl J, Kremer A (1998) Within-population genetic structure in Quercus robur L. and Q. petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7:317–328
Thomas BR, Macdonald SE, Hicks M, Adams DL, Hodgetts RB (1999) Effects of reforestation methods on genetic diversity of lodgepole pine: an assessment using microsatellite and randomly amplified polymorphic DNA markers. Theor Appl Genet 98:793–801. doi:10.1007/s001220051136
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Weising K, Gardner RC (1999) A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 42:9–19
Wright S (1943) Isolation by distance. Genetics 28:114–138
Wright S (1951) The genetic structure of populations. Ann Eugen 15:323–354
Yazdani R, Muona O, Rudin D, Szmidt AE (1985) Genetic structure of a Pinus sylvestris L. seed-tree stand and naturally regenerated understory. For Sci 31:430–436
Acknowledgments
This project was supported by the International Plant Genetic Resource Institute, Bioversity International, formerly IPGRI (project D06C Fellowships). Parvin Salehi Shanjani received Vavilov-Frankle Fellowshio (2003) from Bioversity International, formerly IPGRI.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Salehi Shanjani, P., Vendramin, G.G. & Calagari, M. Genetic diversity and differentiation of Fagus orientalis Lipsky in Hyrcanian forests revealed by nuclear and chloroplast microsatellite markers. Conserv Genet 11, 2321–2331 (2010). https://doi.org/10.1007/s10592-010-0118-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0118-4