
Abstract
Zeaxanthin is a high-value carotenoid that is used in nutraceuticals, cosmetics, food, and animal feed industries. Zeaxanthin is chemically synthesized or purified from microorganisms as a natural product; however, increasing demand requires development of alternative sources such as heterologous biosynthesis by recombinant bacteria. For this purpose, we molecularly engineered Escherichia coli to optimize the synthesis of zeaxanthin from lycopene using fusion protein-mediated substrate channeling as well as by the introduction of tunable intergenic regions. The tunable intergenic regions approach was more efficient compared with protein fusion for coordinating expression of lycopene β-cyclase gene crtY and β-carotene 3-hydroxylase gene crtZ. The influence of the substrate channeling effect suggests that the reaction catalyzed by CrtZ is the rate-limiting step in zeaxanthin biosynthesis. Then Pantoea ananatis, Pantoea agglomerans and Haematococcus pluvialis crtZ were compared. Because P. ananatis crtZ is superior to that of P. agglomerans or H. pluvialis for zeaxanthin production, we used it to generate a recombinant strain of E. coli BETA-1 containing pZSPBA-2(P37-crtZPAN) that produced higher amounts of zeaxanthin (11.95 ± 0.21 mg/g dry cell weight) than other engineered E. coli strains described in the literature.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Carotenoids are a class of terpenoid pigments with multiple physiological and nutritional functions in numerous organisms. It contains C30, C40 and C50 carotenoids. Carotenoids are produced by the food, medical, and cosmetic industries for use as pigments as well as for their potential beneficial effects on human health [34]. Zeaxanthin (3,3′-dihydroxyl-β-carotene) is a yellow oxygenated carotenoid composed of 40 carbon atoms that is used as a food additive and a feed additive for fish (color enhancement for the flesh) and poultry (yolk and skin pigmentation) [27, 34]. Zeaxanthin plays a critical role in preventing age-related macular degeneration [25, 38] and cancer [28] and may protect against age-related cataract formation [25]. Because of increasing market demand [5], it is important to develop new methods for producing zeaxanthin.
Zeaxanthin is found naturally in yellow vegetables and fruits such as corn, orange peppers, mangoes, pink grapefruit, apricots, peaches, cantaloupe, avocados, and marigold flowers [34]. However, plants cannot be used to deliver a stable supply of zeaxanthin, because the costly production method generates low yields. To avoid shortages and price fluctuations, a stable source of affordable zeaxanthin is required. Chemically synthesized zeaxanthin can be produced on a commercial scale; however, it is only used as a pigment and not as a food additive or pharmaceutical because of its low activity [34]. Microorganisms provide an alternative and attractive source of zeaxanthin. For example, zeaxanthin is produced by microorganisms such as the microalgae Dunaliella salina [19] and Chlorella saccharophila [37], the marine bacteria Flavobacterium multivorum [8], Synechocystis sp. strain PCC 6803 [21], Paracoccus zeaxanthinifaciens sp. nov. [6], Sphingomonas natatoria [39], Muricauda sp. YUAB-SO-11 and YUAB-SO-45 [31], Mesoflavibacter zeaxanthinifaciens [3], and Zeaxanthinibacter enoshimensis [4].
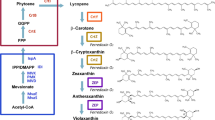
Metabolic engineering is a powerful technique used to improve the metabolic activity of microorganisms. Heterologous expression of carotenoid genes in E. coli to produce specific carotenoids is a particularly attractive alternative. Generic scheme for zeaxanthin synthesis is presented in Fig. 1. In E. coli, the isoprenoid precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are synthesized from pyruvate and glyceraldehyde-3-phosphate via the 2-C-methyl-d-erythritol 4-phosphate pathway. Lycopene production by E. coli is achieved by introducing the crtE, crtB, and crtI genes. The cyclization of lycopene leads to the formation of β-carotene by lycopene β-cyclase (encoded by crtY). The hydroxylation of each ring of β-carotene at the C-3 position by β-carotene hydroxylase encoded by crtZ produces zeaxanthin [34]. There are a few papers on the metabolic engineering of E. coli to produce zeaxanthin [1, 2, 24, 29, 33]. Albrecht et al. [2] reported the highest level of zeaxanthin production of 1.6 mg/g dry cell weight (DCW). However, this value is one-third of that produced by the zea1 strain of the photosynthetic microalga Dunaliellasalina (6 mg zeaxanthin/g DCW) [19]. Thus, further research must be conducted to increase zeaxanthin yields of E. coli.

Zeaxanthin biosynthetic pathway. dxs, E. coli 1-deoxyxylulose-5-phosphate synthase gene; dxr, E. coli 1-deoxy-d-xylulose 5-phosphate reductoisomerase gene; ispH, E. coli 4-hydroxy-3-methylbut-2-enyl diphosphate reductase gene; idi, E. coli IPP isomerase gene; ispA: geranyltranstransferase gene; crtE, P. ananatis geranylgeranyl diphosphate synthase gene; crtB, P. ananatis phytoene synthase gene; crtI, P. ananatis phytoene desaturase gene; crtY, lycopene β-cyclase gene; crtZ, β-carotene 3-hydroxylase gene
In the present study, we optimized the biosynthetic pathway leading from lycopene to zeaxanthin in a recombinant strain of E. coli to produce 11.95 mg/g DCW of zeaxanthin, which is the highest value reported to our knowledge.
Materials and methods
Strains, primers and plasmids
The strains and plasmids used in this study are listed in Table 1. Escherichia coli DH5α was used for plasmid construction. Escherichia coli CBW 12241(ΔiclR, PT5-appY), designated E. coli LYCOP [9], was used as the starting strain. The primers used in this study are listed in Supplementary Table 1.
Construction of the expression plasmid
The detailed procedure used to construct the Biobrick vectors pZSBP and pZSABP is shown in Fig. 2. The plasmid pMD-BP was prepared by Shengong, Inc (Shanghai, China) by synthesizing and ligating the sequence 5′-ACGCGTCCTAGGGAATTCAAAAGATCTCTTACATGAAAAAGGTTCTTGACATTTTAAATCCATGTGGTATATGTCATTTTTCTAGCTAGCAAAGCATGCGAGCTTGGTACCGGGCCCAGCCCCGGGGCGGCCGCTAGTACTCTGCAGGGCGATATCGGATCCAAATCTAGAGTCGACGTGCACGACGTC-3′ to pMD-18 (simple). The sequence comprises the strong promoter P37 flanked by BglII/NheI sites, respectively, as well as the multiple cloning site (MCS; SphI, KpnI, ApaI, SmaI, NotI, ScaI, PstI, and EcoRV). The terminator was amplified from pEC-XK99E [20] using primers rrnF and rrnR. The PCR product was digested using EcoRI and SalI and ligated into the EcoRI/SalI sites of pMD-BP to generate pMD-rrnB. The terminator fragment was excised from pMD-rrnB using BglII and SalI and ligated to BamHI-SalI-digested pMD-BP to generate pMD-BP-rrnB. The pSC101 origin of replication was amplified from pZS21MCS [23] using the primers oriF and oriR. The PCR product was digested using EcoRI and SalI. The DNA fragment containing the P37 promoter, MCS, and terminator was excised from pMD-BP-rrnB using EcoRI and SalI. The above two fragments were ligated to generate pZSBP. The amp gene was amplified from pMD-BP and inserted into the MluI/SacI of pZSBP to generate pZSABP. The plasmids pZSBP and pZSABP were used to assemble the components of the BglBrick vector [22] and ePathBrick vectors [41].

Construction of the Biobrick vectors pZSBP and pZSABP
To increase translation efficiency [35], the optimized sequences (TTTCGGAATTAAGGAGGTAATAAAT) of the 5′-untranslated regions (5′-UTR) were designed using UTR Designer (http://sbi.postech.ac.kr/utr_designer) and were added upstream of the initial ATG codon of each gene. P. ananatis crtY and crtZ were amplified from P. ananatis LMG 20103 using primers dCrtY-F/dCrtY-R, dCrtZ-F/dCrtZ-R, respectively. The two PCR products were ligated to pQE30 to generate pQE-crtYZ.
To construct pQE-crtY-L-crtZ, a linker encoding (G4S)3 was inserted between the two genes, and the TAA stop codon of crtY was deleted. The crtY gene containing a segment of the linker was amplified from pQE-crtYZ using primers CrtY-F/CrtY-R and ligated to pQE30 to generate pQE-crtYL. The crtZ gene containing a segment of the linker was amplified from pQE-crtYZ using the primers CrtZ-F/dCrtZ-R and ligated to pQE-crtYL to generate pQE-crtY-L-crtZ.
After codon optimization for E. coli codon usage by the combined application of Optimizer [32] and UTR Designer [35], the optimized P. agglomerans and H. pluvialis crtZ genes (Supplementary Table 2 and Table 3) were synthesized by GENEWIZ, Inc (Suzhou, China) and ligated to pUC57 to obtain pUC-crtZPAG and pUC-crtZHP, respectively. The genes were ligated to the ApaI/SmaI sites of pZSAPB to generate pZSAPB-crtZPAG and pZSAPB-crtZHP, respectively. P. ananatis crtZ was amplified from pQE-crtYZ using primers crtZ-F1 and dcrtZ-R and ligated to pBAD33 or pZSAPB to generate pBAD-crtZPAN or pZSAPB-crtZPAN. The plasmids pZSAPB-2(P37-crtZPAN), pZSAPB-2(P37-crtZPAG), or pZSAPB-2(P37-crtZHP) were constructed from pZSAPB-crtZPAN, pZSAPB-crtZPAG, or pZSAPB-crtZHP, respectively, using the standard BglBrick cloning strategy with compatible BglII and BamHI sites.
Construction and screening of libraries of tunable intergenic regions (TIGRs)
TIGRs were synthesized using PCR to assemble oligonucleotides into chimeric DNA sequences as described by Pfleger et al. [30]. Briefly, 40 mmols of an equimolar oligonucleotide (A, B, C and D in Supplementary Table 1) mixture was added to a mixture containing 2.5 units of Primer Star DNA Polymerase (Takara, Dalian, China). The assembly was conducted over 35 cycles of PCR for 10 s at 98 °C, 30 s at 72 °C, and 20 + 5 s/cycle at 72 °C. The assembly products were purified using a nucleotide removal column and amplified using the end-specific primers TIGRS-FW and TIGRS-RW. The amplified libraries were subcloned into the SacI/SalI sites of pQE-crtYZ, E. coli DH5α was electroporated with the ligation products, and the resulting transformants were plated on LB agar containing ampicillin. The transformants were collected from agar plates in 5 ml of PBS.
The plasmid library containing TIGRs was isolated from the above E. coli library and transformed into E. coli LYCOP. Then the resulting transformants were plated on LB agar with ampicillin and isopropyl β-D-1-thiogalactopyranoside (IPTG). The plates were incubated at 37 °C overnight. Deep yellow colonies were inoculated into a 4.6 mL deep-well microplate and then incubated for 48 h at 37 °C and shaken at 200 rpm. Protein expression was induced by adding 2 mmol/L IPTG 6 h after inoculating the microplates.
Chromosomal integration
To replace the promoter of the integration expression plasmid pHKKT5b [17], the P37 promoter was excised from pZSABP using MluI and SphI and then inserted into the MluI/SphI sites of pHKKT5b to generate pHKKP37b. P. ananatis crtY was excised from pQE-crtYZ using EcoRI/SacI and inserted into the EcoRI/SacI sites of pHKKP37b to generate pHKKP37b-crtY, which was integrated into the chromosome of the lycopene producer E. coli LYCOP by direct transformation as described previously [9, 11, 17]. These manipulations generated the β-carotene producer E. coli BETA-1.
Zeaxanthin production
Starter cultures of E. coli containing 5 mL of LB medium supplemented with 5 g/L KAc were incubated overnight at 37 °C and used to inoculate (initial optical density at 600 nm [OD600] = 0.1) a 250-mL Erlenmeyer flask containing 50 mL of SBMSN medium supplemented with 5 g/L KAc. SBMSN medium (pH 7.0) contained 12 g/L peptone, 24 g/L yeast extract, 1.7 g/L KH2PO4, 11.42 g/L K2HPO4, 1 g/L MgCl2·6H2O, 1.42 g/L ammonium oxalate, and 2 g/L Tween-80. The cultures were incubated at 37 °C for 48 h in a rotary shaking incubator set to 150 rpm. Cell growth was measured according to the OD600 and converted into DCW (g/L) using a standard curve.
Quantitative real-time PCR (qRT-PCR)
Total RNA from E. coli cells grown for 36 h in shake flasks was isolated using an RNA extraction kit (Dongsheng Biotech, Guangzhou, China), following the manufacturer’s instructions. The first-strand cDNA was synthesized using an All-in-One™ First-Strand cDNA Synthesis Kit (GeneCopoeia, Guangzhou, China). The qRT-PCR was performed with the All-in-One™ qPCR Mix kit (GeneCopoeia) on an iCycler iQ5 Real-Time PCR system (Bio-Rad Laboratories, California, USA). The template was 100 ng of cDNA. The PCR conditions were: 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s, and extension at 72 °C for 15 s. The primers for qRT-PCR are presented in Supplementary Table 1. pQE-crtYZ was used as standard templates for qPCR of the crtY and crtZ genes. Absolute gene copy numbers for each gene were obtained from standard curves.
Extraction and measurement of zeaxanthin
Cells were extracted with acetone to isolate carotenoids as described previously [9]. Samples of E. coli cultures (250 µL) were harvested by centrifugation at 12,000 rpm for 5 min. The cell pellet was washed with water and extracted with 1 mL of acetone at 55 °C for 15 min with intermittent vortexing. The mixture was then centrifuged at 12,000 rpm for 10 min, and the acetone supernatant was transferred to a new tube. Carotenoids were analyzed using a Shimadzu HPLC system (LC-20A, Shimadzu, Japan) equipped with an Inertsil ODS-SP column (5 μm, 4.6 × 150 mm, GL Sciences Inc, Tokyo, Japan). The mobile phase was acetonitrile-methanol (65:35 v/v) at a flow rate of 1 mL/min. The absorbance of carotenoids at 454 nm was detected using a photodiode array detector (SPD-M20A). The column was calibrated with commercially available zeaxanthin, β-carotene, and lycopene (Sigma-Aldrich, St. Louis, MO, USA).
Enzymatic activity assay
Escherichia coli cells grown for 36 h in shake flasks were harvested by centrifugation during the exponential phase, and washed with 10 mmol/L TE buffer (pH 7.5). Cells were sonically disrupted. Cell debris was removed by centrifugation at 5,000×g and 4 °C for 15 min. The resulting crude extracts were used for enzymatic measurement. The CrtY or CrtZ activity assay mixture (final volume 1 mL) contained 4 μg lycopene or beta-carotene, 0.75 mmol/L ATP, 0.125 mmol/L FeSO4, 1.25 mmol/L ascorbic acid and the crude enzyme extracts (approx. 500 μg of protein) in 100 mmol/L Tris–HCl buffer (pH7.5). These mixtures were incubated by shaking in the dark for 12 h at 30 °C. They were terminated by adding 1 mL methanol containing 6 % (w/v) KOH and heated at 60 °C for 20 min. The remaining substrate and the products formed were extracted from the aqueous incubation mixture with petroleum ether (b.p. 30–60 °C). β-carotene was analyzed by HPLC. CrtY or CrtZ activity was expressed as the formation rate or consumption rate of β-carotene per mg of protein under the above conditions, respectively.
Statistical analysis
All experiments were conducted in triplicate, and the data were averaged and are presented as the mean ± standard deviation. One-way analysis of variance followed by Tukey’s test was used to determine significant differences using the OriginPro (version 7.5) package. Statistical significance was defined as P < 0.05.
Results
In our previous paper [9], we constructed E. coli CBW 12241(ΔiclR, PT5-appY) using triclosan-induced chromosomal evolution, designated E. coli LYCOP, which produces high levels of lycopene (33.43 mg/g DCW). Therefore, we transformed E. coli LYCOP with the expression vector pQE30-crtYZ containing P. ananatis crtY and crtZ and measured zeaxanthin production. We were surprised to find that the resulting strain E. coli LYCOP (pQE-crtYZ) produced β-carotene but not zeaxanthin (Fig. 3a).

HPLC analysis of carotenoid products extracted from a E. coli LYCOP (pQE-crtYZ), b E. coli LYCOP (pQE-crtY-L-crtZ), c E. coli LYCOP (pQE-crtY-TIGR11-crtZ), and d E. coli LYCOP (pQE-crtY-TIGR11-crtZ, pBAD-crtZ)
Wang et al. [40] exploited the substrate channeling effect to metabolically engineer α-farnesene production. Other applications of this technique are reviewed by Zhang [42]. Moreover, substrate channeling occurs naturally in enzyme complexes such as pyruvate dehydrogenase [18]. To determine whether the failure to produce zeaxanthin was caused by unbalanced expression of crtY and crtZ, we generated a recombinant E. coli strain designated LYCOP (pQE-crtY-L-crtZ) that expresses a CrtY–CrtZ chimera. We anticipated that substrate channeling through the fusion protein would deliver β-carotene to CrtZ by preventing the intermediate to diffuse freely. E. coli LYCOP (pQE-crtY-L-crtZ) produced 1.54 mg/g DCW of zeaxanthin (Fig. 3b). This result suggests that the expression of a chimeric protein comprising CrtY and CrtZ, which were fused with a (G4S)3 linker, coordinated the expression of the two enzymes to produce zeaxanthin from lycopene.
Pfleger et al. [30] developed a combinatorial engineering approach for coordinating expression of cascade enzymes. For this purpose, libraries of TIGRs are generated that encode mRNAs with diverse secondary structures with RNase cleavage sites. They demonstrated that the TIGRs approach balances the expression of three genes in an operon that encodes a heterologous mevalonate biosynthetic pathway and increases mevalonate production by a factor of seven. We used this approach to construct a library of TIGRs for balancing the expression of P. ananatis crtY and crtZ. The library of TIGRs was inserted between crtY and crtZ to generate a series of operons that we screened for increased zeaxanthin production. The functional operons from the libraries were selected by transforming E. coli LYCOP with the plasmid library, and yellow colonies were chosen for analysis of carotenoid production.
Out of 100 yellow colonies, the highest yield of zeaxanthin (1.84 mg/g DCW) was generated by E. coli LYCOP (pQE-crtY-TIGR11-crtZ) (Fig. 3c), which was greater than that of the CrtYZ fusion strain E. coli LYCOP (pQE-crtY-L-crtZ) (P < 0.05). This finding indicates that the TIGR approach is more efficient than protein fusion for coordinating the expression of crtY and crtZ.
Although the protein fusion and TIGR approaches produced zeaxanthin, the respective strains produced β-carotene as the predominant product (Fig. 3b, c), suggesting that CrtZ may be the key enzyme for synthesizing zeaxanthin. Therefore, E. coli LYCOP (pQE-crtY-TIGR11-crtZ) was transformed with pBAD-crtZ. As we expected, the resulting strain E. coli LYCOP (pQE-crtY-TIGR11-crtZ, pBAD-crtZ) produced more zeaxanthin (2.72 mg/g DCW) (Fig. 3d). Moreover, when P. ananatis crtY was integrated into the chromosome of E. coli LYCOP chromosome to generate E. coli BETA-1, the latter converted all the lycopene into β-carotene (Fig. 4a). Therefore, the reaction catalyzed by CrtZ is the rate-limiting step in zeaxanthin biosynthesis.

HPLC analysis of carotenoid products extracted from a E. coli BETA-1, b E. coli BETA-1 [pZSAPB-2(P37-crtZPAN)], c E. coli BETA-1 [pZSAPB-2(P37-crtZPAG)], and d E. coli BETA-1 [pZSAPB-2(P37-crtZHP)]
To investigate the effect of the CrtY-CrtZ chimera on zeaxanthin production, the mRNA copy numbers of the crtY and crtZ genes and enzymatic activities in the strains were determined. The results are presented in Table 2. Table 2 shows that fusion protein-mediated and TIGR-mediated substrate channeling results in the increases of the transcription levels of the crtY and crtZ genes. The expression levels of the crtY and crtZ genes in E. coli LYCOP (pQE-crtY-L-crtZ) are about 10 and 42 times higher than those in E. coli LYCOP (pQE-crtYZ). The expression levels of the crtY and crtZ genes in E. coli LYCOP (pQE-crtY-TIGR11-crtZ) are about 121 and 476 times higher than those in LYCOP (pQE-crtYZ). The activities show the similar trend to the transcription levels in these strains.
To overcome the bottleneck imposed by CrtZ, we determined the effect of overexpressing codon-optimized P. ananatis, P. agglomerans, or H. pluvialis crtZ on zeaxanthin production. For this purpose, we constructed pZSAPB-2(P37-crtZPAN), pZSAPB-2(P37-crtZPAG), and pZSAPB-2(P37-crtZHP) using BglBrick assembly technology. Each plasmid contained two copies of the cognate crtZ under the control of the P37 promoter. Escherichia coli BETA-1 harboring pZSAPB-2(P37-crtZPAN) produced the highest yield of zeaxanthin. (11.95 ± 0.21 mg/g DCW, 43.46 ± 0.57 mg/L) (Table 3), and zeaxanthin was the predominant carotenoid (Table 3; Fig. 4b). However, the strains expressing P. agglomerans or H. pluvialis crtZ produced β-carotene as the predominant carotenoid (Table 3; Fig. 4c, d).
Discussion
After introducing a foreign metabolic pathway into host, it is essential that the relative levels of the enzymes be coordinated in such a way that no intermediate in the pathway accumulates to toxic levels. Substrate channeling is a powerful tool for balancing expression of genes. It is a process of transferring the product of one enzyme to an adjacent cascade enzyme. Such phenomena can occur in vivo, in vitro, or ex vivo. In addition to enhanced reaction rates through substrate channeling, numerous potential benefits include protection of unstable substrates, circumvention of unfavorable equilibrium and kinetics imposed, forestallment of substrate competition among different pathways, regulation of metabolic fluxes, mitigation of toxic metabolite inhibition, and so on [42]. In this study, we first compared the effect of fusion protein-mediated and TIGR-mediated substrate channeling on zeaxanthin production. This study demonstrated that the substrate channeling occurs in the zeaxanthin synthesis pathway from lycopene (Fig. 3a–c). Substrate channeling resulted in enhancing the transcription levels of the crtY and crtZ genes and their activities (Table 2). It had a stronger influence on the crtZ than the crtY. Pfleger et al. observed the same result. TIGR sequences showed a stronger influence on the expression of the gene 3′ to the TIGR than the gene 5′ to the TIGR [30]. The transcription level of the crtZ and its activity were much lower than those of the crtY in E. coli LYCOP (pQE-crtYZ), resulting in no zeaxanthin to be detected. Among E. coli LYCOP (pQE-crtYZ), E. coli LYCOP (pQE-crtY-L-crtZ), E. coli LYCOP (pQE-crtY-TIGR11-crtZ) and E. coli LYCOP (pQE-crtY-TIGR11-crtZ, pBAD-crtZ), zeaxanthin production was correlated with the transcription level of the crtZ and its activity. Our study proves that the TIGRs approach was more efficient compared with protein fusion for coordinating expression of crtY and crtZ and the reaction catalyzed by CrtZ is the rate-limiting step in zeaxanthin biosynthesis. These results also demonstrate that higher expression level of crtZ would be required for zeaxanthin production. It is consistent with that reported by Nishizaki et al. [29]. They thought that crtZ expression levels several times greater would be required for complete conversion to zeaxanthin [29].
After integration of the crtY under the control of P37 promoter into E. coli LYCOP chromosome, the predominant carotenoid was changed into β-carotene from lycopene (Fig. 4a). It indicates that the expression of one copy of the crtY is enough to concert lycopene to β-carotene.
Of the three CrtZ enzymes compared in this study, P. ananatis CrtZ was the most efficient for zeaxanthin production. Fraser et al. [12] reported that Erwinia uredovora (now classified as P. ananatis) CrtZ exhibited the highest for converting β-carotene to zeaxanthin compared with those of Agrobacterium aurantiacum and Alcaligenes sp. strain PC-1. The CrtZ activities of P. ananatis, Brevundimonas sp. SD212, Paracoccus sp. PC1, Paracoccus sp. N81106, marine bacterium P99-3, and P450 monooxygenase (CYP175A1) from Thermus thermophilus HB27 for converting β-carotene to zeaxanthin are not significantly different [10]. Of the β-carotene hydroxylases from P. ananatis, Chlorella zofingiensis, Arabidopsis thaliana, H. pluvialis, and tomato, H. pluvialis β-carotene hydroxylase coordinates most efficiently with β-carotene ketolase from Chlamydomonas reinhardtii to produce astaxanthin formation [16].
Zeaxanthin production by microorganisms is summarized in Table 4. The zeaxanthin yield achieved in the present study is higher by a factor of 7.5 than that previously reported using metabolically engineered E. coli [2]. Paracoccus zeaxanthinifaciens produces the highest yield of zeaxanthin (19.5 mg/g DCW) [36], which is higher by a factor of 1.6 than that achieved in this study. Therefore, further optimization of E. coli BETA-1 [pZSPBA-2(P37-crtZPAN)] is required.
In conclusion, the protein fusion and TIGR approaches were used successfully to balance the expression of the crtY and crtZ, although the TIGR approach is more efficient. The results of substrate channeling suggest that CrtZ catalyzes the rate-limiting step in zeaxanthin biosynthesis. Of P. ananatis, P. agglomerans and H. pluvialis crtZ, P. ananatis crtZ is the best suited for zeaxanthin production. The yield of zeaxanthin produced by E. coli BETA-1 harboring pZSPBA-2(P37-crtZPAN) is significantly higher compared with engineered strains of E. coli reported to date.
References
Albermann Trachtmann N, Sprenger GA (2010) A simple and reliable method to conduct and monitor expression cassette integration into the Escherichia coli chromosome. Biotechnol J 5(1):32–38
Albrecht M, Misawa N, Sandmann G (1999) Metabolic engineering of the terpenoid biosynthetic pathway of Escherichia coli for production of the carotenoids β-carotene and zeaxanthin. Biotechnol Lett 21(9):791–795
Asker D, Beppu T, Ueda K (2007) Mesoflavibacter zeaxanthinifaciens gen. nov., sp. nov., a novel zeaxanthin-producing marine bacterium of the family Flavobacteriaceae. Syst Appl Microbiol 30(4):291–296
Asker D, Beppu T, Ueda K (2007) Zeaxanthinibacter enoshimensis gen. nov., sp. nov., a novel zeaxanthin-producing marine bacterium of the family Flavobacteriaceae, isolated from seawater off Enoshima Island, Japan. Int J Syst Evol Microbiol 57(4):837–843
BCC R (2011) The Global Market for Carotenoids. http://www.marketresearch.com/BCC-Research-v374/Global-Carotenoids-6558319
Berry A, Janssens D, Humbelin M, Jore JPM, Hoste B, Cleenwerck I, Vancanneyt M, Bretzek W, Mayer AF, Lopez-Ulibarri R, Shanmugam B, Swings J, Pasamontes L (2003) Paracoccus zeaxanthinifaciens sp nov., a zeaxanthin-producing bacterium. Int J Syst Evol Microbiol 53:231–238
Beuttler H, Hoffmann J, Jeske M, Hauer B, Schmid RD, Altenbuchner J, Urlacher VB (2011) Biosynthesis of zeaxanthin in recombinant Pseudomonas putida. Appl Microbiol Biotechnol 89(4):1137–1147
Bhosale P, Larson AJ, Bernstein PS (2004) Factorial analysis of tricarboxylic acid cycle intermediates for optimization of zeaxanthin production from Flavobacterium multivorum. J Appl Microbiol 96(3):623–629
Chen YY, Shen HJ, Cui YY, Chen SG, Weng ZM, Zhao M, Liu JZ (2013) Chromosomal evolution of Escherichia coli for the efficient production of lycopene. BMC Biotechnol 13:6
Choi SK, Matsuda S, Hoshino T, Peng X, Misawa N (2006) Characterization of bacterial beta-carotene 3,3′-hydroxylases, CrtZ, and P450 in astaxanthin biosynthetic pathway and adonirubin production by gene combination in Escherichia coli. Appl Microbiol Biotechnol 72(6):1238–1246
Cui YY, Ling C, Zhang YY, Huang J, Liu JZ (2014) Production of shikimic acid from E. coli through chemically inducible chromosomal evolution and cofactor metabolic engineering. Microb Cell Factories 13:21
Fraser PD, Miura Y, Misawa N (1997) In vitro characterization of astaxanthin biosynthetic enzymes. J Biol Chem 272(10):6128–6135
Gierhart DL (1995) Zeaxanthin-containing compositions produced by flavobacterium multivorum. U.S. patient 5427783
Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177(14):4121–4130
Haldimann A, Wanner BL (2001) Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183(21):6384–6393
Huang JC, Zhong YJ, Liu J, Sandmann G, Chen F (2013) Metabolic engineering of tomato for high-yield production of astaxanthin. Metab Eng 17:59–67
Huang MT, Chen YY, Liu JZ (2014) Chromosomal engineering of Escherichia coli for the efficient production of coenzyme Q10. Chin J Chem Eng 22(5):559–569
Huang X, Holden HM, Raushel FM (2001) Channeling of substrates and intermediates in enzyme-catalyzed reactions. Ann Rev Biochem 70:149–180
Jin ES, Feth B, Melis A (2003) A mutant of the green alga Dunaliella salina constitutively accumulates zeaxanthin under all growth conditions. Biotechnol Bioeng 81(1):115–124
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104(1–3):287–299
Lagarde D, Beuf L, Vermaas M (2000) Increased production of zeaxanthin and other pigments by application of genetic engineering techniques to Synechocystis sp strain PCC 6803. Appl Environ Microbiol 66(1):64–72
Lee TS, Krupa RA, Zhang FZ, Hajimorad M, Holtz WJ, Prasad N, Lee SK, Keasling JD (2011) BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J Biol Eng 5:1–14
Lutz R, Bujard H (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25(6):1203–1210
Misawa N, Nakagawa M, Kobayashi K, Yamano S, Nakamura K, Harashima K (1990) Elucidation of the Erwinia uredovora carotenoid biosynthetic pathway by functional analysis of gene products expressed in Escherichia coli. J Bacteriol 172(12):6704–6712
Moeller SM, Jacques PF, Blumberg JB (2000) The potential role of dietary xanthophylls in cataract and age-related macular degeneration. J Am Coll Nutr 5:522–527
Mohamed I, Mearns AS, Fraser K, Hodgson R (2013) Biological production of zeaxanthin and carotenoid biosynthesis control. U.S. patient 8361743
Nelis JH, De Leenheer PA (1991) Microbial sources of carotenoid pigments used in foods and feeds. J Appl Bacteriol 70:181–191
Nishino H, Murakoshi M, Tokuda H, Satomi Y (2009) Cancer prevention by carotenoids. Arch Biochem Biophys 483(2):165–168
Nishizaki T, Tsuge K, Itaya M, Doi N, Yanagawa H (2007) Metabolic engineering of carotenoid biosynthesis in Escherichia coli by ordered gene assembly in Bacillus subtilis. Appl Environ Microbiol 73(4):1355–1361
Pfleger BF, Pitera DJ, Smolke CD, Keasling JD (2006) Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat Biotechnol 24(8):1027–1032
Prabhu S, Rekha PD, Young CC, Hameed A, Lin SY, Arun AB (2013) Zeaxanthin production by novel marine isolates from coastal sand of India and its antioxidant properties. Appl Biochem Biotechnol 171(4):817–831
Puigbò P, Guzmán E, Romeu A, Garcia-Vallvé S (2007) OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res 35:W126–W131
Ruther A, Misawa N, Boger P, Sandmann G (1997) Production of zeaxanthin in Escherichia coli transformed with different carotenogenic plasmids. Appl Microbiol Biotechnol 48(2):162–167
Sajilata MG, Singhal RS, Kamat MY (2008) The carotenoid pigment zeaxanthin—a review. Compr Rev Food Sci F 7(1):29–49
Seo SW, Yang JS, Kim I, Yang J, Min BE, Kim S, Jung GY (2013) Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab Eng 15:67–74
Shepherd D and Daseek J (1976) Production of zeaxanthin. U.S. patent 3951742
Singh D, Puri M, Wilkens S, Mathur AS, Tuli DK, Barrow CJ (2013) Characterization of a new zeaxanthin producing strain of Chlorella saccharophila isolated from New Zealand marine waters. Bioresour Technol 143:308–314
Snodderly DM (1995) Evidence for protection against age-related macular degeneration by carotenoids and antioxidant vitamins. Am J Clin Nutr 62(6 Suppl):1448S–1461S
Thawornwiriyanun P, Tanasupawat S, Dechsakulwatana C, Techkarnjanaruk S, Suntornsuk W (2012) Identification of newly zeaxanthin-producing bacteria isolated from sponges in the gulf of Thailand and their zeaxanthin production. Appl Biochem Biotechnol 167(8):2357–2368
Wang C, Yoon SH, Jang HJ, Chung YR, Kim JY, Choi ES, Kim SW (2011) Metabolic engineering of Escherichia coli for α-farnesene production. Metab Eng 13(6):648–655
Xu P, Vansiri A, Bhan N, Koffas MAG (2012) ePathBrick: a synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth Biol 1(7):256–266
Zhang YH (2011) Substrate channeling and enzyme complexes for biotechnological applications. Biotechnol Adv 29(6):715–725
Acknowledgments
We are grateful to the National Natural Science Foundation of China (Grant No. 30970089, 21276289), the Natural Science Foundation of Guangdong Province (No. S2011010001396), and the Key Project of the Scientific and Technical Innovation of Higher Education of Guangdong Province (2012CXZD0002) for financial support.
Conflict of interest
The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: Metabolic Engineering.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, XR., Tian, GQ., Shen, HJ. et al. Metabolic engineering of Escherichia coli to produce zeaxanthin. J Ind Microbiol Biotechnol 42, 627–636 (2015). https://doi.org/10.1007/s10295-014-1565-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1565-6