
Abstract
Alzheimer’s disease (AD) is a type of neurodegenerative malady that is associated with the accumulation of amyloid plaques. Metal ions are critical for the development and upkeep of brain activity, but metal dyshomeostasis can contribute to the development of neurodegenerative diseases, including AD. This review highlights the association between metal dyshomeostasis and AD pathology, the feasibility of rebalancing metal homeostasis as a therapeutic strategy for AD, and a survey of current drugs that action via rebalancing metal homeostasis. Finally, we discuss the challenges that should be overcome by researchers in the future to enable the practical use of metal homeostasis rebalancing agents for clinical application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metals can be can broadly classified into toxicological metals and biometals based on their physiological functions, or lack thereof [1]. Toxicological metals including lead, mercury, aluminum, and cadmium have no beneficial biological function and are toxic to the human body [1]. Interestingly, these metals are concentrated in the brain after absorption into the bloodstream [1]. Meanwhile, biometals are metal ions that are essential for normal body function, including brain function. Critically, metal dyshomeostasis has been linked to the etiology of a range of neurodegenerative diseases, including Alzheimer’s disease (AD) in particular [2, 3].
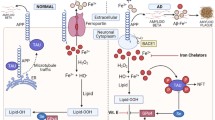
AD is a progressive neurodegenerative disease that afflicts almost 36 million individuals worldwide [4,5,6]. AD is recognized by the presence of amyloid-beta (Aβ) plaque deposits in the brain. Critically, the Aβ misfolding has been found to be significantly affected by the presence of metals, which are found in both the interior and periphery of established AD plaques [7,8,9,10]. Recently, there has been renewed attention given to the function of metal dyshomeostasis as a causative agent for AD. A number of key studies have shown that re-establishing proper metal ion balance in the brain can halt Aβ aggregation, dissemble amyloid plaques, and slow down AD-related cognitive decline in both AD patients and AD transgenic mice (Fig. 1) [11,12,13,14,15]. Here, we review the recent literature on the role of metal ion imbalance on the pathogenesis of AD. We also discuss new possible strategies for treating AD that rely on restoring metal balance in the brain. Finally, we provide our critical view and outlook of the challenges that must be overcome by researchers seeking to progress metal-targeting drugs into the clinic.

a Normal metal homeostasis is essential for healthy brain function. b Metal dyshomeostasis can lead to AD
Metal homeostasis in brain regulation
Metals such as zinc, iron and copper are essential for the biologically vital processes of the human body, including catalysis, stabilization of protein structure, signal transmission, and metabolism [16, 17]. Hence, living systems have developed intricate strategies for regulating the levels of ions and tightly control the entry, exit and storage of metal ions both within the cell and within sub-cellular organelles [18]. Metals can be imported into the brain through two pathways, either via the blood–brain barrier (BBB), or through the brain–cerebrospinal fluid barrier. Metals pass through the BBB in the form of free ions, or they can be carried by ion-specific transporters, or alternatively, they can traverse through the BBB by complexing with cysteine or other amino acids [19, 20]. Within the brain, metal ions are mostly concentrated within membrane metalloproteins or inside synaptic vesicles [21]. The concentration of metal ions can fluctuate in different areas of the brain [22] or subcellular compartments. Astrocytes in the brain help to maintain metal homeostasis [23,24,25], as they are the first kind of cell to interact with metals when they pass through the BBB. Also, astrocytes possess large amounts of metal-binding biomolecules, including metallothioneins and glutathione, which enable them to sequester metal ions and limit their neurotoxicity [26].
Due to the critical role of metals in the brain, the tight regulation of metal homeostasis in the brain is essential. For instance, the correct development of the brain is dependent on appropriate copper regulation [27], as dysfunction copper levels can trigger development defects and abnormalities of the brain. Copper and zinc also play important functions in neurotransmission in the central nervous system (CNS) [28]. Lastly, biometals regulate the potentiation of synapses in the long term, as well as the pharmacology of synaptic receptors [29].
The pathogenic hypothesis of AD
The “metal hypothesis” describes the theory that an imbalance in the levels and localization of metal ions contributes to AD pathology [30]. This theory posits that the binding of metals to Aβ can affect peptide conversion and promote its aggregation into fibrils, eventually forming plaques [30] (Fig. 2). In the amyloid precursor protein (APP) sequence, there are 2 or 4 metal-binding sites, depending on the metal ion recognized [31, 32]. APP has sites that are selective for binding to zinc and copper, and these sites also regulate redox activity to cause aggregation of Aβ even at low levels [33, 34]. Meanwhile, Aβ has both strong and weak-affinity selective sites for binding metals. They can bind equal ratios of zinc and copper, but copper can completely replace zinc in the binding site under acidosis [35, 36]. Aβ reduces metal ions by delivering electrons to O2, with the concomitant production of hydrogen peroxide.

The pathophysiology of AD, showing the production of Aβ monomers from APP, which aggregates into oligomers and eventually plaques
Interestingly, while the degree of deposits of Aβ is age related, there does not appear to be a relationship between Aβ production and age. This may suggest that other changes that are dependent on age, such as aberration of metal homeostasis, may instead be responsible for the pathogenesis of Aβ. As both zinc and copper and zinc modulate neurotransmission [30], deficiencies in metal balance can adversely affect transmission across synapses, via altering the reuptake or storages of metals in the synaptic cleft. Taken together, these data support the metal hypothesis of AD [30], which is based on the idea that the pathogenesis of AD is driven by the interaction of Aβ with particular metal ions. Additionally, a consensus has emerged on the view that transition metals can trigger oxidation stress, leading to another pathological factor driving AD. Accumulating evidence in the literature has ensured that the metal hypothesis of AD is now well established in the scientific community [37,38,39,40,41,42,43].
Metal dyshomeostasis and pathophysiology of AD
The deposition in the brain of Aβ plaques is a key hallmark of AD. The plaques consist of aggregated Aβ peptides ranging between 39 and 43 amino acids in length. These peptides are generated from the transmembrane precursor, APP [44]. Unbalanced metal ion concentrations can disrupt the normal activity of enzymes that regulate the cleavage of APP, leading to aberrant Aβ formation [45]. The 1–40 and 1–42 isoforms are the most abundant isoforms of Aβ. The latter isoform has a greater propensity for fibrillation and, thus, plaque formation [46, 47]. Amyloid fibrillation is reversible in the early stages (Fig. 2). However, whether neurotoxicity is primarily dependent on plaque formation or alternatively to monomeric Aβ toxicity, is still not fully known [48]. In contrast, some studies have shown the neuroprotective effect of monomeric Aβ [49]. Experiments have implicated oxidative stress in AD pathology [48], which could promote the apoptotic death of neurons either via caspase-3-dependent or independent pathways [50]. The presence of copper, iron, and zinc in Aβ aggregates has also been recently linked to neurotoxicity [51, 52]. Studies have shown that these metal ions can interact with and induce the aggregation of Aβ. Moreover, the redox activity of metal ions can trigger a cascade that leads to the production of reactive oxygen species (ROS) [53, 54]. Another mechanism that has been implicated in the pathogenesis of AD is the failure of the ubiquitin–proteasome system (UPS) [55, 56]. Metal dyshomeostasis can lead to the abnormal functioning of the UPS and trigger neurodegeneration via inhibiting self-polyubiquitination reactions [57]. Metal dyshomeostasis has also been suggested to induce CNS damage via modulating autophagy [55, 58,59,60,61,62]. In cellulo experiments also showed that biometal dyshomeostasis is connected to mitochondrial dysfunction [63, 64] and lysosomal storage disorders [65,66,67] in neurons. This suggests the possibility of targeting metal dyshomeostasis in the brain as a potential therapeutic approach.
Targeting metal dyshomeostasis
The putative role of metal dyshomeostasis in promoting the pathology of AD suggests that rebalancing metal homeostasis may be a potential strategy for AD therapy [68]. Metal chelators can sequester metal ions and remove them from the site of the lesion, thus preventing them from participating in redox chemistry or promoting Aβ aggregation. Meanwhile, other compounds can target specific compartments or organelles where metal ions are either relatively abundant or deficient [1]. The following sections discuss recent examples of compounds that target metal dyshomeostasis as potential therapeutic agents for AD.
Metal chelators
Metal chelators are molecules that can capture and bind metal ions via forming two or more coordinate bonds to a single metal ion. They can deplete the total pool of bioavailable metals extracellularly or compete with endogenous ligand as for metal ions as ionophores [1]. 8-Hydroxyquinolines (8HQ), a lipophilic molecule from plants, has been reported to exhibit potent anti-AD effects via chelating metal ions (Fig. 3) [25]. Clioquinol (CQ), a derivative of (8HQ), has entered into the phase II clinical trials for AD therapy [1]. PBT2, another drug for AD treatment in phase II clinical trials, was documented to promote Aβ degradation and phosphorylation of GSK3 through its metal chelator activity [69]. Deferiprone (DFP), an oral iron chelator that has been previously used for the treatment of thalassemia syndrome, exhibits neuroprotective action via alleviating the phosphorylation levels of Aβ and tau without affect the generation of ROS [70]. Trientine (TETA), a tetradentate chelator, inhibits β-secretase (BACE1) and slows amyloidosis via targeting the AGE/RAGE/NF-κB signaling cascade in a murine model of AD [71]. d-penicillamine, a Cu/Zn chelator used as a clinical drug for Wilson’s disease and rheumatoid arthritis [72,73,74], can delay AD via reducing serum oxidative stress [75]. Desferrioxamine (DFO), a preferential iron chelator, was shown to impede APP holoprotein translation via IRE in the 5′ UTR of the APP transcript and significantly improve some cognitive functions in AD patients injected intramuscularly [76]. The preclinical chelator DP-109 has been successfully tested on AD models. In 3-month mouse experiments with Tg2576 mice, DP-109 appeared to induce the increase the solubility of Aβ in the cerebrum, thereby decreasing the amount of Aβ aggregates [77]. Moreover, several metal chelating drugs (e.g., metformin and cyclodipeptides), already approved and used for other purposes, have been reported to be effectively repurposed for the treatment of neurodegenerative diseases, including AD [78].

Metal chelating agents for AD
Although several metal ion chelators have been approved for clinical use for metal overexposure diseases such as for Wilson’s disease [79], lead toxicity [80], and rheumatoid arthritis, to date no chelators have been approved for use in treating AD. Obstacles to the use of metal chelators are the potential adverse effects resulting from the removal of essential metal ions from the brain, as well as their poor BBB permeability due to their hydrophilic nature [61, 62]. Hence, further research is needed into the development of more selective metal ion chelators that can effectively enter the brain to target metal dyshomeostasis without causing systemic effects.
Aβ aggregation inhibitors
An increasing amount of evidence supports the hypothesis that metal ions alter the kinetic pathway of Aβ, directing its aggregation away from a more stable fibrillar structure and toward more neurotoxic structures [81]. It is also believed that fibrils and plaques during AD development result from metal binding to Aβ, causing its aggregation [30]. Thus, inhibition of Aβ aggregation is also a potential strategy to combat metal dyshomeostasis-induced AD.
Natural product-based Aβ aggregation inhibitors
Natural products are a source of chemical scaffolds with abundant activity profiles and moderate toxicity [82, 83]. Several natural products have been explored for their anti-AD activity (Fig. 4). Curcumin and its analogs such as calebin-A and dimethoxycurcumin have shown activity against fibril formation and extension, and promoted the destabilization of pre-aggregated Aβ peptides in SH-SY5Y cells and in vivo [84,85,86]. (−)-Epigallocatechin-3-gallate (EGCG), a polyphenolic constituent of green tea, has been studied for the prevention of age-related AD. A number of cell and animal experiments have shown that EGCG impeded Aβ-induced cell death at micromolar concentrations via decreasing soluble and insoluble levels of Aβ40 and Aβ42 and alleviating plaque load [87,88,89]. Bilobalide, a compound extracted from the Chinese medicinal plant Gingko biloba, reduced Aβ-stimulated apoptosis via downregulating ROS and blocking NF-kB activation [90, 91]. Ginkgolides A, B, C, J and M were also active against AD in part via anti-inflammatory and antioxidative properties [92, 93]. Ginkgolide A prevented Aβ-induced depolarization of cortical neurons by targeting the NMDA receptor [92]. Ginkgolide B reduced Aβ42-induced oxidative damage and restored the long-term antioxidant activities of enzymes in SH-SY5Y cells [94]. Meanwhile, ginkgolide J can enhance memory via inhibiting Aβ42-induced cell death in rodent hippocampal neurons [95].

Natural product-based Aβ aggregation inhibitors
Peptide-based Aβ aggregation inhibitors
Peptide inhibitors have also been used to develop potent pharmacological agents for AD treatment due to their high specificity, low toxicity, BBB permeability, and high chemical and biological diversity [96]. To date, many peptide-based aggregation inhibitors have been developed, which can be classified several subgroups based on their design principle (Fig. 5). Peptide fragments that consist of the hydrophobic core of Aβ [such as residues (15–22), (16–23), (17–24), (25–35)] or the C-terminal of Aβ [such as residues (25–35), (28–38), (39–42)] have been reported to block Aβ aggregation via binding to full-length Aβ [96,97,98]. Interestingly, the ability of the Aβ-derived peptide inhibitors to bind to Aβ and block its aggregation depends on their hydrophobicity, which helps them to incorporate into the β-sheet structure of Aβ. A metalloporphyrin–peptide conjugate is an effective inhibitor of amyloid‐β peptide fibrillation and cytotoxicity [99].

Classifications of peptide aggregation inhibitors of Aβ aggregation inhibitors
Alternatively, neuroprotective peptides that are not based on the Aβ sequence have also been reported. NAP (amino acid sequence: NAPVSIPQ), a peptide drug in phase II clinical trials, was documented to prevent formation of fibrils via abrogating the assembly and inducing disaggregation of Aβ [100]. A 12-mer peptide (PWRWQLWWHNWS), which was identified using the phage display technique, selectively bound to Aβ (1–10) and thus interdicted Aβ fibrillation by maintaining a steady-state equilibrium between monomeric Aβ monomer and soluble plaques, leading to an increase in the proportion of soluble Aβ [101]. An endogenous dipeptide, carnosine (Fig. 6), interacts with monomeric Aβ via impeding intermolecular salt bridge formation, thus blocking the aggregation event. This dipeptide significantly reduced Aβ accumulation and greatly relieved AD- and age-related mitochondrial dysfunction in a transgenic mice model of AD [102, 103].

Aβ sequences and their peptide inhibitors
Natural amino acid-containing Aβ fragments, while effectively inhibiting Aβ aggregation, themselves have high risk of self-associating into fibrils and also exhibit low resistance to cellular proteolytic enzymes [96]. To overcome these problems, many modified peptides have been developed. SEN304 (Fig. 6), a modified derivative of the KLVFF sequence, inhibited Aβ aggregation via directly binding to Aβ40 and Aβ42, retarded β-sheet formation, and induced formation of oligomers in a nontoxic form conformation [104]. SEN606, a derivative of SEN304, was reported to exhibit similar nanomolar activity in preclinical trials. AMY-1 and AMY-2, two peptide analogs of the hydrophobic interior of Aβ, bound avidly to fibrils and abrogated its further assembly via forming a blocking surface [105]. Meanwhile, tong-type mimic peptides (AFBP, AFBP-1, and AFBP-2) mimic the turn motif of Aβ oligomers and could form β-sheets with oligomerized Aβ and to inhibit Aβ aggregation [106].
Metal-based Aβ aggregation inhibitors
Transition metal complexes have also emerged as a viable alternative for the treatment of AD [107, 108]. Organometallic complexes can display great structural diversity of geometrical shapes via variations in assembly of the metal center and its co-ligands [83], allowing the effective targeting of the active sites of proteins or enzymes through shape-specific interactions [107]. The high-affinity metal binding site in Aβ peptides binds with several metal ions such as zinc, copper, and iron to mediate peptide aggression and toxicity [108, 109]. Therefore, occupying this targeting site on Aβ peptides may be a possible therapeutic strategy for AD treatment. Based on this idea, three platinum (Pt) complexes (complexes 1–3) containing phenanthroline ligands were developed as Aβ inhibitors [110] (Fig. 7). Another cisplatin-based complex 4 was found to interrupt the binding between Aβ16 peptides and Cu(II) via binding to the histidine imidazole moiety of Aβ16 [111]. Similarly, an Ru-based complex 5 was developed that could bind to both Aβ28 and Aβ42 via interacting with His-13 and His-14 [112]. Interestingly, a Pt/Ru dual metal core complex 6 also exhibited selective binding to Aβ and impeded amyloid fibril formation via binding to Aβ42 in a ratio of 2:1 [113]. Two Cu complexes containing bis(thiosemicarbazone) (7 and 8) have also been shown to reduce the progression of AD. Unlike the above metal complexes, these two metal complexes exhibited their bioactivity by reducing the oxidation state of the intracellular copper from Cu2+ to Cu+, thus abrogating their ability to bind to Aβ peptides [114]. Two group 9 metal-based complexes (9 and 10) were identified by our group which slowed the aggregation of Aβ40 via binding to His imidazole [107]. Interestingly, complex 10 (rhodium core) was more active than complex 9 (iridium core), suggesting that the interaction with the metal complex with Aβ is strongly metal dependent [107]. Another report described the ability of metal complexes with cyclam glycoconjugates to protect against metal dyshomeostasis-induced amyloid aggregation [115].

Metal-based Aβ aggregation inhibitors
Multifunctional agents against AD
It is believed that many factors contribute to the pathogenesis of AD. Pharmacological agents that target only one factor are often incapable of exerting sufficient therapeutic effect to reverse the progression of AD [116]. Thus, many multifunctional agents have been studied for anti-AD activity in cellulo or even in vivo (Fig. 8). Myricetin, a natural polyphenol, exhibited neuroprotection via regulating Aβ conformation and reducing the enzyme activity of secretases [117]. Donepezil is an inhibitor of Aβ aggregation and BACE1 enzyme, which activates the production of Aβ. Donepezil also binds to sigma-1 receptors, which have anti-amnesic activity [118,119,120]. Apart from natural products, many synthetic compounds have also been developed as multifunctional agents against AD. A multifunctional compound (4n) was produced by hybridizing coumarin, an acetylcholinesterase (AChE) and Aβ aggregation inhibitor, and dithiocarbamate, an AChE inhibitor. The compound exhibited much higher activity against AChE inhibition than either individual molecule and greatly reduced Aβ aggregation [121]. The combination of AChE inhibitors with an N-methyl-d-aspartic acid (NMDA) receptor antagonist is the current recommended standard for AD treatment [122]. A hybrid compound, memagal, formed from the hybridization of galantamine, AChE inhibitor, and memantine, an NMDA receptor antagonist, showed multifunctional activity in cellulo [122]. A 1-benzylamino-2-hydroxyalkyl derivative (11) showed anti-AD activity via combining inhibition against butyrylcholinesterase, BACE1, β-amyloid aggregation, and tau aggregation in a mouse model [123]. In addition, based on drug repurposing, several antipsychotic drugs such as pimozide, benperidol, and anisopirol were also found to inhibit multiple targets involved in AD [124].

Multifunctional inhibitors against AD
Challenges for targeting metal dyshomeostasis against AD
With dramatic rise in the number of AD patients, developing the drugs which can prevent, or cure AD is urgently needed. However, even though many compounds have shown efficacy in treating AD in animal models or clinical trials, few have succeeded in the clinic in terms of showing sustained therapeutic effect. There are many challenges for identifying novel agents against AD. Firstly, some of compounds may have side effects for patients. For example, metal chelators without selectivity might lead to brain impairment via removing essential metal ions [61]. Poor BBB permeability is another obstacle for targeting metal dyshomeostasis against AD. Some compounds are hydrophilic and might exhibit good therapeutic effects in vitro or in cellulo, but they have almost no activity in vivo due to their inability to cross the BBB [62]. Therefore, developing efficient technologies to deliver drugs across the BBB are also urgently needed [125]. Recently, several compounds in clinical trials against AD have been declared failures, which suggest that once AD has entered certain stage, it may be irreversible and cannot be repaired by targeting metal dyshomeostasis [126, 127]. These failures have prompted some researchers turn to AD prevention in the early stages of disease [126, 127]. Last but not least, AD is a multifactor disease, and targeting metal dyshomeostasis via a single pathway alone may not be a guarantee of efficacy in the clinic [128]. Currently, the first diagnosis and therapy for AD patients used separate therapeutic and diagnostic agents, which may make patients miss the optimal therapeutic time window and greatly reduced the efficacy.
Concluding remarks
Although more than a century has elapsed since the first diagnosis of AD, the development of practical treatments for AD is still difficult [129]. Metal ions play critical roles in various critical neurological processes and, therefore, an aberration in their homeostasis can have catastrophic consequences [130]. However, according to the current research, metal ion dyshomeostasis not only leads to neurotoxicity and Aβ aggregation but can also lead to changes in apoptosis [49] and autophagy [54,55,56], mitochondrial dysfunction [57, 58], and lysosomal storage disorders [59,60,61] in neurons. Elucidating the precise mechanisms in which metal homeostasis is affected in each disease of interest is central to the development of new pharmacological agents. It is also extremely critical to appreciate the different functions of various cell types in regulating metal homeostasis, in order to be able to direct the therapeutic modalities to the appropriate region. [131]. Moreover, more effort should be devoted to reversing the onset of AD at an early stage, since the neurological damage caused by AD is irreversible once the disease enters into late stage [126, 127]. Another avenue of investigation could be the combination of diagnostic and therapeutic functions into a single modality, known as a “theranostic”, in order to reduce side effects and potential drug–drug interactions [132]. Finally, although extensive research has implicated the involvement of metal ions in AD, their precise mechanism in the neuropathogenesis of the disease is still unclear [133]. In this context, rebalancing metal homeostasis via developing multifunctional agents has shown great potential in both animal models of AD and in early-stage clinical trials.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-beta
- BBB:
-
Blood–brain barrier
- CNS:
-
Central nervous system
- APP:
-
Amyloid precursor protein
- ROS:
-
Reactive oxygen species
- 8HQ:
-
8-Hydroxyquinoline
- CQ:
-
Clioquinol
- DFP:
-
Deferiprone
- TETA:
-
Trientine
- DFO:
-
Desferrioxamine
- EGCG:
-
(−)-Epigallocatechin-3-gallate
- BACE1:
-
β-Secretase
- AChE:
-
Acetylcholinesterase
- NMDA:
-
N-Methyl-d-aspartic acid
References
Duce JA, Bush AI (2010) Prog Neurobiol 92:1–18
Liu G, Huang W, Moir RD, Vanderburg CR, Lai B, Peng Z, Tanzi RE, Rogers JT, Huang X (2006) J Struct Biol 155:45–51
Zatta P, Lucchini R, van Rensburg SJ, Taylor A (2003) Brain Res Bull 62:15–28
Budimir A (2011) Acta Pharm 61:1–14
Cristóvão JS, Santos R, Gomes CM (2016) Oxid Med Cell Longev 2016:9812178
Zhu YP, Feng Y, Liu T, Wu YC (2015) Integr Med Int 2:63–72
Squitti R, Salustri C (2011) Alzheimer's disease pathogenesis-core concepts, shifting paradigms and therapeutic targets. In: Monte SDL (ed) IntechOpen, Hampshire, pp 369–402
Rivera-Mancía S, Pérez-Neri I, Ríos C, Tristán-López L, Rivera-Espinosa L, Montes S (2010) Chem Biol Interact 186:184–199
Savelieff MG, Lee S, Liu Y, Lim MH (2013) ACS Chem Biol 8:856–865
Gupta UC, Gupta SC (2011) Curr Nutr Food Sci 7:221–231
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K (2008) Neuron 59:43–55
Choi JS, Braymer JJ, Nanga RP, Ramamoorthy A, Lim MH (2010) Proc Natl Acad Sci USA 107:21990–21995
Chen YC, Chiu YJ, Lin CH, Hsu WC, Wu JL, Huang CH, Lin CW, Yao CF, Huang HJ, Lo YS, Chen CM, Wu YR, Chang KH, Lee-Chen GJ, Mei Hsieh-Li H (2019) J Alzheimers Dis 67:737–756
Xie Y, Liu Q, Zheng L, Wang B, Qu X, Ni J, Zhang Y, Du X (2018) Mol Nutr Food Res 62:1800107
Kawada H, Blessing K, Kiyota T, Woolman T, Winchester L, Kador PF (2015) J Alzheimers Dis 44:297–307
Kepp KP (2012) Chem Rev 112:5193–5239
Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A (2001) Oncogene 20:1594
Zheng W, Monnot AD (2012) Pharmacol Ther 133:177–188
Yokel RA (2006) J Alzheimers Dis 10:223–253
Choi BS, Zheng W (2009) Brain Res 1248:14–21
Ximenes-da-Silva A (2016) Front Neurosci 10:233
Ergen K, Ince H, Düzova H, Karakoç Y, Emre MH (2013) Balkan Med J 30:105
Scheiber IF, Mercer JF, Dringen R (2014) Prog Neurobiol 116:33–57
Dringen R, Bishop GM, Koeppe M, Dang TN, Robinson SR (2007) Neurochem Res 32:1884–1890
Tiffany-Castiglioni E, Hong S, Qian Y (2011) Int J Dev Neurosci 29:811–818
Brown DR (2004) Neurobiol Dis 15:534–543
Hatori Y, Yan Y, Schmidt K, Furukawa E, Hasan NM, Yang N, Liu N, Sockanathan S, Lutsenko S (2016) Nat Commun 7:10640
Mathie A, Sutton GL, Clarke CE, Veale EL (2006) Pharmacol Ther 111:567–583
Opazo CM, Greenough MA, Bush AI (2014) Front Aging Neurosci 6:143
Bush AI, Tanzi RE (2008) Neurotherapeutics 5:421–432
Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R (2003) J Biol Chem 278:17401–17407
Ciuculescu ED, Mekmouche Y, Faller P (2005) Chem Eur J 11:903–909
Lee MC, Yu WC, Shih YH, Chen CY, Guo ZH, Huang SJ, Chan JC, Chen YR (2018) Sci Rep 8:4772
Matheou CJ, Younan ND, Viles JH (2016) J Mol Biol 428:2832–2846
Hsu HW, Bondy SC, Kitazawa M (2018) Toxicol Sci 163:338–345
Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI (2000) J Neurochem 75:1219–1233
Davenward S, Bentham P, Wright J, Crome P, Job D, Polwart A, Exley C (2013) J Alzheimers Dis 33:423–430
McLachlan DR, Fraser PE, Dalton AJ (1992) Ciba Found Symp 169:87–89
Loy R, Tariot PN (2002) J Mol Neurosci 19:301–307
Bush AI (2008) J Alzheimers Dis 15:223–240
Bush AI (2013) J Alzheimers Dis 33:S277–S281
Kepp KP (2017) Coord Chem Rev 351:127–159
Palanimuthu D, Poon R, Sahni S, Anjum R, Hibbs D, Lin HY, Bernhardt PV, Kalinowski DS, Richardson DR (2017) Eur J Med Chem 139:612–632
Reitz C (2012) Int J Alzheimers Dis 2012:369808
Postina R (2012) J Neurochem 120:46–54
Polanco JC, Li C, Bodea LG, Martinez-Marmol R, Meunier FA, Götz J (2018) Nat Rev Neurol 14:22
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J (2010) Cell 142:387–397
Hölscher C (2019) EBioMedicine 39:17–18
Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A (2009) J Neurochem 29:10582–10587
Atwood CS, Huang X, Moir RD, Tanzi RE, Bush AI (2018) In metal ions in biological systems. Routledge, Abingdon, pp 309–364
Kim AC, Lim S, Kim YK (2018) Int J Mol Sci 19:128
Garza-Lombó C, Posadas Y, Quintanar L, Gonsebatt ME, Franco R (2018) Antioxid Redox Signal 28:1669–1703
Nam E, Derrick JS, Lee S, Kang J, Han J, Lee SJC, Chung SW, Lim MH (2018) ACS Chem Neurosci 9:2655–2666
Pohanka M (2018) Bratisl Lek Listy 119:535–543
Grasso G, Santoro AM, Lanza V, Sbardella D, Tundo GR, Ciaccio C, Marini S, Coletta M, Milardi D (2017) Coord Chem Rev 347:1–22
Santoro AM, Monaco I, Attanasio F, Lanza V, Pappalardo G, Tomasello MF, Cunsolo A, Rizzarelli E, De Luigi A, Salmona M, Milardi D (2016) Sci Rep 6:33444
Arena G, Bellia F, Frasca G, Grasso G, Lanza V, Rizzarelli E, Tabbì G, Zito V, Milardi D (2013) Inorg Chem 52:9567–9573
Pellacani C, Costa L (2018) Environ Pollut 235:791–805
Alsousi AA, Igwe OJ (2019) Exp Cell Res 374:19–28
Song GL, Chen C, Wu QY, Zhang ZH, Zheng R, Chen Y, Jia SZ, Ni JZ (2018) Metallomics 10:1107–1115
Guo C, Liu JL, Fan YG, Yang ZS, Wang ZY (2018) Front Neurosci 12:632
Cao J, Hou J, Ping J, Cai D (2018) Mol Neurodegener 13:64
Grubman A, White AR, Liddell JR (2014) Mitochondrial metals as a potential therapeutic target in neurodegeneration. Br J Pharmacol 171:2159–2173
Nam E, Han J, Suh JM, Yi Y, Lim MH (2018) Curr Opin Chem Biol 43:8–14
Koh JY, Kim HN, Hwang JJ, Kim YH, Park SE (2019) Mol Brain 12:18
Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, Pastores GM, Rubinsztein DC, Nixon RA, Duchen MR, Mallucci GR, Kroemer G, Levine B, Eskelinen EL, Mochel F, Spedding M, Louis C, Martin OR, Millan MJ (2018) Nat Rev Drug Discov 17:660
Chen LL, Huang YJ, Cui JT, Song N, Xie J (2019) ACS Chem Neurosci 10(2):863–871
White AR, Kanninen K, Crouch P (2015) Front Aging Neurosci 7:127
Crouch PJ, Savva MS, Hung LW, Donnelly PS, Mot AI, Parker SJ, Greenough MA, Volitakis I, Adlard PA, Cherny RA (2011) J Neurochem 119:220–230
Prasanthi JRP, Schrag M, Dasari B, Marwarha G, Dickson A, Kirsch WM, Ghribi O (2012) J Alzheimer’s Dis 30:167–182
Wang CY, Xie JW, Xu Y, Wang T, Cai JH, Wang X, Zhao BL, An L, Wang ZY (2013) Antioxid Redox Signal 19:2024–2039
Weiss K, Manolaki N, Zuin M, Kruse C, Dhawan A (2018) J Hepatol 68:S106–S107
Litwin T, Członkowska A, Socha P (2019) Clinical and translational perspectives on WILSON DISEASE. Academic Press, Cambridge, pp 357–364
Smolen JS, Aletaha D (2015) Nat Rev Rheumatol 11:276
Squitti R, Rossini PM, Cassetta E, Moffa F, Pasqualetti P, Cortesi M, Colloca A, Rossi L, Finazzi-Agro’ A (2002) Eur J Clin Investig 32:51–59
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR (2002) J Biol Chem 277:45518–45528
Lee JY, Friedman JE, Angel I, Kozak A, Koh JY (2004) Neurobiol Aging 25:1315–1321
Lanza V, Milardi D, Di Natale G, Pappalardo G (2018) Curr Med Chem 25:525–539
Palumbo CS, Schilsky ML (2019) Ann Transl Med 7(Suppl 2):S65
Gracia RC, Snodgrass WR (2007) Am J Health Syst Pharm 64(1):45–53
Hane F, Leonenko Z (2014) Biomolecules 4:101–116
Yang GJ, Lei PM, Wong SY, Ma DL, Leung CH (2018) Molecules 23:3194
Yang GJ, Ko CN, Zhong HJ, Leung CH, Ma DL (2019) Cancers 11:92
Goozee KG, Shah TM, Sohrabi HR, Rainey-Smith SR, Brown B, Verdile G, Martins RN (2016) Br J Nutr 115:449–465
Tang M, Taghibiglou C (2017) J Alzheimers Dis 58:1003–1016
Dey A, Bhattacharya R, Mukherjee A, Pandey DK (2017) Biotechnol Adv 35:178–216
Mori T, Koyama N, Tan J, Segawa T, Maeda M, Town T (2019) J Biol Chem 294:2714–2731
Xicota L, Rodriguez-Morato J, Dierssen M, de la Torre R (2017) Curr Drug Targets 18:174–195
Chang X, Rong C, Chen Y, Yang C, Hu Q, Mo Y, Zhang C, Gu X, Zhang L, He W, Cheng S, Hou X, Su R, Liu S, Dun W, Wang Q, Fang S (2015) Exp Cell Res 334:136–145
Rojas C, Rojas-Castaneda J, Rojas P (2016) Curr Top Nutraceutical Res 14(1):1–16
Jiang M, Li J, Peng Q, Liu Y, Liu W, Luo C, Peng J, Li J, Yung KK, Mo Z (2014) J Neuroinflammation 11:167
Kuo LC, Song YQ, Yao CA, Cheng IH, Chien CT, Lee GC, Yang WC, Lin Y (2018) J Agric Food Chem 67:81–89
Bate C, Tayebi M, Williams A (2008) Mol Neurodegener 3:1
Gill I, Kaur S, Kaur N, Dhiman M, Mantha AK (2017) J Alzheimers Dis 60:S25–S40
Vitolo O, Gong B, Cao Z, Ishii H, Jaracz S, Nakanishi K, Arancio O, Dzyuba SV, Lefort R, Shelanski M (2009) Neurobiol Aging 30:257–265
Goyal D, Shuaib S, Mann S, Goyal B (2017) ACS Comb Sci 19:55–80
Dułak D, Banach M, Gadzała M, Konieczny L, Roterman I (2018) Acta Biochim Pol 65:595–604
Pizzi A, Dichiarante V, Terraneo G, Metrangolo P (2018) Pept Sci 110(5):e23088
Villari V, Tosto R, Di Natale G, Sinopoli A, Tomasello MF, Lazzaro S, MicaliN Pappalardo G (2017) ChemistrySelect 2:9122–9129
Shiryaev N, Jouroukhin Y, Giladi E, Polyzoidou E, Grigoriadis NC, Rosenmann H, Gozes I (2009) Neurobiol Dis 34:381–388
Wang F, Zhou X-L, Yang Q-G, Xu W-H, Wang F, Chen Y-P, Chen G-H (2011) PLoS One 6:e27649
Corona C, Frazzini V, Silvestri E, Lattanzio R, La Sorda VR, Piantelli M, Canzoniero LM, Ciavardelli D, Rizzarelli E, Sensi SL (2011) PLoS One 6:e17971
Attanasio F, Convertino M, Magno A, Caflisch A, Corazza A, Haridas H, Esposito G, Cataldo S, Pignataro B, Milardi D, Rizzarelli E (2013) ChemBioChem 14:583–592
Amijee H, Bate C, Williams A, Virdee J, Jeggo R, Spanswick D, Scopes DI, Treherne JM, Mazzitelli S, Chawner R (2012) Biochemistry 51:8338–8352
Singh A, Hasan A, Tiwari S, Pandey LM (2018) CNS Neurol Disord Drug Targets 17:571–589
Hamada Y, Miyamoto N, Kiso Y (2015) Bioorg Med Chem Lett 25:1572–1576
Man BYW, Chan HM, Leung CH, Chan DSH, Bai LP, Jiang ZH, Li HW, Ma DL (2011) Chem Sci 2:917–921
Lu L, Zhong HJ, Wang M, Ho SL, Li HW, Leung CH, Ma DL (2015) Sci Rep 5:14619
Valensin D, Gabbiani C, Messori L (2012) Coord Chem Rev 256:2357–2366
Barnham KJ, Kenche VB, Ciccotosto GD, Smith DP, Tew DJ, Liu X, Perez K, Cranston GA, Johanssen TJ, Volitakis I, Bush AI, Masters CL, White AR, Smith JP, Cherny RA, Cappai R (2008) Proc Natl Acad Sci USA 105:6813–6818
Sasaki I, Bijani C, Ladeira S, Bourdon V, Faller P, Hureau C (2012) Dalton Trans 41:6404–6407
Valensin D, Anzini P, Gaggelli E, Gaggelli N, Tamasi G, Cini R, Gabbiani C, Michelucci E, Messori L, Kozlowski H, Valensin G (2010) Inorg Chem 49:4720–4722
Kumar A, Moody L, Olaivar JF, Lewis NA, Khade RL, Holder AA, Zhang Y, Rangachari V (2010) ACS Chem Neurosci 1:691–701
Price KA, Crouch PJ, Lim S, Paterson BM, Liddell JR, Donnelly PS, White AR (2011) Metallomics 3:1280–1290
Lanza V, D’Agata R, Iacono G, Bellia F, Spoto G, Vecchio G (2015) J Inorg Biochem 153:377–382
Zhang HY (2005) FEBS Lett 579:5260–5264
Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H (2008) J Neurosci Res 86:368–377
Meunier J, Ieni J, Maurice T (2006) Br J Pharmacol 149:998–1012
Dubois B, Tolosa E, Katzenschlager R, Emre M, Lees AJ, Schumann G, Pourcher E, Gray J, Thomas G, Swartz J, Hsu T, Moline ML (2012) Mov Disord 27(10):1230–1238
Montero-Odasso M, Speechley M, Chertkow H, Sarquis-Adamson Y, Wells J, Borrie M, Vanderhaeghe L, Zou GY, Fraser S, Bherer L, Muir-Hunter SW (2019) Eur J Neurol 26(4):651–659
Jiang N, Huang Q, Liu J, Liang N, Li Q, Li Q, Xie SS (2018) Eur J Med Chem 146:287–298
Rodda J, Carter J (2012) BMJ 344:e2986
Panek D, Więckowska A, Jończyk J, Godyń J, Bajda M, Wichur T, Pasieka A, Knez D, Pišlar A, Korabecny J, Soukup O, Sepsova V, Sabaté R, Kos J, Gobec S, Malawska B (2018) ACS Chem Neurosci 9:1074–1094
Kumar S, Chowdhury S, Kumar S (2017) BMC Neurosci 18(1):76
Dong X (2018) Theranostics 8:1481
Hsu D (2017) A Marshall G. Curr Alzheimer Res 14:426–440
Panza F, Solfrizzi V, Imbimbo BP, Logroscino G (2014) Expert Opin Biol Ther 14:1465–1476
Sharma A, Pachauri V, Flora SJS (2018) Front Pharmacol 9:1247
Savelieff MG, Nam G, Kang J, Lee HJ, Lee M, Lim MH (2018) Chem Rev 119:1221–1322
Kenche VB, Barnham KJ (2011) Br J Pharmacol 163:211–219
Konttinen H, Lejavová K, Malm T, Kanninen KM (2017) Biometals in neurodegenerative diseases. Elsevier, Amsterdam, pp 195–215
Wang X, Wang X, Guo Z (2018) Coord Chem Rev 362:72–84
Lee HJ, Savelieff MG, Kang J, Brophy MB, Nakashige TG, Lee SJC, Nolan EM, Lim MH (2018) Metallomics 10:1116–1127
Acknowledgements
This work is supported by Hong Kong Baptist University (FRG2/17-18/003, China), the Health and Medical Research Fund (HMRF/14150561, China), the National Natural Science Foundation of China (21575121 and 21775131, China), the Hong Kong Baptist University Century Club Sponsorship Scheme 2018 (China), the Interdisciplinary Research Matching Scheme (RC-IRMS/16-17/03, China), Interdisciplinary Research Clusters Matching Scheme (RC-IRCs/17-18/03, China), Collaborative Research Fund (C5026-16G, China), SKLEBA and HKBU Strategic Development Fund (SKLP_1718_P04, China), the Funded by The Science and Technology Development Fund, Macau SAR (File no. 077/2016/A2 and SKL-QRCM-2017-2019) and the University of Macau (MYRG2016-00151-ICMS-QRCM and MYRG2018-00187-ICMS, China).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, G., Liu, H., Ma, DL. et al. Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem 24, 1159–1170 (2019). https://doi.org/10.1007/s00775-019-01712-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-019-01712-y