
Abstract
Essential trace metals like zinc (Zn), iron (Fe), and copper (Cu) play an important physiological role in the metabolomics and healthy functioning of body organs, including the brain. However, abnormal accumulation of trace metals in the brain and dyshomeostasis in the different regions of the brain have emerged as contributing factors in neuronal degeneration, Aβ aggregation, and Tau formation. The link between these essential trace metal ions and the risk of AD has been widely studied, although the conclusions have been ambiguous. Despite the absence of evidence for any clinical benefit, therapeutic chelation is still hypothesized to be a therapeutic option for AD. Furthermore, the parameters like bioavailability, ability to cross the BBB, and chelation specificity must be taken into consideration while selecting a suitable chelation therapy. The data in this review summarizes that the primary intervention in AD is brain metal homeostasis along with brain metal scavenging. This review evaluates the impact of different trace metals (Cu, Zn, Fe) on normal brain functioning and their association with neurodegeneration in AD. Also, it investigates the therapeutic potential of metal chelators in the management of AD. An extensive literature search was carried out on the “Web of Science, PubMed, Science Direct, and Google Scholar” to investigate the effect of trace elements in neurological impairment and the role of metal chelators in AD. In addition, the current review highlights the advantages and limitations of chelation therapies and the difficulties involved in developing selective metal chelation therapy in AD patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
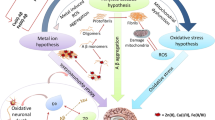
The field of metallo-biology is wide, with diseases marked by abnormalities in metal transport proteins (Bush 2003). Overexpression of various metal ions and transport proteins plays an important role in the development of Alzheimer’s disease (AD). Conventionally, a number of metals have been suggested to be relevant to AD, each with varying amounts of preclinical and clinical evidence (Wang and Wang 2017). AD is a neurodegenerative condition of the central nervous system (CNS) that is complex and diverse and affects almost 36 million individuals worldwide (Fang et al. 2013; Cristóvão et al. 2016). In 2030 and 2050, these figures will be 78 and 138 million, respectively, as per World Alzheimer’s report (Gauthier et al. 2021). Clinically, progressive memory loss, cognitive dysfunction, language misinterpretation, and personality changes are all manifestations of AD (Xu et al. 2016; Zhang et al. 2019). The development of amyloid-beta (Aβ) plaques in the brain is a major pathological feature of AD. The plaques consist primarily of Aβ peptides and of Zn2+, Cu2+, and Fe2+ trace metals (Bagheri et al. 2018). The cleavage induced by β and γ-secretases produces Aβ from amyloid precursor protein (APP), in which cleavage by β-secretase is thought to be the rate-limiting step. Furthermore, Aβ aggregation may result in the formation of oligomers and plaques, with the oligomers being the more toxic form of Aβ (Dubois et al. 2016; Elmaleh et al. 2019). Amyloid oligomers have also been linked to tau-dependent microtubule disintegration (Taylor et al. 2021). Recent research suggests that tau hyperphosphorylation may play a dual role in AD neurodegeneration either by making cells more resistant to acute apoptosis or by increasing intracellular tau accumulation, causing a variety of cellular dysfunctions, including endoplasmic reticulum (ER) stress (Wang et al. 2020a, b). The presence of metals, which are present in both the interior and periphery of known AD plaques, has been found to have a major effect on Aβ misfolding (Yang et al. 2019). There are currently no effective medications that can be used to safely reverse the mental deterioration of affected patients (Kabir et al. 2021a). Pharmacological techniques aiming at decreasing brain metal ion and targeting Aβ-metal ion interactions may offer a huge potential for chelation therapy as the demand for novel and more effective medications for AD treatment continue to develop.
Iron (Fe) has the ability to serve as an electron donor or acceptor, making it an essential component of a broad variety of metabolic pathways, including cellular energy output, proliferation, and differentiation, as well as a cofactor for many enzymes (Hentze et al. 2010). The most prevalent transition metal in the brain is Fe, which plays a key role in neurotransmitter synthesis, myelination, and mitochondrial activity (Hare et al. 2013; Huang et al. 2018). In a healthy brain, the Fe content is about 0.04 mg/g fresh tissue, with a concentration of 720 µM (Dalvi et al. 2021). Many neurodegenerative conditions, including AD, are thought to be caused by a build-up of Fe in the brain, which causes cell death (Viktorinova and Durfinova 2021).
Zinc (Zn) is a trace metal that is required by humans and many other living species. It is the second most abundant transition metal after Fe. As a cofactor for over 300 enzymes and metalloproteins, Zn modulates gene transcription and the antioxidant response. Zn content in the brain is estimated to be 150 μM, which is ten times higher than that of the serum (Wang et al. 2020a, b). Zn dysregulation impairs mental and physical development and learning skills in childhood (Prasad 2013). Excessive zinc reduces Cu and Fe absorption, boosting ROS generation in the mitochondria, altering metabolic enzyme activities, and triggering apoptosis (Brzozowska 1989; Furuta et al. 2016). At micromolar concentrations, Zn inhibits Aβ induced neurotoxicity by preferentially precipitating aggregation intermediates (Garai et al. 2007). However, at high concentrations, Zn binding to Aβ may promote fibrillar Aβ aggregation and cause toxicity both in vitro and in vivo (Lovell et al. 1999; Bishop and Robinson 2004; Rezaei-Ghaleh et al. 2011).
Copper (Cu) is an essential trace element for a variety of biological functions in living cells. It plays an important role in a variety of metabolic processes in the brain, including serving as an active site for cuproenzymes such as superoxide dismutase (SOD), cytochrome oxidase, ceruloplasmin (Cp), and tyrosinase (Barnham et al. 2004). The majority of Cu in the body is concentrated in the organs with high metabolic activity such as the brain, kidneys, heart, and liver. A total of 95% of Cu is bound to Cp, and the unbounded Cu acts as an oxidant, catalyzing the formation of highly reactive hydroxyl radicals, which can damage DNA, proteins, and lipids (Uriu-Adams and Keen 2005). The Cu and Aβ oligomer complexes are capable of penetrating the neurons and can induce oxidative stress in different neuronal sub-compartments. Excess Cu plays a crucial role in the etiology of AD (Baldari et al. 2020a).
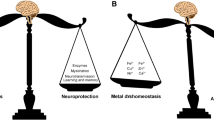
Metal ion homeostasis is critical for sustaining normal brain processes. Changes in the complex balance of metal ions in the brain are closely associated with Aβ deposition in AD patients (Adlard and Bush 2018a). In the course of a variety of neurodegenerative illnesses, including AD, metal ions build improperly in the brain as aging progresses (Kabir et al. 2021b). Metal dyshomeostasis has been related to the etiology of a variety of neurodegenerative disorders, most notably AD (Wang et al. 2020a, b). Based on the mislocalization of metal ions in AD patients, several clinical trials involving supplementation, chelation, or regulation of metal ions have been performed (Adlard and Bush 2018a; Fulgenzi et al. 2020).
A chelator is a chemical molecule that can selectively bind to a specific ion forming a stable complex ring-like structure. Dietary supplements, radiopharmaceuticals, cleaning chemicals, cosmetics, plastics, fertilizers, aquaculture growth supplements, removal of toxic metals from the soil, and metal chelation therapy are just a few of the applications for metal chelating agents (Kim et al. 2019). Extracellularly, they may deplete the total pool of bioavailable metals or compete for metal ions as ionophores with endogenous ligands (Gromadzka et al. 2020). As a result, metal chelation therapy may now be considered a promising clinical treatment option for AD. Iron chelation therapy is a well-established treatment for Fe overload in thalassemia, sickle cell disease, and myelodysplasia patients who need regular blood transfusions (Killick 2017). Since iron chelation therapy prevents neuronal death in a variety of animal AD models, it can also be extended for the treatment of AD (Rao et al. 2020; Shahandeh et al. 2020). Like iron chelators, copper and zinc chelating agents are one of the most promising methods for regulating physiological Cu and Zn concentrations (Baldari et al. 2020a).
Recent research studies that have evaluated the role of metal levels in different stages of AD pathogenesis have only been studied in a few studies. Although there have been causal ties between theoretical grounds of chelation chemistry for treatments in AD, there are still missing links, a lack of relevant data, and strong conclusions about the benefits of metal chelation therapy in AD. It is reported that, when utilized to treat CNS illnesses, chelation therapy presents a number of general and particular problems (Shahandeh et al. 2020). Hence, the current paper discusses the association of the brain’s physiological transition metals along with the pathophysiology of AD. Furthermore, the review also provides insights on iron, copper, and zinc chelation therapies and the associated challenges with these metal chelation therapies.
Metal Homeostasis in Brain Regulation
Metals can reach the brain through one of two routes: the blood–brain barrier (BBB) or the brain-cerebrospinal fluid barrier (BCSF). Metals can pass across the BBB as free ions by being carried by ion-specific transporters or by forming complexes with cysteine or other amino acids (Yokel 2006; Choi and Zheng 2009). Metal ion concentrations vary in different parts of the brain (Skalny et al. 2019). Metal ions are often concentrated in membrane metalloproteins or synaptic vesicles in the brain (Das et al. 2021). Metal-binding biomolecules, such as metallothioneins and glutathione, are abundant in astrocytes, allowing them to sequester metal ions and reduce neurotoxicity (Gerhold et al. 2021). As they are the first type of cell to associate with metals when they move through the BBB, astrocytes in the brain aid in metal homeostasis (Tiffany-Castiglioni et al. 2011; Scheiber et al. 2014). Metals such as Zn, Fe, and Cu are important for the biologically critical processes of the human body, including catalysis, protein structure stabilization, signal transmission, and metabolism. Because of the vital function of metals in the brain, strict control of metal homeostasis in the brain is important (Guanjun et al. 2019).
Role of Copper, Iron, and Zinc in the Pathophysiology and Neuropathology of AD
Iron (Fe2+,Fe3+)
Axonal projections ranging from the ventral hippocampus to the substantia nigra via the medial prefrontal cortex and the route of the thalamus, amygdala, and medial prefrontal cortex have recently been discovered to transport Fe across brain regions (Lei et al. 2019). While it has been shown that Fe translocation in this channel might affect anxiety behavior, the discovery also opens up a novel trafficking system in which other pathways may occur and be disrupted in AD (Wang et al. 2019b). Between ferrous and ferric, iron’s valence state changes. This trait is necessary for physiology, but it can also be harmful as a cause of oxidative stress, specifically in an aerobic environment. As a result, Fe is closely regulated in the brain, where both Fe deficiency and excess can cause brain dysfunction (Bao et al. 2021; Peng et al. 2021). In the early stages of life, Fe deficiency slows down neurodevelopment and Fe overload causes early onset AD (also known as familial AD) (Wang et al. 2019a; Georgieff et al. 2019). Age-dependent Fe accumulation in the brain, on the other hand, is an unavoidable result of aging (Ijomone et al. 2020) and may lead to a variety of neurodegenerative disorders, including AD (Tuo and Guo 2019). Lipofuscin also known as “aging pigment,” which contains exceptionally elevated concentrations of Fe as well as other metal ions controlled by oxidized peptide fragments, accumulates next to mitochondria in older neurons, yet its pathophysiological significance is uncertain. According to a major recent review, Fe accumulation in the inferior temporal cortex was only seen in subjects diagnosed clinically with AD and reported post-mortem by standardized standards (Ayton et al. 2020). Because of small sample sizes, inconsistencies in the clinical and pathological diagnosis of AD, Fe depletion by fixatives, and low detection limits, reports of elevated Fe in post-mortem tissue were inconsistent in previous research (van der Weerd et al. 2020; Loef and Walach 2021).
The most selective MRI modality for tissue Fe is quantitative susceptibility mapping (QSM), according to experts (Du et al. 2018). Only those with elevated Fe on MRI and increased Aβ on positron emission tomography exhibited a significant negative link between entorhinal cortex volume and age (Foster et al. 2020). Fe accumulation in AD was found to be associated with cognitive dysfunction, tangle deposition, and Aβ deposition using QSM, indicating that Fe in CSF could be useful as a biomarker for AD progression (Lei et al. 2021).
Significant evidence reveals that in AD, the precise regulation of brain Fe homeostasis has broken down, which is consistent with the link between brain Fe levels and neurodegeneration. Fe pathways are prominently agitated in AD brain tissue, according to recent unbiased single-cell transcriptomics and proteomic studies (Zhou et al. 2020; Bai et al. 2020). The canonical Fe-controlling genes have been linked to the risk of AD in genetic studies. The TF gene coding polymorphism Pro570Ser has been linked to an increased risk of AD with an odds ratio of 1.2 (van Rensburg et al. 1993). The apolipoprotein variant (APOE-4), which has been linked to elevated brain Fe levels, is a significant genetic risk factor for AD (Fan et al. 2019). According to recent research, the canonical Fe-associated genes interfere with the APOE-4 risk allele, increasing the risk of AD (Tisato et al. 2018). Cp, a protein that promotes cellular Fe export, has been shown to be downregulated in AD brain tissue, and transferrin protein levels have been found to be elevated in the frontal cortex of AD patients (Ashraf et al. 2020; D’Mello and Kindy 2020).
Through the regulation of Fe transporter levels, hepcidin (a peptide hormone) regulates dietary Fe absorption, recycling by macrophages, and release from hepatic stores, resulting in a decrease in Fe plasma levels (Yiannikourides and Latunde-Dada 2019). Hepcidin, a protein that disrupts the ferroportin (Fe exporter), has been discovered to be repressed in AD cortical tissue (Qian and Ke 2020). Hepcidin is expressed in a variety of brain cells, and its function in brain homeostasis is complicated by the fact that it is a high expression in astrocytes and can indirectly alter Fe homeostasis in neurons since it is expressed at BBB (Vela 2018). In hippocampal neurons, hepcidin pre-treatment reduces the secretion of inflammatory cytokines caused by the Aβ peptide, and also the toxicity of astrocytes and microglia-conditioned media was reduced (Urrutia et al. 2017). Indeed, in the APP/PS1 transgenic mouse model for AD, overexpression of hepcidin in astrocytes by an adeno-affiliated virus (AAV) relieved neuropathology and cognition. (Overexpression of hepcidin by astrocytes decreases Fe entry into the brain and Fe accumulation in neurons resulting in less neuronal death in the cortex and hippocampus) (Xu et al. 2020).
The proteins APP and tau, which cause AD proteinopathy, have been related to the metabolism of Fe (Ganguly et al. 2017). Tau-mediated APP trafficking regulates neuronal iron (Fe2+) export in normal healthy conditions. Tau transports APP load here to the surface of neurons, where it engages with and stabilizes ferroportin, allowing Fe to be exported from neurons. Fe export from neurons is impaired by a reduction in soluble tau or a familial AD (FAD) mutation in APP, resulting in the retention of Fe. When β-site amyloid precursor protein cleaving enzyme 1(BACE1) cleaves APP, ferroportin fails surface stabilization resulting in the buildup of Fe (Fig. 1) (Tsatsanis et al. 2019).

Diagrammatic illustration of abnormal iron (Fe2+, Fe3+) accumulation/metabolism and ferroptosis in AD
Another physiological interaction between APP and Fe homeostasis is with heme-oxygenase 1 (HO-1), an intracellular enzyme responsible for the breakdown of heme into free Fe2+, carbon monoxide, and biliverdin. While HO-1 can alleviate oxidative stress by reducing the load of pro-oxidant heme, excessive HO-1 activity can produce oxidative stress and promote ferroptosis by exposing the cytoplasm to excessive Fe2+. HO-1 levels have repeatedly been found to be higher in astrocytes AD-affected brain tissue, but lower in plasma and CSF (Schipper et al. 2019). It has been demonstrated that APP inhibits HO-1 and HO-2, with the FAD mutant APP species binding with higher affinity (Takahashi et al. 2000). Tau protein has also been shown to facilitate Fe export in conjunction with APP. Soluble tau levels have been shown to be lower in AD patients, and in mice, this loss induces Fe accumulation and, as a result, neurodegeneration, which can be rescued with iron chelation or antioxidant supplementation (Lei et al. 2012; Singh et al. 2019). Fe can also boost APOE secretion by influencing its expression at both the posttranscriptional and transcriptional stages in neurons and astrocytes (Xu et al. 2016). Overall, these findings suggest that dyshomeostasis of Fe is linked to the proteins most involved in AD pathology.
Fe can play a role in the pathology of AD in a variety of ways. Elevated concentrations of Fe in senile plaques and co-localization with tangles suggest that Fe can play a role in plaque and tangle formation (Telling et al. 2017; Everett et al. 2018). It induces Aβ aggregation in cell-free structures, resulting in neuronal toxicity (Galante et al. 2018). The toxicity of Aβ-iron (Fe-Aβ) complexes may also be due to the specific structure of the induced aggregation of Aβ, which is likely to trigger cell death pathways (Kuperstein and Yavin 2004; Liu et al. 2011). Accumulation of Fe (e.g., from aging or from BACE1 processing of APP) increases the reaction of cytoplasmic Fe2+ with H2O2 to produce the hydroxyl radical (HO, Fenton chemistry), which then interacts with PUFA-containing membrane phospholipids, producing lipid peroxides and triggering lipid radical propagation, causing ferroptosis. This process is induced by auto-oxidation, but Fe-dependent ferroptosis can also be triggered by arachidonate lipoxygenase 15 (ALOX15) mediated phospholipid peroxidation (Fig. 1) (Doll and Conrad 2017).
Since phosphate groups have a high affinity for Fe3+, hyperphosphorylated tau can be isolated from post-mortem AD brain samples using this affinity (Ping et al. 2020). So, while tau phosphorylation separates tau from microtubules, increased cytosolic Fe3+, as seen in aging and AD, can neutralize the charge on the phosphates and promote aggregation. Finally, the gliosis that characterizes AD pathology can play a role in harmful Fe-mediated reactions (Peters et al. 2018; Lei et al. 2021).
These findings show that Fe may play a role in AD pathogenesis either by accumulating in the tissue, binding to the amyloid or tangle proteinopathy, causing the proteinopathy, or cooperating with the proteinopathy. As a result, targeting Fe as a therapeutic strategy for AD may be a viable option.
Copper (Cu+, Cu2+)
Higher Cu deposition has been observed in the brains of AD patients. Cu transporter 1 (CTR1) regulates Cu uptake, and ATP7A and ATP7B regulate Cu efflux in all cells (Curnock and Cullen 2020). Cu2+ is taken up by CTR1 and exported by ATP7A/B in neurons. Extracellular Cu2+ can be entrapped by Aβ oligomer, which then instills in the membrane, generating a catalytic complex that produces H2O2. Due to its permeability, H2O2 can deplete antioxidants like GSH and denature SOD. Levels of Cu in bulk tissue are reduced, which leads to Cp activity being reduced. However, in AD-affected tissue, the fraction of cytoplasmic-free Cu+ increases, which could lead to tau hyperphosphorylation through Cyclin-dependent kinase 5 (CDK5) or glycogen synthase kinase-3 (GSK3) activation (Lei et al. 2021). ATP7B gene mutation causes an increase in the Cu non-bound to Cp, generally observed in Wilson’s disease. A recent study compared the healthy control with patients suffering from frontotemporal lobar degeneration (FTLD) and AD. It revealed that ATP7B mutation causes a decline of Cu-ATPase function in AD specifically and not in other types of dementia. Therefore, ATP7B mutation also tends to increase the risk of AD (Rongioletti et al. 2018). The role of Cu in the normal neuronal function and repercussions on its dysregulation is illustrated in Fig. 2 (Mercer et al. 2017).

Copper (Cu+, Cu2+) dysregulation pathways in AD
During neurotransmission, the concentration of Cu in the synapse increases temporarily. Cu released at micromolar amounts from copper-containing vesicles affects synaptic functioning as synaptic depolarization develops (Wang et al. 2020b). Once released into the synaptic cleft, it can act as a high-affinity blocker of the N-methyl-D-aspartate receptor (NMDA) and glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA), resulting in a significant reduction of glutamate-mediated neurotransmission (Dodani et al. 2014). Cu may also affect neurotransmission and neuronal excitability via altering the activity of Gaba aminobutyric acid (GABA) and P2X receptors, either directly or indirectly. Additionally, Cu has the ability to regulate the trafficking of synaptic vesicles and protein interactions, whereas neurotransmission can affect Cu trafficking and delivery in neuronal cells (Wang et al. 2020a, b).
The copper-Aβ (Cu-Aβ) interaction was initially discovered in 1994, when Cu2+ was found to significantly increase the production of soluble dimers of Aβ at neutral pH (Bush et al. 1994). APP is a metal-binding protein with two Zn2+ and Cu2+ binding domains in its N-terminal. It has also been shown to convert oxidized Cu2+ to reduced Cu+. APP dimerization and trafficking, as well as APP expression, processing, and production of Aβ, are all associated with Zn2+ and Cu2+ (Kawahara et al. 2020). In cortical neuronal cultures obtained from BL6Jx129sv mouse, the Cu-Aβ interaction could form a catalytic redox-cycling complex which integrates into the lipid membrane and recruits substrates like cholesterol to produce hydrogen peroxide and enhance oxidative stress, both of which induce neurotoxicity (Huang et al. 1999; Opazo et al. 2002; Curtain et al. 2003). In vitro, Cu binds to tau protein, promotes aggregation, and can produce hydrogen peroxide, similar to the effect of binding Aβ (Su et al. 2007; Martic et al. 2013).
Cu levels have been shown to be lower in AD-affected brain tissue than in healthy control tissue (Rembach et al. 2013; Xu et al. 2017a). Despite a decrease in total Cu in AD-affected tissue, the proportion of loosely bound exchangeable Cu ions improved, indicating a disruption in the typical Cu coordination environment of the tissue. Cu distribution is deficient in the cells, but excess Cu is trapped in the extracellular plaques. This excess Cu can promote Aβ production and tau hyperphosphorylation along with the aggregation of vulnerable peptides (Wang et al. 2020a, b; Lei et al. 2021). Copper chelation solubilized Aβ from the insoluble portion of AD-affected brain tissue, suggesting that peptide aggregation could be reverted by chelation, indicating proof of concept of pharmacological targeting of the metal center for reverting amyloid development (Cherny et al. 1999). A copper chelator inhibited Aβ accumulation and overexpression in Caenorhabditis elegans, protecting the organism from Cu poisoning (Ejaz et al. 2020; Pretsch et al. 2020). Treatment with Cu, copper chelation therapy, as well as reducing the Cu transporters (Ctr) CtrlB or CtrlC or overexpressing the cellular Cu-exporter DmATP7, reduces the generation of Aβ oligomers and the amount of oxidative stress in an Aβ transgenic drosophila model, improving motor deficits and increasing lifespan (Lang et al. 2013). Cu-targeting chelation therapies have been shown to enhance memory deficits in the Tg2576 mouse (Ceccom et al. 2012; Esmieu et al. 2019).
Transgenic mice models for AD have shown low levels of Cu in their brain when compared with controls (Kabir et al. 2021b). Similarly, the post-mortem reports of the patients suffering from AD indicated lower levels of Cu when compared to the normal brains (Liu et al. 2019; Huat et al. 2019). According to several studies, increasing brain Cu levels had been suggested to suppress amyloid pathology and therefore was suggested to be a therapeutic strategy in the treatment of AD (Bayer et al. 2003; Phinney et al. 2003). However, based on these findings, a phase 2 randomized clinical trial of copper orotate supplementation in minimally affected AD patients was conducted. This Cu treatment did not show any significant effect (Kessler et al. 2008). Also, oral administration of Cu in a triple-transgenic mouse (mutant APP/PSEN/Tau) model of AD, increased tau hyperphosphorylation by activating CDK5 (Kitazawa et al. 2009). Furthermore, Cu-enriched drinking water has been shown to exacerbate cognitive dysfunction and worsen neurodegeneration in wild-type rats and Tg2576 and 3xTg-AD mice (Behzadfar et al. 2017; Yao et al. 2018; Chen et al. 2019).
Zinc (Zn2+)
In the brain, 80 to 90% of Zn is tightly associated with proteins in order to produce enzymatic activity or structural stability, and the proteome has identified roughly 2800 Zn-binding proteins. A small amount of Zn is concentrated in synaptic vesicles (> 100 M) at glutamatergic nerve terminals. It is released synaptically upon the neuronal activity, influencing synaptic transmission and a range of biological functions (Paoletti et al. 2009). Zn has high flux at the synapse and contributes to synaptic plasticity, and Zn at presynaptic and postsynaptic sites modulates long-term potentiation (LTP) in the hippocampus. Synaptic Zn turnover is an important factor for maintaining cognitive balance. This vigorous turnover of Zn decreases with aging, implying that Zn dysregulation may play a role in cognitive decline (Datki et al. 2020).
Metallothionein 3 (MT3) is a key participant in maintaining Zn homeostasis in the brain, where Zn is chelated as metal thiolate clusters. The Zn in MT3 can interchange with Cu in the Cu-Aβ complex, reducing the oxidative damage caused by the Cu-Aβ complex, and Zn2+ can also preserve sulfhydryl groups in cells from oxidation (Wang et al. 2020b). Therefore, Zn or MT3 downregulation, as seen in AD neurons, may result in oxidative damage. Under pathological situations, excess Zn is released from presynaptic neurons and astrocytes, inducing NADPH-oxidase activation and ROS formation in neurons, along with microglial activation and neuronal death (Furuta et al. 2016). Moreover, since Zn2+ suppresses numerous enzymes, an abundance of Zn can induce a number of metabolic diseases by interfering with related enzyme activity. Zn2+ is extremely sensitive to the mitochondrial respiratory chain, and an increase in Zn2+ in mitochondria increases ROS production. Excessive Zn, which can be produced as a result of increased metalloprotein release, can enhance Aβ production and deposition (Wang et al. 2020a, b). Depending on the Aβ/Zn ratio, Zn2+ can stimulate various types of Aβ aggregates: stoichiometric Zn concentrations stimulate non-fibrillar aggregates nourished in the reversible alpha-helical conformation, whilst substoichiometric Zn accumulation induces fibrillar, Beta-sheet–enriched clusters as a result of seeding (Lei et al. 2021). Zn levels in plaques in AD can reach 1 mM, demonstrating that Zn2 + aggregates soluble Aβ in vivo. Zn levels are higher in plaques of APP/PS1 mice as determined by Timm’s stain and X-ray fluorescence microscopy, plaques of Tg2576 mice as determined by metallomic imaging mass spectrometry, and plaques inside the amygdala of old macaques as measured by metallomic imaging mass spectrometry (Lei et al. 2021).
Zn2+ enters the cytoplasm of neurons through ZIPs, while ZnTs regulate outflow from the cytoplasm. Although there are several varieties of ZIPs and ZnTs expressed in neurons, ZnT3 has been linked to cognitive decline and amyloid production in AD. ZnT3 elevates Zn2+ in glutamatergic synaptic vesicles, which are then taken up by unknown energy-dependent mechanisms. Mitochondrial energy declines with age, resulting in sluggish extracellular Zn2+ reuptake. When estrogen levels drop, as they do during menopause, ZnT3 protein levels rise, likely boosting Zn2+ release. Extra-cellular Zn2+ bonds to Aβ, enabling it to clump and then become trapped within the amyloid. Intracellularly, metallothioneins, being the primary Zn2 + -buffering peptides, regulate free Zn2+ levels, while metallothionein III levels in neurons are reduced in AD. By activating CDK5, GSK3, ERK1/2, or JNK kinases and suppressing PP2A activity, increased cytoplasmic-free Zn2+ promotes tau phosphorylation (Lei et al. 2021). The dysregulation of Zn is shown in Fig. 3.

Diagrammatic illustration of zinc (Zn2+) dysregulation pathways in AD
Changes in the expression of various Zn transporter (ZnT) proteins have been observed in studies of postmortem brain tissue from AD patients. In the frontal cortex of AD patients as well as APP/PS1 mice, ZnT10 protein levels were reduced compared to the control (Xu et al. 2019). ZnT6 protein levels were shown to be higher in the hippocampus and parahippocampal gyrus region of pathologically proven AD patients. Also, ZnT1 levels were considerably lower in the same area (Lyubartseva et al. 2010). There have been multiple reports of Zn interacting with the other key proteins linked to AD. Zn has been shown to increase the expression of presenilin 1 and impact the stability of apolipoprotein E (ApoE), specifically ApoE4 (Uddin et al. 2019). Zn can control the production of Aβ by modifying the secretases accountable for it. The activities of beta-secretase, a disintegrin, and metalloprotease 10 that are essential for APP cleavage into the non-amyloidogenic cascade are performed by a Zn metalloproteinase, and alteration of its Zn binding domain prevents it from working. In vitro studies have reported the reduction of β-secretase activity by an increase in the APP-C99 fragment dimerization, implying that Zn consumption could limit Aβ production (Hoke et al. 2005; Gerber et al. 2017).
Chelation Therapy Targeting Excessive Accumulation of Essential Metals in the Brain
As mentioned earlier, metal dyshomeostasis is considered to play an important role in the pathology of AD, and the rebalancing of metal homeostasis may be a therapeutic option for the management of AD (Ben-Shushan and Miller 2021). Chelation therapy has been suggested as a treatment for reducing abnormal accumulations of critical heavy metals like Fe, Cu, and Zn, as well as non-essential and toxic metals like lead (Pb), mercury (Hg), and cadmium (Cd) (De Benedictis et al. 2019). Metal chelators can sequester metal ions and transport them away from the lesion site, preventing them from engaging in redox chemistry or facilitating Aβ aggregation (Chaves et al. 2021). They have the ability to trap and bind metal ions by forming two or more coordinate bonds with a single metal ion (Guanjun et al. 2019). Apart from simple chelating agents, multifunctional molecules with various targets have emerged as intriguing treatment alternatives to conventional AD treatment (Sharma et al. 2018). These multifunctional compounds have one or a few molecules with chelating capabilities. By removing metal ions from circulation and lowering intracellular concentrations of redox-active metal ions, multifunctional drugs generally fulfill the chelation principle. Also, metal protein attenuating compounds possessing lower affinities with metal ions can control the generation of ROS species (Ritchie et al. 2004). These compounds not only help to limit metal-protein interaction, but they also assist to solubilize Aβ protein and protect it from oxidative stress-induced damage (Fasae et al. 2021). As a result, the evolution and development of multitarget ligands have become a significant focus for therapeutic candidates for AD (Rana et al. 2018; Wang et al. 2018).
Iron chelators have therapeutic potential beyond their current use in those who require chronic blood transfusions, as our understanding of the accumulation of Fe and ferroptosis in neurodegenerative disease has improved. Although the pathophysiological mechanisms underlying part of Fe in the development of AD are still being unraveled,the accumulation of Fe has been strongly linked to the disease’s pathogenesis. Deferoxamine, deferiprone, and deferasirox are the three FDA-approved iron chelators currently available. The properties of these chelators are listed in Table 1.
Deferoxamine, a potent iron chelator, has been shown in various animal studies to enhance memory by inhibiting amyloidogenic APP processing and Aβ aggregation. In P301L tau transgenic mice, intranasal deferoxamine increases efficiency in the radial arm water maze, stabilizes HIF-1α, and phosphorylates GSK3 (Fine et al. 2012). It also reduces synapse loss in APP/PS1 transgenic mice’s brains by upregulating the P38/HIF-1α pathway (Guo et al. 2015). In 1991, a 2-year, single-blind study including 48 patients evaluated the role of deferoxamine (i.m.) for aluminum chelation as a therapy for AD. The effects of deferoxamine (125 mg) administered twice daily for a period of 24 months were compared to the oral placebo and no-treatment group. According to their findings, deferoxamine decreased the rate of clinical progression of AD and also reduced the rate of cognitive decline in AD patients as measured by home-behavioral assessment. There was a significant reduction in the rate of cognitive decline in the treatment group when compared to the placebo and no-treatment group (McLachlan et al. 1991). It has been shown to inhibit APP holoprotein translation via IRE in the 5′-untranslated region of the APP transcript and enhance certain cognitive functions in AD patients when injected intramuscularly (Rogers et al. 2002). On AD models, the preclinical chelator DP-109 has been found to be effective. It tended to improve the solubility of Aβ in the cerebrum in 3-month mouse experiments with Tg2576 mice, reducing the amount of Aβ aggregates (Lee et al. 2004). Deferiprone, a lipid-soluble iron chelator, is one that can cross the blood-neural barrier and chelate inadequately compartmentalized Fe within the CNS. In phase 2 investigations, deferiprone, an orally accessible brain permeable iron chelator, reduced brain iron in children with neurodegeneration caused by a mutant pantothenate kinase (Neurodegeneration with brain iron accumulation type 1) (Ayton and Bush 2019; Klopstock et al. 2019). Also, clinical benefits of using deferiprone were reported in adults with Parkinson’s disease in phase 2 studies (Devos et al. 2014; Martin-Bastida et al. 2017). The 3D study (deferiprone to delay dementia 2017–2021) is a randomized, placebo-controlled, multicenter study that aims to see whether treating AD patients with deferiprone (30 mg kg−1 day−1) for 52 weeks will slow cognitive decline or not is still going on (NCT03234686) (Adlard and Bush 2018b).
Cu and Zn levels have been shown to be modulated by various chelating drugs through various mechanisms. Penicillamine and trientine form complexes which can be excreted in the urine. Cu biliary excretion is aided by tetrathiomolybdate (Baldari et al. 2020a). The most common properties of copper and zinc chelating agents are summarized in Table 2.
Clioquinol and PBT2, which target the metal ion in Cu-Aβ beta complexes (as well as zinc—Aβ complexes), have been explored in preclinical animal models of AD. These are ionophores that facilitate Zn2+ and Cu2+ uptake. Clioquinol and PBT2 both counteract cognitive impairment in aging ZnT3 knockout mice and normal mice (Adlard et al. 2015; Lei et al. 2021). Tau phosphorylation and aggregation are said to be aided by free Zn2+. PBT2 interacts with accessible Zn2+ and Cu2+ to increase Aβ aggregate dissolution, absorption, and degradation. PBT2 also inhibits Aβ from binding Zn2+ and Cu2+, neutralizing the metal ion’s charge and enabling it to pass through cell membranes passively. This promotes the recycling of Zn2+ and Cu2+ from the synaptic cleft, harmonizing functional fluxes and intracellular metal accumulation.
Cu comprising bis-thiosemicarbazone can act as an ionophore, transport Cu in neurons, and lower the level of Aβ in cell culture and animal models of AD (Pyun et al. 2022). It can also enhance neurite elongation and prevent cognitive deficits in APP and PS1 mice (Bica et al. 2014; Haque et al. 2019). CuATSM is a small synthetic molecule that can pass the blood–brain barrier and transport copper to cells bearing damaged mitochondria. Chemically, it is a bis-thiosemicarbazone complex with two methyl groups on the di-imine backbone. In the absence of a methyl group, it is termed CuGTSM. CuGTSM and CuATSM have been explored in animal models of AD. CuGTSM has a substantially lower affinity for Cu2+ than CuATSM, but it is far more effective in correcting cognitive impairment in an APP/PS1 transgenic mice model (Crouch et al. 2009). This advantage may be due to the increased dissociation of Cu after cellular uptake, which has an effect on GSK3β activity (Crouch et al. 2009). PET imaging with 64CuGTSM and 64CuATSM showed substantially increased Cu uptake in the brains of an APP/PS1 transgenic mouse model as compared to wild-type controls for 64CuGTSM but not for 64CuATSM. Furthermore, no association to amyloid plaques was seen after the treatment of AD brain regions (Fodero-Tavoletti et al. 2010). CuATSM had little effect on the APP/PS1 mouse model, but its PET radioligand identifies neurodegeneration in Parkinson’s disease and amyotrophic lateral sclerosis patients, and it has recently been shown to interact with the lipid peroxyl groups formed during ferroptosis (Southon et al. 2020). Therefore, copper chelating agents or metal protein attenuating compounds may be a possible therapeutic alternative for AD patients.
Pro-Chelators and Pharmacological Properties of Molecules with Multi-Functionality
Pro-chelators are compounds that improve the targeting of metal-binding agents in AD (Gleason and Bush 2021). This can be achieved by attaching moieties to improve BBB permeability and providing a group that can be activated by external triggers (ROS, enzymes) to promote metal ion binding (Wang and Franz 2016). One study developed stimulus-responsive pro-chelators in neurodegeneration. BSIH, a salicylaldehyde isonicotinoyl hydrazone chelator masked with a boronic acid pinacol ester has been synthesized. BSIH is a ROS-responsive pro-chelator that is activated by hydrogen peroxide to release free phenol and activate metal binding (Charkoudian et al. 2006). Furthermore, an enzymatic approach for the activation of chelators was developed. In this study, the β-secretase substrate, when activated by an enzyme, produces an ATCUN binding motif possessing a high affinity for Cu. This compound, upon activation, sequesters Cu from Aβ plaque due to which Cu-promoted Aβ accumulation can be reversed (Folk and Franz 2010). One study conjugated deferiprone derivatives to glucose, concealing its chelating groups to prevent systemic metal binding but facilitate the crossing of the BBB via glucose receptors (Schugar et al. 2007). Another study described the attachment of carbohydrate moieties to chelators to aid in BBB transport via glucose transporters while restricting metal ion binding until the carbohydrate moieties were removed by a β-glucosidase enzyme (Storr et al. 2007). A series of 8-hydroxyquinoline pro-chelator derivatives with a hydroxyl group substituted with tetra-O-acetyl- and tetra-O-benzyl-β-dglucopyranoside and galactopyranosides was described. In this work, deglycosylation of glycosyl-substituted 8-hydroxyquinoline derivatives was produced by the addition of Cu2+, Zn2+, and Fe3+ (Chang and Ho 2006).
Owing to the multi-factorial nature of AD, various compounds and molecules with multiple functions have been designed. Pyclen (tetraazamacrocycles) and its derivatives have been described as antioxidants with the ability to suppress metal-induced Aβ formation, metal ion chelation, and protection against ROS-induced cell death (Green et al. 2010; Lincoln et al. 2013). In vitro biological investigation of series deferiprone-resveratrol hybrids revealed strong antioxidant activity, good inhibitory activity against self-induced Aβ aggregation in the micro-molar range, and a robust metal chelating capacity (Xu et al. 2017b). Multifunctional compounds based on the natural product coumarin’s ring structure fused to a metal binding moiety were created. The prime molecule from this series was able to bind Cu2+ demonstrated by UV–Vis spectroscopy and was also able to inhibit MAO-A and MAO-B (Huang et al. 2015). Another study broadened a family of coumarin-fused metal chelating compounds by joining the scaffolds at a different points on the coumarin ring. The best molecule from this series was able to inhibit MAO-A and MAO-B more potently and also revealed Cu2+ binding activity along with radical scavenging and anti-oxidant activity (Wang et al. 2015). The study focusing on multi-target directed ligand properties synthesized a novel series of compounds by integrating pharmacophores of resveratrol and clioquinol, which demonstrated excellent multi-target directed ligand properties with significant ability to bind Cu2+ with potential anti-oxidant activity (Mao et al. 2014). In their design of multifunctional molecules, one study used the aurone scaffold, onto which they directly installed metal chelating groups using the incorporation technique. The best molecule from this series was able to inhibit MAO-A and MAO-B with inhibition of Cu2+ induced Aβ aggregation (Li et al. 2016). Pyridoxine and resveratrol hybrids containing mannich base moieties were produced and found to preferentially inhibit acetylcholinesterase, MAO-B, and display antioxidant activity (Yang et al. 2017). Thus, multi-functional compounds not only assist in inhibiting metal-protein interaction, but they also help to solubilize Aβ protein and hence can be promising candidates in AD therapy.
Opportunities and Challenges in Essential Metal Chelation Therapy in AD Patients
Chelation therapy has a range of distinctive challenges when used to treat CNS diseases (Shahandeh et al. 2020). The chelator of choice must be well absorbed through the digestive system and should bind only to the specific metal ion and not to other biologically important divalent metals such as calcium (Ca2+) and magnesium (Mg2+). Also, chelation therapy has some toxic effects. In certain cases, iron chelation therapy has been shown to cause retinal toxicity. The incidence of retinal pigment epithelium mottling in patients receiving chronic deferoxamine for conditions such as transfusion-related hemosiderosis ranged from 1.2 to 9% (Cohen et al. 1990; Baath et al. 2008; Di Nicola et al. 2015). Deferoxamine at high doses has been linked to ocular toxicity (Dunaief 2006). The causes of retinal toxicity in some patients, as well as the mechanism by which it occurs, are unknown. The retinal pigment epithelium tends to be the primary site of deferoxamine ocular toxicity (Haimovici et al. 2002; Klettner et al. 2010). These findings highlight the importance of routine ocular monitoring and screening for chelation therapy patients. Furthermore, iron chelation as a therapeutic intervention for retinal disease would necessitate careful dose titration and/or the use of new compounds with better safety profiles. Current FDA-approved iron chelating agents which have been on the market for nearly 50 years have certain adverse effects. However, they have been beneficial in extending the life expectancy of thalassemia and haemochromatosis patients (Vitrano et al. 2017). The prescription of metal chelators must be based on the risk-to-benefit ratio (Origa 2017). Cu combines with other metals in amyloid plaques, as a result, there is an overall deficiency of Cu in the neuronal cells but an overload in the extracellular plaques. According to recent research, intracellular Cu deficit increases Aβ formation, whereas extracellular Cu2+ pooling promotes Aβ precipitation. Eventually, neither copper chelation nor copper supplementation is likely to lead to anticipated benefits (Lei et al. 2021). D-penicillamine, ionophore PBT2, and clioquinol have been used in small sample-size clinical trials in AD patients and have reported several adverse effects as depicted in Table 3. However, larger trials are still required to demonstrate the cognitive efficacy of these chelators.
In 2018, the National Institute on Aging-Association Alzheimer’s (NIA-AA) presented A/T/N diagnostic criteria, which included Aβ, p-tau, t-tau in CSF, and positron emission tomography (PET). However, due to the invasiveness of lumbar puncture for CSF measurement and the lack of popularity of PET, researchers are looking for biomarkers that are minimally invasive, easy to collect, and cost-effective (Feng et al. 2021). Xu et al. used inductively coupled plasma mass spectrometry to detect seven critical metals (including iron, copper, and zinc) and selenium in plasma samples from AD and matched control cases to see if metal dysregulation might be used to generate biomarkers in AD. Plasma metal levels did not differ between cases and controls in the entire research group or among female patients. In men, however, there was some evidence that Zn levels in AD were trending upward as compared to controls. In short, these findings imply that changes in plasma metal levels associated with AD may differ between genders (Xu et al. 2018).
Releasing Cu trapped by amyloid (or tangles) or facilitating their uptake into tissue may have a number of beneficial effects in AD, but based on the data, it is difficult to assign the recorded benefits to metal ions. In comparison to Fe, where a type of controlled cell lethality called ferroptosis could be at play, impaired extracellular Cu turnover could likely lead to amyloid formation and neurophysiological dysfunction; however, it is not yet clear how this could propel neurodegeneration. A vast number of contradictory findings have been presented on the changes or role of Zn in AD. Zinc supplementation caused Aβ deposition as well as decreased spatial memory in APP/PS1 mice, but had no effect on Tg2576 mice (Vilella et al. 2020). Dietary Zn deprivation, on the other hand, increased plaque size in APP/PS1 mice (Stoltenberg et al. 2007). In a drosophila model of AD overexpression Aβ, eye damage was seen, which was increased by zinc or copper supplementation but restored by zinc/copper chelators (Hua et al. 2011). Nutritional Zn deficiency is common in the elderly and worsens age-related cognitive loss in rodents, but zinc supplementation can assist (Sandusky-Beltran et al. 2017). In addition, Zn therapy was observed to delay hippocampal-dependent memory deficits and reduce Aβ in adult 3xTg-AD mice (Corona et al. 2010). A meta-analysis of clinical studies using zinc supplementation, on the other hand, found no indication of efficacy in the treatment of AD (Corona et al. 2010; Loef et al. 2012).
Releasing Zn and Cu trapped by amyloid (or tangles) or increasing their uptake into tissue could have a number of beneficial effects in AD, albeit based on the evidence, assigning the asserted benefits to each metal ion is problematic. In contrast to Fe, where a type of regulated cell lethality known as ferroptosis may be at work, while low extracellular Zn and Cu turnover may contribute to the amyloid accumulation and neurophysiological dysfunction, it is unclear how this may promote neurodegeneration.
Concluding Remarks
The physiological processes of the brain rely on metal ion homeostasis. Metal ions and their transporters have been found to be unbalanced in AD patients and animal models of AD. Furthermore, the homeostasis of these metals in the brain changes with age, which could explain why age is the leading risk factor for AD. Abnormal metal accumulation in various CNS regions can disrupt mitochondrial function, resulting in oxidative stress, which can lead to the excessive production of reactive oxygen species (ROS) and consequently initiate a cascade of pathogenic events in the brain. Elevated or unbalanced metal ions can generate or aggravate Aβ overproduction, tau hyperphosphorylation, and Aβ/tau aggregation in AD by regulating particular protein kinases and/or phosphatases or β-, α-, γ-secretases or producing oxidative stress. The plaques in AD are mostly made up of Aβ peptides and trace metals like Zn2+, Cu2+, and Fe2+. Metal chelators reduce the overall pool of bioavailable metals or compete for metal ions as ionophores with the endogenous ligand. As a result, metal chelation therapy for AD may now be regarded as a promising clinical treatment option. However, employing excessive doses of chelators still carries the danger of aggravating the illness. More clinical studies are needed to provide a more comprehensive view of intracellular metal trafficking, particularly in mitochondria, and a mechanistic interpretation of the effects of chelators, in order to improve our understanding of these neurodegenerative disorders and develop new therapeutic strategies. Also, measuring metal co-regulation in plasma may provide a useful depiction of metal disturbances occurring in the AD brain and hence could be useful as plasma-based biomarkers. However, only a limited number of studies are available on these metal-co-regulating biomarkers in body fluids. Thus, more research is required to explore biomarker-based diagnostic strategies for early prediction and diagnosis of AD. Furthermore, more research is also needed to establish the metal chelation process in vivo and to develop potential anti-AD medicines that not only sequester metal ions but also limit Aβ aggregation by competing with metal ions, reducing metal-induced oxidative damage and neurotoxicity.
Data Availability
Not applicable.
References
Adlard PA, Bush AI (2018a) Metals and Alzheimer’s disease: how far have we come in the clinic? J Alzheimer’s Dis 62:1369–1379. https://doi.org/10.3233/JAD-170662
Adlard PA, Bush AI (2018b) Metals and Alzheimer’s disease: how far have we come in the clinic? J Alzheimer’s Dis 62:1369–1379
Adlard PA, Parncutt J, Lal V et al (2015) Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol Dis 81:196–202. https://doi.org/10.1016/j.nbd.2014.12.012
Ashraf A, Ashton NJ, Chatterjee P et al (2020) Plasma transferrin and hemopexin are associated with altered Aβ uptake and cognitive decline in Alzheimer’s disease pathology. Alzheimers Res Ther 12. https://doi.org/10.1186/S13195-020-00634-1
Ayton S, Bush AI (2019) Decreasing iron neurotoxicity in pantothenate kinase-associated neurodegeneration. Lancet Neurol 18:616–617
Ayton S, Wang Y, Diouf I et al (2020) Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry 25:2932–2941. https://doi.org/10.1038/S41380-019-0375-7
Baath JS, Lam W-C, Kirby M, Chun A (2008) Deferoxamine-related ocular toxicity. Retina 28:894–899. https://doi.org/10.1097/IAE.0b013e3181679f67
Bagheri S, Squitti R, Haertlé T et al (2018) Role of copper in the onset of Alzheimer’s disease compared to other metals. Front Aging Neurosci 9. https://doi.org/10.3389/FNAGI.2017.00446
Bai B, Wang X, Li Y et al (2020) Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105:975-991.e7. https://doi.org/10.1016/j.neuron.2019.12.015
Baldari S, Di Rocco G, Toietta G (2020a) Current biomedical use of copper chelation therapy. Int J Mol Sci 21:1069. https://doi.org/10.3390/ijms21031069
Baldari S, Rocco G Di, Toietta G (2020b) Current biomedical use of copper chelation therapy. Int J Mol Sci 2020b, Vol 21, Page 1069 21:1069. https://doi.org/10.3390/IJMS21031069
Bao WD, Pang P, Zhou XT et al (2021) Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ 28:1548–1562. https://doi.org/10.1038/S41418-020-00685-9
Barman Balfour JA, Foster RH (1999) Deferiprone Drugs 58:553–578. https://doi.org/10.2165/00003495-199958030-00021
Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidatives stress. Nat Rev Drug Discov 3:205–214
Bayer TA, Schäfer S, Simons A et al (2003) Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Aβ production in APP23 transgenic mice. Proc Natl Acad Sci U S A 100:14187–14192. https://doi.org/10.1073/pnas.2332818100
Behzadfar L, Abdollahi M, Sabzevari O et al (2017) Potentiating role of copper on spatial memory deficit induced by beta amyloid and evaluation of mitochondrial function markers in the hippocampus of rats. Metallomics 9:969–980. https://doi.org/10.1039/C7MT00075H
Ben-Shushan S, Miller Y (2021) Neuropeptides: roles and activities as metal chelators in neurodegenerative diseases. J Phys Chem B 125:2796–2811. https://doi.org/10.1021/ACS.JPCB.0C11151
Bica L, Liddell JR, Donnelly PS et al (2014) Neuroprotective copper bis(thiosemicarbazonato) complexes promote neurite elongation. PLoS One 9:e90070. https://doi.org/10.1371/journal.pone.0090070
Bishop GM, Robinson SR (2004) The amyloid paradox: amyloid-β-metal complexes be neurotoxic and neuroprotective. Brain Pathol 14:448–452. https://doi.org/10.1111/J.1750-3639.2004.TB00089.X
Brzozowska A (1989) Interaction of iron, zinc and copper in the body of animals and humans. Rocz Panstw Zakl Hig 40:302–312
Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci 26:207–214. https://doi.org/10.1016/S0166-2236(03)00067-5
Bush AI, Pettingell WH, Paradis MD, Tanzi RE (1994) Modulation of A beta adhesiveness and secretase site cleavage by zinc. J Biol Chem 269:12152–12158. https://doi.org/10.1016/S0021-9258(17)32694-7
Ceccom J, Coslédan F, Halley H et al (2012) Copper chelator induced efficient episodic memory recovery in a non-transgenic Alzheimer’s mouse model. PLoS One 7:e43105. https://doi.org/10.1371/journal.pone.0043105
Chang CW, Ho TL (2006) A new glycosylation method based on 8-quinolyl glycosides. J Chinese Chem Soc 53:1567–1570. https://doi.org/10.1002/JCCS.200600204
Charkoudian LK, Pham DM, Franz KJ (2006) A pro-chelator triggered by hydrogen peroxide inhibits iron-promoted hydroxyl radical formation. J Am Chem Soc 128:12424–12425. https://doi.org/10.1021/JA064806W
Chaves S, Várnagy K, Santos MA (2021) Recent multi-target approaches on the development of anti- Alzheimer’s agents integrating metal chelation activity. Curr Med Chem 28:7247–7277. https://doi.org/10.2174/0929867328666210218183032
Chen C, Jiang X, Li Y et al (2019) Low-dose oral copper treatment changes the hippocampal phosphoproteomic profile and perturbs mitochondrial function in a mouse model of Alzheimer’s disease. Free Radic Biol Med 135:144–156. https://doi.org/10.1016/j.freeradbiomed.2019.03.002
Cherny RA, Legg JT, McLean CA et al (1999) Aqueous dissolution of Alzheimer’s disease Aβ amyloid deposits by biometal depletion. J Biol Chem 274:23223–23228. https://doi.org/10.1074/jbc.274.33.23223
Choi BS, Zheng W (2009) Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res 1248:14–21. https://doi.org/10.1016/J.BRAINRES.2008.10.056
Cohen A, Martin M, Mizanin J et al (1990) Vision and hearing during deferoxamine therapy. J Pediatr 117:326–330. https://doi.org/10.1016/S0022-3476(05)80556-6
Corona C, Masciopinto F, Silvestri E et al (2010) Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis 110(1):e91–e91. https://doi.org/10.1038/cddis.2010.73
Cristóvão JS, Santos R, Gomes CM (2016) Metals and neuronal metal binding proteins implicated in Alzheimer’s disease. Oxid Med Cell Longev
Crouch PJ, Lin WH, Adlard PA et al (2009) Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc Natl Acad Sci U S A 106:381–386. https://doi.org/10.1073/pnas.0809057106
Curnock R, Cullen PJ (2020) Mammalian copper homeostasis requires retromer-dependent recycling of the high-affinity copper transporter 1. J Cell Sci 133. https://doi.org/10.1242/JCS.249201
Curtain CC, Ali FE, Smith DG et al (2003) Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-β peptide with membrane lipid. J Biol Chem 278:2977–2982. https://doi.org/10.1074/jbc.M205455200
D’Mello SR, Kindy MC (2020) Overdosing on iron: elevated iron and degenerative brain disorders. Exp Biol Med (maywood) 245:1444–1473. https://doi.org/10.1177/1535370220953065
Dalvi T, Dewangan B, Agarwal G et al (2021) Design, synthesis and in-vitro evaluation of fluorinated triazoles as multi-target directed ligands for Alzheimer disease. Bioorg Med Chem Lett 42. https://doi.org/10.1016/J.BMCL.2021.127999
Das N, Raymick J, Sarkar S (2021) Role of metals in Alzheimer’s disease. Metab Brain Dis 36:1627–1639. https://doi.org/10.1007/S11011-021-00765-W
Datki Z, Galik-Olah Z, Janosi-Mozes E et al (2020) Alzheimer risk factors age and female sex induce cortical Aβ aggregation by raising extracellular zinc. Mol Psychiatry 2511(25):2728–2741. https://doi.org/10.1038/s41380-020-0800-y
De Benedictis CA, Vilella A, Grabrucker AM (2019) The role of trace metals in Alzheimer’s disease. Alzheimer’s Dis 85–106. https://doi.org/10.15586/ALZHEIMERSDISEASE.2019.CH6
Devos D, Moreau C, Devedjian JC et al (2014) Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxidants Redox Signal 21:195–210. https://doi.org/10.1089/ars.2013.5593
Di Nicola M, Barteselli G, Dell’Arti L et al (2015) Functional and structural abnormalities in deferoxamine retinopathy: a review of the literature. Biomed Res Int 2015
Dodani SC, Firl A, Chan J et al (2014) Copper is an endogenous modulator of neural circuit spontaneous activity. Proc Natl Acad Sci U S A 111:16280–16285. https://doi.org/10.1073/pnas.1409796111
Doll S, Conrad M (2017) Iron and ferroptosis: a still ill-defined liaison. IUBMB Life 69:423–434. https://doi.org/10.1002/iub.1616
Drew SC (2017) The case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease. Front Neurosci 11. https://doi.org/10.3389/FNINS.2017.00317
Du L, Zhao Z, Cui A et al (2018) Increased iron deposition on brain quantitative susceptibility mapping correlates with decreased cognitive function in Alzheimer’s disease. ACS Chem Neurosci 9:1849–1857. https://doi.org/10.1021/ACSCHEMNEURO.8B00194
Dubois B, Hampel H, Feldman HH et al (2016) Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement 12:292–323. https://doi.org/10.1016/J.JALZ.2016.02.002
Dunaief JL (2006) Iron induced oxidative damage as a potential factor in age-related macular degeneration: the Cogan lecture. Investig Ophthalmol vis Sci 47:4660–4664. https://doi.org/10.1167/iovs.06-0568
Ejaz HW, Wang W, Lang M (2020) Copper toxicity links to pathogenesis of Alzheimer’s disease and therapeutics approaches. Int J Mol Sci 21:1–33. https://doi.org/10.3390/IJMS21207660
Elmaleh DR, Farlow MR, Conti PS et al (2019) Developing effective Alzheimer’s disease therapies: clinical experience and future directions. J Alzheimer’s Dis 71:715–732. https://doi.org/10.3233/JAD-190507
Esmieu C, Guettas D, Conte-Daban A et al (2019) Copper-targeting approaches in Alzheimer’s disease: how to improve the fallouts obtained from in vitro studies. Inorg Chem 58:13509–13527. https://doi.org/10.1021/ACS.INORGCHEM.9B00995
Everett J, Collingwood JF, Tjendana-Tjhin V et al (2018) Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale 10:11782–11796. https://doi.org/10.1039/c7nr06794a
Fan J, Tao W, Li X et al (2019) The contribution of genetic factors to cognitive impairment and dementia: apolipoprotein E gene, gene interactions, and polygenic risk. Int J Mol Sci 20:. https://doi.org/10.3390/IJMS20051177
Fang L, Gou S, Fang X et al (2013) Current progresses of novel natural products and their derivatives/ analogs as anti-Alzheimer candidates: an update. Mini Rev Med Chem 13:870–887. https://doi.org/10.2174/1389557511313060009
Farr AC, Xiong MP (2021) Challenges and opportunities of deferoxamine delivery for treatment of Alzheimer’s disease, Parkinson’s disease, and intracerebral hemorrhage. Mol Pharm 18:593–609. https://doi.org/10.1021/ACS.MOLPHARMACEUT.0C00474
Fasae KD, Abolaji AO, Faloye TR et al (2021) Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer’s disease: limitations, and current and future perspectives. J Trace Elem Med Biol 67. https://doi.org/10.1016/J.JTEMB.2021.126779
Feng L, Li J, Zhang R (2021) Current research status of blood biomarkers in Alzheimer’s disease: diagnosis and prognosis. Ageing Res Rev 72. https://doi.org/10.1016/J.ARR.2021.101492
Fine JM, Baillargeon AM, Renner DB et al (2012) Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1α, and phosphorylates GSK3β in P301L tau transgenic mice. Exp Brain Res 219:381–390. https://doi.org/10.1007/s00221-012-3101-0
Fodero-Tavoletti MT, Villemagne VL, Paterson BM et al (2010) Bis (thiosemicarbazonato) Cu-64 complexes for positron emission tomography imaging of Alzheimer’s disease. J Alzheimer’s Dis 20:49–55. https://doi.org/10.3233/JAD-2010-1359
Folk DS, Franz KJ (2010) A prochelator activated by beta-secretase inhibits Abeta aggregation and suppresses copper-induced reactive oxygen species formation. J Am Chem Soc 132:4994–4995. https://doi.org/10.1021/JA100943R
Foster CM, Kennedy KM, Daugherty AM, Rodrigue KM (2020) Contribution of iron and Aβ to age differences in entorhinal and hippocampal subfield volume. Neurology 95:e2586–e2594. https://doi.org/10.1212/WNL.0000000000010868
Fulgenzi A, Vietti D, Ferrero ME (2020) EDTA chelation therapy in the treatment of neurodegenerative diseases: an update. Biomedicines 8:269. https://doi.org/10.3390/biomedicines8080269
Furuta T, Ohshima C, Matsumura M et al (2016) Oxidative stress upregulates zinc uptake activity via Zrt/Irt-like protein 1 (ZIP1) in cultured mouse astrocytes. Life Sci 151:305–312. https://doi.org/10.1016/J.LFS.2016.03.025
Galante D, Cavallo E, Perico A, D’Arrigo C (2018) Effect of ferric citrate on amyloid-beta peptides behavior. Biopolymers 109. https://doi.org/10.1002/BIP.23224
Ganguly G, Chakrabarti S, Chatterjee U, Saso L (2017) Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des Devel Ther 11:797–810. https://doi.org/10.2147/DDDT.S130514
Garai K, Sahoo B, Kaushalya SK et al (2007) Zinc lowers amyloid-beta toxicity by selectively precipitating aggregation intermediates. Biochemistry 46:10655–10663. https://doi.org/10.1021/BI700798B
Gauthier S, Rosa-Neto P, Morais J, Webster C (2021) World Alzheimer report 2021: journey through the diagnosis of dementia
Georgieff MK, Krebs NF, Cusick SE (2019) The benefits and risks of iron supplementation in pregnancy and childhood. Annu Rev Nutr 39:121–146. https://doi.org/10.1146/ANNUREV-NUTR-082018-124213
Gerber H, Wu F, Dimitrov M et al (2017) Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-β peptide production *. J Biol Chem 292:3751–3767. https://doi.org/10.1074/JBC.M116.754101
Gerhold D, Kim HH, Tong Z-B (2021) Biomarkers of neurotoxicity inform mechanisms of vulnerability and resilience in dopaminergic neurons. Handb Neurotox 1–15. https://doi.org/10.1007/978-3-030-71519-9_183-1
Gleason A, Bush AI (2021) Iron and ferroptosis as therapeutic targets in Alzheimer’s disease. Neurotherapeutics 18:252–264. https://doi.org/10.1007/S13311-020-00954-Y
Green DE, Bowen ML, Scott LE et al (2010) In vitro studies of 3-hydroxy-4-pyridinones and their glycosylated derivatives as potential agents for Alzheimer’s disease. Dalton Trans 39:1604–1615. https://doi.org/10.1039/B918439B
Gromadzka G, Tarnacka B, Flaga A, Adamczyk A (2020) Copper dyshomeostasis in neurodegenerative diseases-therapeutic implications. Int J Mol Sci 21:1–35. https://doi.org/10.3390/IJMS21239259
Guanjun Y, Yang G-J, Liu H et al (2019) Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy screening BRD4 inhibitors view project rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. Artic JBIC J Biol Inorg Chem 24:1159–1170. https://doi.org/10.1007/s00775-019-01712-y
Guo C, Zhang Y-X, Wang T et al (2015) Intranasal deferoxamine attenuates synapse loss via up-regulating the P38/HIF-1α pathway on the brain of APP/PS1 transgenic mice. Front Aging Neurosci 7:104. https://doi.org/10.3389/fnagi.2015.00104
Haimovici R, D’Amico DJ, Gragoudas ES, Sokol S (2002) The expanded clinical spectrum of deferoxamine retinopathy. Ophthalmology 109:164–171. https://doi.org/10.1016/S0161-6420(01)00947-2
Haque MM, Murale DP, Kim YK, Lee JS (2019) Crosstalk between oxidative stress and tauopathy. Int J Mol Sci 20. https://doi.org/10.3390/IJMS20081959
Hare D, Ayton S, Bush A, Lei P (2013) A delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci 5
Hentze MW, Muckenthaler MU, Galy B, Camaschella C (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142:24–38. https://doi.org/10.1016/J.CELL.2010.06.028
Hoke DE, Tan J-L, Ilaya NT et al (2005) In vitro gamma-secretase cleavage of the Alzheimer’s amyloid precursor protein correlates to a subset of presenilin complexes and is inhibited by zinc. FEBS J 272:5544–5557. https://doi.org/10.1111/J.1742-4658.2005.04950.X
Hua H, Münter L, Harmeier A et al (2011) Toxicity of Alzheimer’s disease-associated Aβ peptide is ameliorated in a Drosophila model by tight control of zinc and copper availability. Biol Chem 392:919–926. https://doi.org/10.1515/BC.2011.084
Huang M, Xie SS, Jiang N et al (2015) Multifunctional coumarin derivatives: monoamine oxidase B (MAO-B) inhibition, anti-β-amyloid (Aβ) aggregation and metal chelation properties against Alzheimer’s disease. Bioorg Med Chem Lett 25:508–513. https://doi.org/10.1016/J.BMCL.2014.12.034
Huang X, Cuajungco MP, Atwood CS et al (1999) Cu(II) potentiation of Alzheimer aβ neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274:37111–37116. https://doi.org/10.1074/jbc.274.52.37111
Huang XT, Liu X, Ye CY et al (2018) Iron-induced energy supply deficiency and mitochondrial fragmentation in neurons. J Neurochem 147:816–830. https://doi.org/10.1111/jnc.14621
Huat TJ, Camats-Perna J, Newcombe EA et al (2019) Metal toxicity links to Alzheimer’s disease and neuroinflammation. J Mol Biol 431:1843–1868. https://doi.org/10.1016/J.JMB.2019.01.018
Ijomone OM, Ifenatuoha CW, Aluko OM et al (2020) The aging brain: impact of heavy metal neurotoxicity. Crit Rev Toxicol 50:801–814. https://doi.org/10.1080/10408444.2020.1838441
Kabir MT, Uddin MS, Jeandet P et al (2021a) Anti-Alzheimer’s molecules derived from marine life: understanding molecular mechanisms and therapeutic potential. Mar Drugs 19. https://doi.org/10.3390/MD19050251
Kabir MT, Uddin MS, Zaman S et al (2021b) Molecular mechanisms of metal toxicity in the pathogenesis of Alzheimer’s disease. Mol Neurobiol 58. https://doi.org/10.1007/S12035-020-02096-W
Kawahara M, Kato-Negishi M, Tanaka KI (2020) Amyloids: regulators of metal homeostasis in the synapse. Molecules 25. https://doi.org/10.3390/MOLECULES25061441
Kessler H, Bayer TA, Bach D et al (2008) Intake of copper has no effect on cognition in patients with mild Alzheimer’s disease: a pilot phase 2 clinical trial. J Neural Transm 115:1181–1187. https://doi.org/10.1007/s00702-008-0080-1
Killick SB (2017) Iron chelation therapy in low risk myelodysplastic syndrome. Wiley Online Libr 177:375–387. https://doi.org/10.1111/bjh.14602
Kim J-J, Kim Y-S, Kumar V (2019) Heavy metal toxicity: an update of chelating therapeutic strategies. Elsevier. https://doi.org/10.1016/j.jtemb.2019.05.003
Kitazawa M, Cheng D, LaFerla FM (2009) Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J Neurochem 108:1550–1560. https://doi.org/10.1111/j.1471-4159.2009.05901.x
Klettner A, Koinzer S, Waetzig V et al (2010) Deferoxamine mesylate is toxic for retinal pigment epithelium cells in vitro, and its toxicity is mediated by p38. Cutan Ocul Toxicol 29:122–129. https://doi.org/10.3109/15569521003745685
Klopstock T, Tricta F, Neumayr L et al (2019) Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: a randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol 18:631–642. https://doi.org/10.1016/S1474-4422(19)30142-5
Krishnan HS, Bernard-Gauthier V, Placzek MS et al (2018) Metal protein-attenuating compound for PET neuroimaging: synthesis and preclinical evaluation of [11C]PBT2. Mol Pharm 15:695–702. https://doi.org/10.1021/ACS.MOLPHARMACEUT.7B00936
Kuperstein F, Yavin E (2004) Pro-apoptotic signaling in neuronal cells following iron and amyloid beta peptide neurotoxicity. J Neurochem 86:114–125. https://doi.org/10.1046/j.1471-4159.2003.01831.x
Lang M, Fan Q, Wang L et al (2013) Inhibition of human high-affinity copper importer Ctr1 orthologous in the nervous system of Drosophila ameliorates Aβ42-induced Alzheimer’s disease-like symptoms. Neurobiol Aging 34:2604–2612. https://doi.org/10.1016/j.neurobiolaging.2013.05.029
Lawson MK, Valko M, Cronin MTD, Jomová K (2016) Chelators in iron and copper toxicity. Curr Pharmacol Reports. https://doi.org/10.1007/s40495-016-0068-8
Lee JY, Friedman JE, Angel I et al (2004) The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human β-amyloid precursor protein transgenic mice. Neurobiol Aging 25:1315–1321. https://doi.org/10.1016/j.neurobiolaging.2004.01.005
Lei P, Ayton S, Bush AI (2019) Axonal dispatch of iron in neuronal signaling. Nat Chem Biol 15:1135–1136. https://doi.org/10.1038/S41589-019-0394-3
Lei P, Ayton S, Bush AI (2021) The essential elements of Alzheimer’s disease. J Biol Chem 296:100105. https://doi.org/10.1074/JBC.REV120.008207
Lei P, Ayton S, Finkelstein DI et al (2012) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18:291–295. https://doi.org/10.1038/nm.2613
Li Y, Qiang X, Luo L et al (2016) Synthesis and evaluation of 4-hydroxyl aurone derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem 24:2342–2351. https://doi.org/10.1016/J.BMC.2016.04.012
Lincoln KM, Gonzalez P, Richardson TE et al (2013) A potent antioxidant small molecule aimed at targeting metal-based oxidative stress in neurodegenerative disorders. Chem Commun (camb) 49:2712–2714. https://doi.org/10.1039/C2CC36808K
Liu B, Moloney A, Meehan S et al (2011) Iron promotes the toxicity of amyloid β peptide by impeding its ordered aggregation. J Biol Chem 286:4248–4256. https://doi.org/10.1074/jbc.M110.158980
Liu P-P, Xie Y, Meng X-Y, Kang J-S (2019) History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther 4:37. https://doi.org/10.1038/s41392-019-0071-8
Loef M, von Stillfried N, Walach H (2012) Zinc diet and Alzheimer’s disease: a systematic review. Nutr Neurosci 15:2–12. https://doi.org/10.1179/1476830512Y.0000000010
Loef M, Walach H (2021) Systematic review copper and iron in Alzheimer’s disease: a systematic review and its dietary implications. cambridge.org. https://doi.org/10.1017/S000711451100376X
Lovell MA, Xie C, Markesbery WR (1999) Protection against amyloid beta peptide toxicity by zinc. Brain Res 823:88–95. https://doi.org/10.1016/S0006-8993(99)01114-2
Lyubartseva G, Smith JL, Markesbery WR, Lovell MA (2010) Alterations of zinc transporter proteins ZnT-1, ZnT-4 and ZnT-6 in preclinical Alzheimer’s disease brain. Brain Pathol 20:343–350. https://doi.org/10.1111/J.1750-3639.2009.00283.X
Mao F, Yan J, Li J et al (2014) New multi-target-directed small molecules against Alzheimer’s disease: a combination of resveratrol and clioquinol. Org Biomol Chem 12:5936–5944. https://doi.org/10.1039/C4OB00998C
Martic S, Rains MK, Kraatz HB (2013) Probing copper/tau protein interactions electrochemically. Anal Biochem 442:130–137. https://doi.org/10.1016/j.ab.2013.07.015
Martin-Bastida A, Ward RJ, Newbould R et al (2017) Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci Rep 7:1–9. https://doi.org/10.1038/s41598-017-01402-2
McLachlan DRC, Kruck TPA, Kalow W et al (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337:1304–1308. https://doi.org/10.1016/0140-6736(91)92978-B
Mercer SW, Wang J, Burke R (2017) In vivo modeling of the pathogenic effect of copper transporter mutations that cause Menkes and Wilson diseases, motor neuropathy, and susceptibility to Alzheimer’s disease. J Biol Chem 292:4113–4122. https://doi.org/10.1074/jbc.M116.756163
Opazo C, Huang X, Cherny RA et al (2002) Metalloenzyme-like activity of Alzheimer’s disease β-amyloid: Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J Biol Chem 277:40302–40308. https://doi.org/10.1074/jbc.M206428200
Origa R (2017) β-Thalassemia. Genet Med 19:609–619
Paoletti P, Vergnano AM, Barbour B, Casado M (2009) Zinc at glutamatergic synapses. Neuroscience 158:126–136. https://doi.org/10.1016/J.NEUROSCIENCE.2008.01.061
Peng Y, Chang X, Lang M (2021) Iron homeostasis disorder and Alzheimer’s disease. Int J Mol Sci 22. https://doi.org/10.3390/IJMS222212442
Peters DG, Pollack AN, Cheng KC et al (2018) Dietary lipophilic iron alters amyloidogenesis and microglial morphology in Alzheimer’s disease knock-in APP mice. Metallomics 10:426–443. https://doi.org/10.1039/C8MT00004B
Phinney AL, Drisaldi B, Schmidt SD et al (2003) In vivo reduction of amyloid-β by a mutant copper transporter. Proc Natl Acad Sci U S A 100:14193–14198. https://doi.org/10.1073/pnas.2332851100
Ping L, Kundinger SR, Duong DM et al (2020) Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. Sci Data 7:1–13. https://doi.org/10.1038/s41597-020-00650-8
Prasad AS (2013) Impact of the discovery of human zinc deficiency on health. 28:257–265. https://doi.org/10.1080/07315724.2009.10719780
Pretsch D, Rollinger JM, Schmid A et al (2020) Prolongation of metallothionein induction combats Aß and α-synuclein toxicity in aged transgenic Caenorhabditis elegans. Sci Rep 10. https://doi.org/10.1038/S41598-020-68561-7
Pyun J, McInnes LE, Donnelly PS et al (2022) Copper bis(thiosemicarbazone) complexes modulate P-glycoprotein expression and function in human brain microvascular endothelial cells. J Neurochem. https://doi.org/10.1111/JNC.15609
Qian ZM, Ke Y (2020) Hepcidin and its therapeutic potential in neurodegenerative disorders. Med Res Rev 40:633–653. https://doi.org/10.1002/MED.21631
Rana M, Cho HJ, Roy TK et al (2018) Azo-dyes based small bifunctional molecules for metal chelation and controlling amyloid formation. Inorganica Chim Acta 471:419–429. https://doi.org/10.1016/J.ICA.2017.11.029
Rao SS, Portbury SD, Lago L et al (2020) The iron chelator deferiprone improves the phenotype in a mouse model of tauopathy. J Alzheimers Dis 78:1783. https://doi.org/10.3233/JAD-209009
Rembach A, Hare DJ, Lind M et al (2013) Decreased copper in Alzheimer’s disease brain is predominantly in the soluble extractable fraction. Int J Alzheimers Dis 2013. https://doi.org/10.1155/2013/623241
Rezaei-Ghaleh N, Giller K, Becker S, Zweckstetter M (2011) Effect of Zinc Binding on β-Amyloid Structure and Dynamics: Implications for Aβ Aggregation. Biophys J 101:1202. https://doi.org/10.1016/J.BPJ.2011.06.062
Říha M, Karlíčková J, Filipský T et al (2013) Novel method for rapid copper chelation assessment confirmed low affinity of D-penicillamine for copper in comparison with trientine and 8-hydroxyquinolines. J Inorg Biochem 123:80–87. https://doi.org/10.1016/J.JINORGBIO.2013.02.011
Ritchie CW, Bush AI, Masters CL (2004) Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin Investig Drugs 13:1585–1592. https://doi.org/10.1517/13543784.13.12.1585
Rogers JT, Randall JD, Cahill CM et al (2002) An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem 277:45518–45528. https://doi.org/10.1074/jbc.M207435200
Rongioletti M, Fostinelli S, Ghidoni R et al (2018) Excess copper in Alzheimer disease but not in frontotemporal lobar degeneration: next-generation sequencing study of ATP7B gene in patients typified by high copper. Am J Clin Pathol 150:63–67. https://doi.org/10.1093/AJCP/AQY092
Sampson EL, Jenagaratnam L, McShane R (2012) Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane database Syst Rev 5. https://doi.org/10.1002/14651858.CD005380.PUB4
Sandusky-Beltran LA, Manchester BL, McNay EC (2017) Supplementation with zinc in rats enhances memory and reverses an age-dependent increase in plasma copper. Behav Brain Res 333:179–183. https://doi.org/10.1016/J.BBR.2017.07.007
Scheiber IF, Mercer JFB, Dringen R (2014) Metabolism and functions of copper in brain. Prog Neurobiol 116:33–57. https://doi.org/10.1016/J.PNEUROBIO.2014.01.002
Schipper HM, Song W, Tavitian A, Cressatti M (2019) The sinister face of heme oxygenase-1 in brain aging and disease. Prog Neurobiol 172:40–70
Schugar H, Green DE, Bowen ML et al (2007) Combating Alzheimer’s disease with multifunctional molecules designed for metal passivation. Angew Chem Int Ed Engl 46:1716–1718. https://doi.org/10.1002/ANIE.200603866
Shahandeh A, Bui BV, Finkelstein DI, Nguyen CTO (2020) Therapeutic applications of chelating drugs in iron metabolic disorders of the brain and retina. J Neurosci Res 98:1889–1904
Sharma A, Pachauri V, Flora SJS (2018) Advances in multi-functional ligands and the need for metal-related pharmacology for the management of Alzheimer disease. Front Pharmacol 9. https://doi.org/10.3389/FPHAR.2018.01247
Singh YP, Pandey A, Vishwakarma S, Modi G (2019) A review on iron chelators as potential therapeutic agents for the treatment of Alzheimer’s and Parkinson’s diseases. Mol Divers 23:509–526. https://doi.org/10.1007/S11030-018-9878-4
Skalny AV, Zaitseva IP, Gluhcheva YG et al (2019) Cobalt in athletes: hypoxia and doping - new crossroads. J Appl Biomed 17:28–28. https://doi.org/10.32725/jab.2018.003
Southon A, Szostak K, Acevedo KM et al (2020) Cu II (atsm) inhibits ferroptosis: implications for treatment of neurodegenerative disease. Br J Pharmacol 177:656–667. https://doi.org/10.1111/bph.14881
Squitti R, Rossini PM, Cassetta E et al (2002) D-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur J Clin Invest 32:51–59
Stoltenberg M, Bush AI, Bach G et al (2007) Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150:357–369. https://doi.org/10.1016/J.NEUROSCIENCE.2007.09.025
Storr T, Merkel M, Song-Zhao GX et al (2007) Synthesis, characterization, and metal coordinating ability of multifunctional carbohydrate-containing compounds for Alzheimer’s therapy. J Am Chem Soc 129:7453–7463. https://doi.org/10.1021/JA068965R
Su XY, Wu WH, Huang ZP et al (2007) Hydrogen peroxide can be generated by tau in the presence of Cu(II). Biochem Biophys Res Commun 358:661–665. https://doi.org/10.1016/j.bbrc.2007.04.191
Takahashi M, Doré S, Ferris CD et al (2000) Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 28:461–473. https://doi.org/10.1016/S0896-6273(00)00125-2
Taylor HBC, Emptage NJ, Jeans AF (2021) Long-term depression links amyloid-β to the pathological hyperphosphorylation of tau. Cell Rep 36. https://doi.org/10.1016/J.CELREP.2021.109638
Telling ND, Everett J, Collingwood JF et al (2017) Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer’s disease. Cell Chem Biol 24:1205-1215.e3. https://doi.org/10.1016/j.chembiol.2017.07.014
Tiffany-Castiglioni E, Hong S, Qian Y (2011) Copper handling by astrocytes: Insights into neurodegenerative diseases. Int J Dev Neurosci 29:811–818. https://doi.org/10.1016/J.IJDEVNEU.2011.09.004
Tisato V, Zuliani G, Vigliano M et al (2018) Gene-gene interactions among coding genes of iron-homeostasis proteins and APOE-alleles in cognitive impairment diseases. PLoS One 13. https://doi.org/10.1371/journal.pone.0193867
Tsatsanis A, Dickens S, Kwok JCF et al (2019) Post translational modulation of β-amyloid precursor protein trafficking to the cell surface alters neuronal iron homeostasis. Neurochem Res 44:1367–1374. https://doi.org/10.1007/s11064-019-02747-y
Tuo Q-Z, Guo Y (2019) Diagnostics and treatments of iron-related CNS diseases. Springer 1173:179–194. https://doi.org/10.1007/978-981-13-9589-5_10
Uddin MS, Kabir MT, Al Mamun A et al (2019) APOE and Alzheimer’s disease: evidence mounts that targeting APOE4 may combat Alzheimer’s pathogenesis. Mol Neurobiol 56:2450–2465. https://doi.org/10.1007/S12035-018-1237-Z
Uriu-Adams JY, Keen CL (2005) Copper, oxidative stress, and human health. Mol Aspects Med 26:268–298. https://doi.org/10.1016/J.MAM.2005.07.015
Urrutia PJ, Hirsch EC, González-Billault C, Núñez MT (2017) Hepcidin attenuates amyloid beta-induced inflammatory and pro-oxidant responses in astrocytes and microglia. J Neurochem 142:140–152. https://doi.org/10.1111/jnc.14005
van der Weerd L, Lefering A, Webb A et al (2020) Effects of Alzheimer’s disease and formalin fixation on the different mineralised-iron forms in the human brain. Sci Rep 10. https://doi.org/10.1038/S41598-020-73324-5
van Rensburg SJ, Carstens ME, Potocnik FCV et al (1993) Increased frequency of the transferrin C2 subtype in Alzheimer’s disease. NeuroReport 4:1269–1271. https://doi.org/10.1097/00001756-199309000-00015
Vela D (2018) The dual role of hepcidin in brain iron load and inflammation. Front Neurosci 12:740
Viktorinova A, Durfinova M (2021) Mini-review: is iron-mediated cell death (ferroptosis) an identical factor contributing to the pathogenesis of some neurodegenerative diseases? Neurosci Lett 745. https://doi.org/10.1016/J.NEULET.2021.135627
Vilella A, Daini E, De Benedictis CA, Grabrucker AM (2020) Targeting metal homeostasis as a therapeutic strategy for Alzheimer’s disease. Alzheimer’s Dis Drug Discov 83–98. https://doi.org/10.36255/EXONPUBLICATIONS.ALZHEIMERSDISEASE.2020.CH5
Vitrano A, Calvaruso G, Lai E et al (2017) The era of comparable life expectancy between thalassaemia major and intermedia: is it time to revisit the major-intermedia dichotomy? Br J Haematol 176:124–130. https://doi.org/10.1111/BJH.14381
Wang L, Yin YL, Liu XZ et al (2020a) Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl Neurodegener 91(9):1–13. https://doi.org/10.1186/S40035-020-00189-Z
Wang L, Yin YL, Liu XZ et al (2020b) Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl Neurodegener 9. https://doi.org/10.1186/S40035-020-00189-Z
Wang P, Wang ZY (2017) Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev 35:265–290. https://doi.org/10.1016/J.ARR.2016.10.003
Wang Q, Franz KJ (2016) Stimulus-responsive prochelators for manipulating cellular metals. Acc Chem Res 49:2468–2477. https://doi.org/10.1021/ACS.ACCOUNTS.6B00380
Wang XQ, Zhao CP, Zhong LC et al (2018) Preparation of 4-flexible amino-2-arylethenyl-quinoline derivatives as multi-target agents for the treatment of Alzheimer’s disease. Molecules 23. https://doi.org/10.3390/MOLECULES23123100
Wang Y, Wu Y, Li T et al (2019a) Iron metabolism and brain development in premature infants. Front Physiol 10. https://doi.org/10.3389/FPHYS.2019a.00463
Wang Z, Zeng YN, Yang P et al (2019b) Axonal iron transport in the brain modulates anxiety-related behaviors. Nat Chem Biol 15:1214–1222. https://doi.org/10.1038/S41589-019-0371-X
Wang ZM, Xie SS, Li XM et al (2015) Multifunctional 3-Schiff base-4-hydroxycoumarin derivatives with monoamine oxidase inhibition, anti-β-amyloid aggregation, metal chelation, antioxidant and neuroprotection properties against Alzheimer’s disease. RSC Adv 5:70395–70409. https://doi.org/10.1039/C5RA13594J
Xu H, Perreau VM, Dent KA et al (2016) Iron regulates apolipoprotein e expression and secretion in neurons and astrocytes. J Alzheimer’s Dis 51:471–487. https://doi.org/10.3233/JAD-150797
Xu J, Church SJ, Patassini S et al (2017a) Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 9:1106–1119. https://doi.org/10.1039/C7MT00074J
Xu J, Church SJ, Patassini S et al (2018) Plasma metals as potential biomarkers in dementia: a case-control study in patients with sporadic Alzheimer’s disease. Biometals 31:267–276. https://doi.org/10.1007/S10534-018-0089-3
Xu P, Zhang M, Sheng R, Ma Y (2017b) Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ 1–42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur J Med Chem 127:174–186. https://doi.org/10.1016/J.EJMECH.2016.12.045
Xu Y, Xiao G, Liu L, Lang M (2019) Zinc transporters in Alzheimer’s disease. Mol Brain 12. https://doi.org/10.1186/S13041-019-0528-2
Xu Y, Zhang Y, Zhang JH et al (2020) Astrocyte hepcidin ameliorates neuronal loss through attenuating brain iron deposition and oxidative stress in APP/PS1 mice. Free Radic Biol Med 158:84–95. https://doi.org/10.1016/j.freeradbiomed.2020.07.012
Xu Z, Yang S, Zhao S et al (2016) Biomarkers for early diagnostic of mild cognitive impairment in type-2 diabetes patients: a multicentre, retrospective, nested case–control study. Elsevier. https://doi.org/10.1016/j.ebiom.2016.02.014
Yang GJ, Liu H, Ma DL, Leung CH (2019) Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem 24:1159–1170. https://doi.org/10.1007/S00775-019-01712-Y
Yang LPH, Keam SJ (2007) Keating GM (2012) Deferasirox. Drugs 6715(67):2211–2230. https://doi.org/10.2165/00003495-200767150-00007
Yang X, Qiang X, Li Y et al (2017) Pyridoxine-resveratrol hybrids Mannich base derivatives as novel dual inhibitors of AChE and MAO-B with antioxidant and metal-chelating properties for the treatment of Alzheimer’s disease. Bioorg Chem 71:305–314. https://doi.org/10.1016/J.BIOORG.2017.02.016
Yao D, Jing T, Niu L et al (2018) Amyloidogenesis induced by diet cholesterol and copper in a model mouse for Alzheimer’s disease and protection effects of zinc and fluvastatin. Brain Res Bull 143:1–8. https://doi.org/10.1016/j.brainresbull.2018.09.003
Yiannikourides A, Latunde-Dada G (2019) A short review of iron metabolism and pathophysiology of iron disorders. Med (basel, Switzerland) 6:85. https://doi.org/10.3390/MEDICINES6030085
Yokel RA (2006) Blood-brain barrier flux of aluminum, manganese, iron and other metals suspected to contribute to metal-induced neurodegeneration. J Alzheimer’s Dis 10:223–253
Zhang Y, Xie J-Z, Xu XY, et al (2019) Liraglutide ameliorates hyperhomocysteinemia-induced Alzheimer-like pathology and memory deficits in rats via multi-molecular targeting. Neurosci Bull 35:724–734. https://doi.org/10.1007/s12264-018-00336-7
Zhou Y, Song WM, Andhey PS et al (2020) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26:131–142. https://doi.org/10.1038/s41591-019-0695-9
Acknowledgements
The authors acknowledge the financial support provided by the Core Research Grant of DST SERB, Government of India (CRG/2020/003316). The authors are also thankful to Shobhaben Pratapbhai Patel School of Pharmacy & Technology Management, SVKM’s NMIMS for providing the infrastructure facility.
Author information
Authors and Affiliations
Contributions
Ginpreet Kaur and Harpal S. Buttar visualized the presented idea and supervised the project. Vinay Chaudhari, Shubhangi Gupta, Siddhi Bagwe-Parab, and Amisha Vora contributed in doing literature searches and preparing the manuscript draft. Ginpreet Kaur and Harpal S Buttar revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chaudhari, V., Bagwe-Parab, S., Buttar, H.S. et al. Challenges and Opportunities of Metal Chelation Therapy in Trace Metals Overload-Induced Alzheimer’s Disease. Neurotox Res 41, 270–287 (2023). https://doi.org/10.1007/s12640-023-00634-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-023-00634-7