
Abstract
Changes in the transition metal homeostasis in the brain are closely linked with Alzheimer’s disease (AD), including intraneuronal iron accumulation and extracellular copper and zinc pooling in the amyloid plague. The brain copper, zinc, and iron surplus are commonly acknowledged characteristics of AD, despite disagreements among some. This has led to the theory that oxidative stress resulting from abnormal homeostasis of these transition metals may be a causative explanation behind AD. In the nervous system, the interaction of metals with proteins appears to be an essential variable in the development or suppression of neurodegeneration. Chelation treatment may be an option for treating neurodegeneration induced by transition metal ion dyshomeostasis. Some clinicians even recommend using chelating agents as an adjunct therapy for AD. The current review also looks at the therapeutic strategies that have been attempted, primarily with metal-chelating drugs. Metal buildup in the nervous system, as reported in the AD, could be the result of compensatory mechanisms designed to improve metal availability for physiological functions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) has long been accepted as the most prevalent type of dementia, constituting 60–70% of all dementia cases globally. As per the Alzheimer’s Association’s 2022 US report, AD-related deaths have surged by 145% from 2000 to 2019 in the USA [1]. AD was associated with 121,499 deaths in 2019, making it the seventh-highest cause of human death for individuals of age 65 years and over. Conversely, the predicted cost to treat and care for this dementia in 2022 was roughly $321 billion USD. By 2050, this sum is predicted to rise to roughly $1.0 trillion USD [2]. In spite of the fact that AD is thought to be a disease of specific protein aggregation in the brain, it is also being researched as a disease of several metal dyshomeostasis. Age-related metal dyshomeostasis consequently has a causal association with the pathogenesis of AD because it is a key factor for AD. A change in the metal ion concentrations in the neurons, as well as the buildup of protein deposits due to metal-induced oxidative damage, serves as an indicator of AD. In the brain, transition metal ions such as copper (Cu), zinc (Zn), and iron (Fe) are essential for the normal operation of enzymes, neurotransmission, and aging. Additionally, mounting research has connected metal dyshomeostasis to a number of neurodegenerative disorders. For example, in AD, the presence of Cu, Zn, and Fe in the plaques is proportional to their localization with Aβ protein in the brain [3]. The field of metallobiology has remarkably contributed in the study of the relationship between metals, metalloproteins, and neurodegenerative disorders, including AD. Aging is the key cause of AD, but there is evidence that AD is also linked to the dysregulation of some trace metal ions such as Cu, Zn, and Fe, as well as other elements like aluminum (Al), manganese (Mn), lead (Pb), and cadmium (Cd). The biochemical and physiological processes of the human body depend on Fe, Zn, Cu, and Mn, which also play important roles in protein regulation and structural activities, cellular signaling, and oxygen transport. Each organ’s metal distribution inside the human body varies [4]. For instance, Fe, which typically originates as heme Fe (Fe attached to hemoglobin inside the red blood cells), is the most predominant trace metal in the brain. Along with helping to transport oxygen, Fe work as a cofactor for numerous enzymes involved in the production of neurotransmitters, including tryptophan and tyrosine hydroxylase. Fe also has a number of other roles in the central nervous system (CNS), such as controlling synaptic plasticity, myelinating neuronal axons, and controlling neuronal energy status [5, 6]. In the humanoid brain, Zn is the second most predominant trace element that modulates synaptic plasticity similarly to Fe. Furthermore, it controls neurogenesis, neuronal migration, and differentiation in addition to participating in neurotransmission [7]. Over 300 enzymes utilize Zn as a cofactor, making it an essential structural and catalytic component of proteins. Additionally, it affords stability to numerous transcription factors. Last but not least, Zn has neuroprotective properties and can protect against oxidative damage [8, 9]. The 3rd most common trace element identified in the brain is Cu. Cu, like Zn, functions as an enzymatic cofactor for a number of other biological enzymes and is essential for the biosynthesis of neurotransmitters [10]. Several biological enzymes, including pyruvate carboxylase, glutamine synthetase, MnSOD, arginase, and threonine phosphatase-1/protein serine, require manganese as a cofactor. As a result, trace metals play a vital role in the regulation of gene expression, the activation of enzymes and defense against reactive oxygen species (ROS). The proper growth, development, and function of the brain depend on them. The blood–brain barrier (BBB) tightly controls the homeostasis of these charged elements, and numerous transport proteins actively move metal ions across the BBB. Any alteration in the balance of metals, which may be brought on by defects in the mechanisms of absorption, bio-distribution and excretion, mutations in the proteins that transport or bind metals, or competition between metals ions for the specific binding sites (such as Zn and Cu), may result in inequity of vital trace metals or improved the levels of trace elementsspecifically non-essential [11, 12].
Transition Metals in AD
Transition metals, such as Cu, Fe, and Zn, are essential for numerous bio-processes and brain neuronal activities. When compared to those seen in healthy brains, Cu, Fe, and Zn are overexposed and assemble in the amyloid plaques of AD patients by up to 5.7-, 2.9-, and 2.8-fold, respectively [13]. The synaptic clefts in the brain need high concentrations of free metal ions for synaptic transmission [14]. Even while metal ions are crucial for many biological functions, a disturbance of the brain's equilibrium can result in neuronal degeneration and death [15]. According to studies, roughly 50% of all proteins need to be combined with metals to form metalloproteins in order to function more effectively [16]. As a result, the CNS needs rather high levels of several metals, including Cu, Fe, and Zn. These metals are useful for regulating gene expression, enzyme activity, neural activity, and cell structure maintenance. A substantial malfunction of numerous enzymes, such as Cu–Zn superoxide dismutase (SOD-1) and cytochrome oxidase (COX), has been linked to a disproportion in the concentration of Cu ions. Through the generation of ROS and oxidative stress, elevated Fe levels in the neutrophils of an AD brain are closely associated with disease [13, 17, 18].
Zinc
Zn ion is one of the most predominant divalent metals in the brain and a non-redox-active essential element that plays a significant role in the chemistry of brain neurons [19, 20]. Apart from Fe, Zn is the most widespread metal in the body, with the olfactory bulb, neocortex, hippocampus, and amygdale regions of the brain having the highest amounts [21, 22]. Either static or labile describes the Zn ion pool in the brain. While Zn is strongly attached to different metalloproteins in the static pool, its protein binding can be easily broken in the labile pool, making Zn more moveable. Additionally, 90% of the Zn in the brain is bound; this Zn primarily serve as a structural constituent of transcription factors like c-Fos and p53 as well as receptors for vitamin D, retinoic acid, and thyroid hormone, all of which comprise Zn-binding motifs. Zn is also necessary for the normal operation of metalloenzymes such as superoxide dismutase-1 (Zn, Cu-dependent SOD) [23]. Zn ions have also been linked to the degeneration of neurons, particularly in AD, where it has been found to play a key role in the formation of amyloid plaques as well as the exact operation and control of a number of Zn proteins [24]. Neurological problems have been linked to both Zn excess and deficiency.
Additionally, Zn plays a dual role, both exacerbating neurotoxicity and offering neuroprotection. In AD, disrupted Zn levels, particularly elevated levels in amyloid plaques, worsen the disease by fostering the aggregation of Aβ peptides and subsequent neuronal harm. Conversely, Zn exhibits neuroprotective qualities: it acts as an antioxidant, regulates synaptic function, and enhances synaptic plasticity, all of which are impaired in AD [25]. Research indicates that Zn supplementation in animal models of AD can mitigate cognitive decline, reduce Aβ accumulation, and improve synaptic function. However, caution is necessary with Zn supplementation, as excessive levels can be toxic. Thus, understanding the optimal dosage and timing of Zn supplementation is crucial for its potential therapeutic benefits in AD, highlighting its intricate and multifaceted role in the disease [26]. The Zn therapy offers a promising approach to tackling AD, supported by its neuroprotective properties and ability to counteract neurodegeneration. Its well-tolerated nature, with minimal gastric issues and no long-term side effects, establishes it as a safe treatment option, reinforced by regular biomarker monitoring to ensure safety and efficacy. With its natural composition and mechanism of inducing metallothionein to regulate Cu absorption, Zn emerges as an appealing therapeutic avenue for individuals with mild cognitive impairment and disrupted Cu levels, reminiscent of Wilson disease (WD). Despite earlier clinical trials falling short of primary endpoints, Zn has shown effectiveness in reducing non-ceruloplasmin Cu levels and enhancing cognitive function without notable adverse effects [27].
Copper
Cu, an essential micronutrient, is crucial for all the aerobic species because it acts as a prosthetic group to speed up the transfer of electrons to key enzymatic processes [28]. The brain needs Cu for healthy growth and operation, and an average adult’s brain contains roughly 7.3% of the body’s total Cu [29]. The nucleus, mitochondria, and cytosol are the three main cell compartments in the brain that contain Cu. However, it is not evenly distributed throughout the CNS. For instance, adult rats’ brains contain high concentrations of Cu in the lateral amygdale, some regions of the midbrain, forebrain, and gray matter, whereas the levels in the hippocampus are modest [30]. But in humans, the hippocampus has higher Cu concentrations than other parts of the brain. Consequently, different species have diverse distribution patterns for Cu. Numerous brain areas may have higher Cu concentrations due to increased Cu demand [30]. Ceruloplasmin, dopamine hydroxylase, cytochrome-c oxidase, and Cu, Zn-dependent superoxide dismutase (Cu-ZnSOD) are just a few of the key enzymes to which Cu can readily bind. Cu is a redox-active solid metal that can exist in oxidized and reduced states. These states are all crucial for various metabolic processes. On the other hand, due to Cu’s redox activity, it can take part in Fenton and Haber–Weiss reactions that result in the production of ROS, which are thought to be a pathological component of neurodegenerative illnesses [31]. Due to Cu’s dual purpose, import, export, and distribution must all be strictly regulated [29]. Similar to Zn, there is a dispute regarding Cu’s contribution to AD’s onset and progression. However, there is general agreement that the interactions between the amyloid precursor protein (APP) and the cleavage result, amyloid-β (Aβ), play a significant role in the link between Cu and AD [32].
Iron
The body and the brain contain the greatest amounts of Fe, a transition metal. All kinds of brain cells, including astrocytes, neurons, microglia, and oligodendrocytes, contain it [33]. The brain requires a lot of energy, but it also needs a lot of Fe. Because it is an integral part of cytochrome oxidase, cytochromes a, b, and c, as well as the Fe-sulfur ion complexes of the electron transport chain (ETC), Fe plays a crucial role in the synthesis of adenosine triphosphate (ATP). Additionally, it functions as a co-factor for essential brain enzymes like tyrosine hydroxylase and tryptophan hydroxylase, which are crucial for the synthesis of neurotransmitters [34]. Both an excess and a shortage of Fe are linked to neurodegeneration, much like Cu and Zn [35]. Motor impairments, reduced dopamine activity, and irregularities in myelinogenesis have all been linked to Fe shortage in the brain [34]. Due to its redox activity, Fe poses a hazard to the brain when present in excess. Similar to Cu, Fe can take part in Fenton reactions that consequence in the production of superoxide and hydroxyl radicals that can interrelate with DNA, lipids, and proteins. Highly controlled systems are in place to maintain brain Fe homeostasis [23].
Transition Metal Dyshomeostasis in AD
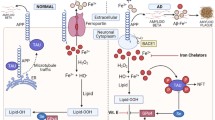
Transition metal dyshomeostasis, involving Cu, Fe, and Zn, is a complicated aspect of AD pathogenesis. The consequence of interactions between the metal ions and the crucial proteins APP and its derivatives, Aβ and tau, metal imbalances in the brain are seen in AD [4, 36] (Fig. 1). This imbalance led to oxidative stress, protein aggregation, and synaptic dysfunction in the CNS, all of which are the most critical components of AD pathology. APP, which is primarily expressed in the humanoid brain, has a role in neurite outgrowth and neuronal cell migration [37]. Between residues 142 and 166 of APP, Cu binds to the protein, playing crucial functions in the protein’s structural stability, including folding stability and homodimerization, as well as expression levels [38]. In order to control the homodimerization of APP, Zn can also cooperate with APP at the location between 170 and 188 positions of amino acid [39]. Furthermore, there is reciprocal regulation between Fe and APP: Fe can directly control the translation of APP, and levels of APP regulate the Fe export in the neurons [40]. The Aβ is resulting from APP in the process of consecutive proteolysis produced by β and γ-secretases. A pathogenic aspect of AD, insoluble amyloid fibrils of Aβ are linked to metals like Fe, Cu, and Zn. By binding to Aβ at the histidine-13 (His-13) and His-6 sites, Zn aids in the constancy of amyloid fibrils [41]. The crucial involvement of Zn in this process has been highlighted by the discovery of high Zn concentrations at the level of senile plaques in postmortem tissues of AD patients and in plaques of hereditary AD animal models. The neurotoxicity of Aβ is thought to be significantly influenced by Cu. Three highly affine His Cu-binding sites are present on the monomeric A: His-6, His-13, and His-14. They have been observed to combine with Cu ions, the N-terminal of an amino group, and aspartate to create a tetragonal complex [42]. Additionally, Cu has a functional role in the production of β-sheet structures, which serve as the building blocks for the poisonous complexes of the fibrillary form of Aβ. Cu chelators were found to be able to stop the buildup of Aβ in transgenic AD animal models, according to a number of investigations [43]. At the binding sites of Glu3, Asp1, and the three His residues (His6, His13, and His14), Fe also interacts with Aβ. This interaction results in the generation of free radicals through Fenton chemistry [6, 44].

Metal ion, oxidative stress, and Aβ-cascade hypotheses in AD. Transition metal ions induce the production of ROS through an oxidative state and subsequently stimulate mitochondrial dysfunction and accumulation of ROS in mitochondria. Metals ion induces the Aβ-aggregation via direct binding to Aβ proteins. Aβ can also show oligomeric alteration to the proto-fibrils and fibrils, which actuate the mitochondria and finally lead to extra ROS production. Ultimately, ROS damages the proteins and other biological molecules in the CNS, increasing the risk of AD
Meta-analyses investigating the complicated relationship between Cu, Fe, and Zn in AD provide a systematic and quantitative approach to consolidating our understanding of their roles within the pathology. These analyses offer crucial insights into how these metals interconnect and influence the progression of AD, predominantly within the context of human serum/plasma and brain specimens [45, 46]. By focusing on the levels of Zn and Fe, future meta-analyses can expand our comprehension of their interplay with Cu, specifically through pathways involving ceruloplasmin and metallothioneins (MTs). Addressing the gaps in present research involves investigating the specific roles of genes encoding enzymes and transporters responsible for regulating Cu balance in AD. By employing a sequencing hypothesis-driven approach, researchers can identify metal-related gene variants in individuals with Cu imbalance, thus illuminating their prevalence and potential contributions to AD pathogenesis [46]. The hypothesis posits that variants in genes like ATP7B may confer a percentage of the risk for intermittent AD. Despite previous genome-wide association studies (GWAS) failing to establish a significant association between AD and single-nucleotide polymorphisms (SNP predominantly decent Aβ-plaque collectors) in the ATP7B chromosomal region, this could be due to the limitations of GWAS in detecting multiple rare variants [47]. Such variants may play a crucial role in explaining the missing heritability of complex diseases like AD. To advance our understanding further, it is imperative to integrate chemical and biochemical scientific studies into future research efforts. This approach will offer valued insights into the molecular mechanisms underlying Cu dysregulation and its implications for AD. By amalgamating findings from meta-analyses with genetic and biochemical studies, researchers can unravel the complex network of interactions between Cu, Zn, and Fe. This integrative approach paves the way for the development of targeted therapeutic interventions for AD. Moreover, meta-analyses have clarified the multifaceted roles of Cu, Fe, and Zn in AD pathology [48].
Numerous scientific studies suggest that Cu, Fe, and Zn dysregulation and metal-catalyzed oxidative damage contribute to AD [6]. Neocortex Cu, Fe, and Zn levels were not supported by a new meta-analysis study that reported on the metal ion levels in the AD brain, while neocortex Cu levels were considerably lower when evaluating quantitative analyses. In the same study, it was discovered that there was a strong scientific publication bias, with research papers mentioning higher Fe levels being referenced far more regularly than those reporting lower or unchanged levels [49]. Nevertheless, there is highly strong indication to support the idea that some Aβ-plaque forms contain Fe, Cu, and Zn, and that intracellular or local metal ions levels may vary [49, 50]. While Aβ-plaque related Fe and Cu ions can redox cycle and generate ROS, Zn does not, but has been described to be a predominant Aβ-plaque collector and a tau-protein hyperphosphorylation inducer [51, 52]. The Fe-export ferroxidase activity of APP may also be inhibited by Zn [18]. There is evidence of considerable brain A-plaque depositions in 20–40% of elderly individuals with normal cognitive function, indicating that not all plaque types are harmful [53]. An additional recent meta-analysis discovered that AD patients had considerably higher serum Cu levels than controls. In clinical research on AD in humans and AD animal models, positive results predominate when metal chelators are administered therapeutically [15].
Cu, is the most important trace element, has been implicated in AD pathology through its involvement in Aβ aggregation and deposition, oxidative stress induction, and synaptic dysfunction. Meta-analyses have revealed that Cu dysregulation may exacerbate neurodegeneration by facilitating Aβ plaque formation and promoting oxidative damage to neurons. Additionally, disruptions in Cu homeostasis have been linked to impaired synaptic transmission, contributing to cognitive decline in AD. Squitti et al. conducted a study to explore Cu dysregulation as a potential susceptibility factor for AD using meta-analysis and replication studies. Their findings demonstrated reduced Cu levels in AD brain specimens, elevated Cu and non-bound ceruloplasmin (Non-Cp) Cu in serum/plasma samples from humans, with no significant change in ceruloplasmin levels [54]. They observed that excess Cu in serum/plasma was linked to a three to four-fold increase in the risk of developing AD. Additionally, they noted a higher frequency of carriers of the ATP7B AG haplotype, associated with the Cu transporter ATPase7B, in the AD group. These results suggest a failure to maintain Cu metabolic balance in AD patients and imply a subset of individuals predisposed to Cu imbalance. The study underscores the potential for precision medicine-based approaches targeting Cu dysregulation in specific AD subtypes [54]. Another study investigates the possible etiological role of Cu imbalance in AD, emphasizing the need for a detailed understanding of the fundamental biochemical mechanisms rather than merely observing correlations. It offers that as the brain ages, there is a shift in Cu balance from bound to loosely bound metal ions, which may lead to mitochondrial dysfunction, neuronal energy depletion, and aggravated protein misfolding, all contributing to neurodegeneration. The findings highlight the importance of integrating Cu imbalance with genetic predispositions, ageing-related changes, metabolic dysregulation, and other pathological processes like neuroinflammation and protein aggregation. Notably, the study highlights the intricate relationship between Cu imbalance and the amyloid hypothesis, suggesting that interactions between Cu and Aβ may play a significant role in AD pathogenesis. This holistic perspective highlights the complexity of AD and the necessity for multifaceted therapeutic approaches targeting Cu homeostasis together with other pathological factors [55].
Similarly, Fe accumulation in the brain has been associated with oxidative stress, neurotoxicity, and tau protein aggregation, all of which are hallmarks of AD pathology. Meta-analyses highlight the role of Fe in catalyzing the production of ROS, leading to neuronal damage and BBB dysfunction, thus exacerbating neurodegeneration in AD. Gong and co-workers performed the meta-analysis and presented valuable insights into the role of Fe and its related proteins in AD. It reveals notable distinctions in Fe levels in blood, ferritin levels in CSF, and lactoferrin levels in serum between AD patients and healthy controls. Specifically, AD patients exhibited lower Fe levels in blood, higher ferritin levels in CSF, and lower lactoferrin levels in serum compared to healthy controls. These findings suggest a potential link between disturbances in Fe metabolism and AD pathology. Elevated ferritin levels in CSF imply possible Fe accumulation in brain tissues, contributing to neurodegeneration, while decreased lactoferrin levels in serum may indicate a risk factor for AD progression. The study emphasizes the need for further research to explore the diagnostic potential of Fe-related biomarkers and investigate the association between Fe status and AD symptoms. Overall, the study underscores the importance of understanding Fe dysregulation in AD and the necessity for continued investigation into its clinical implications [56]. Ayton et al. recently updated the Fe hypothesis of AD by incorporating recent large-scale evidence. While early AD research focused on the potential role of Fe in triggering the disease through its association with proteinopathies, new findings suggest a more complex relationship. Specifically, normal-range brain levels of Fe appear to accelerate disease progression in individuals with underlying proteinopathic neuropathology, representing an independent effect of Fe on neurodegeneration. This hypothesis suggests that tissue Fe levels may influence the likelihood of neurodegeneration in AD, potentially through mechanisms such as ferroptosis. The outcomes of the subsequent study are expected to provide further insights into the role of Fe in AD pathology, offering potential therapeutic targets and diagnostic avenues [57].
Zn, another essential metal, exhibits dual roles in AD pathology, influencing Aβ metabolism and synaptic function. While some studies suggest that Zn may promote Aβ aggregation, others propose that it enhances Aβ clearance from the brain. Moreover, dysregulation of Zn homeostasis has been associated with neurotoxicity and synaptic dysfunction, further contributing to cognitive impairment in AD. Squitti et al. performed a meta-analysis of 27 studies conducted between 1983 and 2014, revealing significant heterogeneity in both the demographic characteristics and methodological approaches of the included studies, which persisted despite our attempts to address them. However, a constant finding across the studies was a decrease in serum Zn levels among AD patients compared to healthy controls. This decrease remained statistically significant even when serum and plasma studies were combined. Interestingly, when we specifically looked at age-matched studies, the difference in Zn levels between AD patients and healthy controls was not significant, suggesting a potential influence of age on Zn metabolism in the context of AD. We hypothesize that the observed reductions in serum Zn may indicate a possible dietary Zn deficiency and propose that alterations in Zn levels could interact with Cu metabolism in AD [58]. These findings have prompted further investigation into the relationship between Zn and Cu dysregulation in AD progression, with the aim of identifying potential therapeutic targets or diagnostic markers.
Mechanisms
Multiple mechanisms have been identified as contributing to AD; however, these changes do not occur in all cases. The pathophysiology of AD is thought to be caused by neuronal extracellular deposition of Aβ peptides (senile/amyloid plaques) and intracellular accumulation of hyperphosphorylated tau protein, which forms neurofibrillary tangles (NFTs). The changes in the protein dynamics and the accumulation of proteins occur due to the deficiency of the ubiquitin-proteosome-autophagy system. However, synaptic disruption is the most common underlying cause of cognitive and behavioral dysfunction in AD cases. The neural dysfunction is linked to higher levels of Aβ and phosphorylated tau, which diminish synaptic strength by aggregating in the dendritic spine and internalizing N-methyl-d-aspartic acid receptors (NMDARs). Other factors, such as oxidative stress, which increases in the brain with age, have been linked to the formation of senile plaques and the deposition of NFTs [59, 60]. Because Aβ causes oxidative stress which further increases Aβ deposition, oxidative stress and Aβ are inevitably intertwined. Further, mitochondrial dysfunction, impaired bioenergetics, dysfunction of neurotrophins, and disruptions of neuronal Golgi apparatus and axonal transport are also factors that contribute to AD. Furthermore, elevated inflammatory cytokine levels and related genes have been associated with the development of AD. These interrelated mechanisms result in neuronal death over a long period of time, which is known as “neurodegeneration” [61]. In the upcoming sub-sections, some well-accoladed hypotheses linked to the etiology of AD have been discussed briefly.
Oxidative Stress
Evidence suggests that clinical and pathological signs of AD, including Aβ accumulation, NFTs development, metabolic changes, and psychological impairments, result from long-term progressive oxidative damage [62,63,64]. Furthermore, oxidative stress is a contributing aspect in the pathophysiology of numerous risk factors of AD. Free radicals-induced damage, as identified in AD, comprises advanced glycation end products (AGEs), nitration, peroxidation of lipids, carbonyl-related post-translational modifications of neurofilament protein, and free carbonyls [65,66,67]. Because the brain membrane has polyunsaturated fatty acids (PUFA), it is highly sensitive to free radicals-induced injury. PUFA enables the elimination of hydrogen ions and increases lipid peroxidation, which is a majorly noticeable characteristic of degenerative change in the AD brain [68]. Furthermore, protein oxidation by increased levels of ROS is another important phenomenon in AD. This is because oxidation of CNS-associated proteins might impair the functions of proteins necessary for neuron and glial cell activities. Two enzymes are particularly susceptible to oxidative variations in AD brains: glutamine synthetase and creatine kinase. Both of these enzymes are significantly decreased in AD brains. This reduction reflects changes in the glutamate level and increased excitotoxicity. Additionally, alterations in the creatine kinase activity due to oxidative stress may impair energy homeostasis in AD [69]. The NFTs are distinguished by protein aggregation and Tau-protein hyperphosphorylation into the paired helical filaments. Phosphorylation is connected to the oxidation via microtubule-associated protein kinase pathway and transcription factor nuclear factor-κB (NF-κB) activation, significantly correlating oxidation with the protein hyperphosphorylation [70].
While it is yet unknown what causes the redox imbalance in AD, it is probable that the phenomenon is mainly reliant on redox-sensitive transition metals like Cu, Zn, and Fe [18, 71]. Dyshomeostasis of these redox-sensitive metals is noteworthy in context with an increase in the oxidative stress parameters, including lipid peroxidation and oxidative damage to NFTs, amyloid plaques, and nucleic acids [72, 73]. The N-terminal metal-binding domain of Aβ and its precursor APP contain high affinity binding sites for Cu and Zn [38, 74]. Cu is a potent carrier of the highly reactive hydroxyl radical (OH•), which subsequently contributes to the increase in oxidative burden and is found in high concentrations in the plaques. This appears to be dependent on the length of Aβ fragments, with Aβ (1–42) being more hazardous and capable of producing hydrogen peroxide (H2O2) and other free radicals than Aβ (1–40) [75]. Furthermore, elevated Zn concentration has been linked to memory and cognitive brain regions such as the neocortex, amygdala and hippocampus, all of which are impaired in AD. The binding of Zn induces a conformational change in Aβ (1–40), which eventually leads to the accumulation of toxic fibrillary Aβ aggregates. Therefore, the immunological/inflammatory response against non-soluble Aβ deposits includes dyshomeostasis of Zn content and uncontrolled cerebral Zn release. Thus, unregulated Zn or Aβ accumulation causes Zn- and Aβ-mediated oxidative stress and neurotoxicity [12, 76]. Recent histochemical studies have shown that prior treatment of tissue sections to Cu and Fe selective chelators inhibits the redox activity in lesions associated with AD. Re-exposing the chelator-treated tissues to Cu or Fe salts can restore activity, implying that the redox changes in AD relies on these metals [77]. As a result, it is likely that Fe and Cu accumulation is a key source of ROS generation, which is accountable for more widespread oxidative stress in AD. Further, free Fe has been associated with redox changes in-vivo, resulting in the formation of ROS. Neurodegenerative disorders, including AD have been linked to abnormally elevated levels of free Fe and oxidative stress [78, 79].
Aβ disrupts healthy mitochondrial function and induces redox imbalance [80]. Aberrant mitochondrial functioning is also associated with the etiology of AD, as increased mitochondrial DNA (mtDNA) oxidation is one of the early signs of the disease [81]. Actually, age-related dysfunctionality of mitochondria might be among the initial events in the pathophysiology of sporadic, late-onset AD. As per the mitochondrial cascade theory, age-related reduced mitochondrial activity impacts APP expression and processing, resulting in Aβ oligomers that subsequently aggregate to form plaques in AD [82, 83]. Decreased efficiency of electron transport caused by alteration of oxidative phosphorylation (OXPHOS) increases ROS formation, primarily at complex I and complex III sites [84]. These ROS negatively affect mitochondrial macromolecules when they are released into the environment. Besides increasing ROS production at the mitochondrial level, the peptide Aβ also prevents ROS clearance. In fact, studies have demonstrated that Aβ can prevent oxidative damage by inhibiting mitochondrial superoxide dismutase (MnSOD), the enzyme most crucial for scavenging superoxide radicals [85, 86]. Aβ can also bind to and inhibit mitochondrial alcohol dehydrogenase, commonly known as Aβ binding alcohol dehydrogenase (ABAD). ABAD plays a beneficial role in the body by detoxifying aldehydes, including 4-hydroxinonenal. The interaction between Aβ peptide and ABAD disrupts the enzyme’s detoxifying activity, causes ROS production, lipid peroxidation, and mitochondrial dysfunction [87]. It is known that ROS or lipid peroxidation products can upregulate uncoupling proteins (UCP2 and UCP3) to lower proton motive force and decrease mitochondrial membrane potential and ATP synthesis. Eventually, it results in mitochondrial uncoupling and a decrease in ROS formation from mitochondria [88]. As a result, UCP expression and activation are considered defensive mechanisms against oxidative stress. These protective phenomenons seem to be defective in the AD brains, where UCP2, 4, and 5 are downregulated [89]. The activation of UCP2 and UCP4 proteins in response to superoxide exposure was prevented in SH-SY5Y neuroblastoma cells over-expressing APP or APP mutant; although the mechanistic pathways were unknown, it indicated that Aβ deposition could contribute to permanent neuronal alterations [90].
Neuroinflammation
Neuroinflammation is an immune response characterized by glial cell activation and the generation of inflammatory mediators [91]. Several research findings prove neuroinflammation is a key player in AD pathology. Further, inflammatory mediators have been linked to the development of moderate cognitive deficit to apparent AD. Misfolded and aggregated proteins interact to the pattern recognition receptors (PRR) on microglia and astrocytes, eliciting an innate immune response demonstrated by the production of pro-inflammatory cytokines including IL-1, IL-6, and IL-18, chemokines like C–C motif chemokine ligand 1 (CCL1), CCL5, and C-X-C motif chemokine ligand 1 (CXCL1) [92,93,94]. Microarray analysis conducted by Cribbs and colleagues on young, old, and AD cases revealed an upsurge of the innate immune activity in the ageing brains and a minor rise in the associated genes, implying that inflammation is involved in the pathogenesis of AD [95]. Neuroinflammation in AD is a persistent response because microglia are already “primed” and hence extremely susceptible to subsequent insults, resulting in an immediate switch to the harmful M1 phenotype [96]. This microglial priming is presumably caused by a combination of activators, including chronic Aβ exposure, neuronal debris and chronic vascular alterations such as cerebrovascular dysfunction and cerebral micro-infarcts. Aβ may bind to many microglial receptors, causing the release of not only inflammatory mediators, but also a considerable number of ROS (•OH and O2− •), nitric oxide (•NO) and tumor necrosis factor-α (TNF-α) [97, 98]. The formation and secretion of inflammatory mediators can be responsible for the loss of synaptic plasticity, neuronal damage, and perturbations in the neurogenesis [99]. The TNF-α causes neurodegeneration by activating TNF-α- receptor 1 (TNFR1) and recruiting caspase 8. IL-1 induces loss of synaptic plasticity by increasing prostaglandin E2 production, which eventually increases release of presynaptic glutamate and activates postsynaptic NMDA receptors [100, 101].
Furthermore, the complement system can be activated by enhancing the phagocytic activity of microglia, which eventually may result in erroneous synaptic pruning [102]. Anti-inflammatory cytokines such as IL-1 receptor antagonists, IL-4, IL-10, and IL-11 are also involved in the neuroinflammatory cascade and may be a component of the complex mechanism for avoiding exaggeration of neuroinflammation [103]. However, neuroinflammation is a persistent phenomenon that does not get ameliorated on its own and is seen as a critical cause of the neurodegenerative diseases. The deposition of Aβ itself may be enough to cause an inflammatory response, which then promotes to cognitive loss and the onset and progression of AD. Neuroinflammation in AD is not a passive process activated by the continuously aggregating senile plaques and NFTs, but rather is equally responsible for the plaques and tangles formation [104]. The importance of neuroinflammation is reinforced by findings that immunological receptor genes, such as TREM22 and CD33, are linked to AD [105, 106]. Considering the possibility that Aβ deposition follows “cognitive deficits” by decades, exogenous or intrinsic factors may be able to affect the innate immune response that results from Aβ exposed microglia. Thus, modifiable environmental risk factors for AD, such as systemic inflammation, obesity, and traumatic brain injury, may influence risk via prolonged neuroinflammation.
The NLR family pyrin domain containing 3 (NLRP3) inflammasome is a protein complex that resides in the cytoplasm. It has recently been discovered to be implicated in the neuronal damage and innate immune response [108]. Recently, it has been shown that Aβ activates NLRP3 in microglia and astrocytes. This phenomenon leads to the production of caspase 1 and release of cytokines such as IL-1 and IL-18, which causes permanent damage. Conversely, inhibiting the NLRP3 inflammasome reduces Aβ accumulation and has a neuroprotective effect in a transgenic AD mice model [109,110,111]. Hence, accumulating evidence indicate Aβ as a key player in AD-related inflammation and oxidative stress (Fig. 2).

Interplay of Metal Ions in Oxidative Stress and Protein Activity. A Redox-active metal ions (Cu2+ and Fe3+) and associated complexes (along with Aβ) generate ROS via Fenton-like reactions, while Zn.2+ inhibits mitochondrial oxidative phosphorylation, leading to ROS production. SOD1 modification by ROS contributes to oxidative stress. B Aggregated Ab may serve as a reservoir for metal ions, influencing their bioavailability. Metal ion dyshomeostasis and/or oxidative stress can disrupt critical biological functions, such as ATP production, ROS breakdown, and maintenance of metal ion homeostasis, affecting protein activity. Proteins implicated include cytochrome c oxidase (CcO), Cu/Zn superoxide dismutase (SOD1), ceruloplasmin (Cp), metallothioneins (MTs), and hemeoxygenase 1 (HO1). [Reproduced from Lee et al. with kind permission of the Copyright holder, Royal Society of Chemistry, 2014] [107]
Protein Misfolding and Aggregation
Protein misfolding is a cellular phenomenon which is frequently observed over the course of a cell’s existence. Several phenomenons, including translational error, inappropriate protein modification, stress, and genetic mutations ultimately results in protein misfolding and aggregation [112]. Comparable to other neurodegenerative disorders, the primary cause of AD is the aggregation of brain proteins. Specific protein aggregation, including those of Aβ, phosphorylated tau and synuclein, is typically linked to AD.
Aβ Aggregation
In healthy neurons, alpha (α) and gamma (γ) secretase enzymes degrade the APP. This reaction generates some soluble polypeptides, which can later be broken down and recycled in the cell. In contrast, if the β-secretase and γ-secretase collaborate, the situation goes wrong. This digestive processing results in the formation of 40–42 amino acids long Aβ peptide fragments. These fragments come together to form oligomers, which can subsequently combine to form harmful insoluble Aβ plaques in the vicinity of neurons [113]. When Aβ plaques arise, it can cause significant problems in the cell. Aβ plaques may bind to the healthy neurons and interrupt their signalling pathways [114, 115]. When neurons fail to transmit impulses, the brain may be severely injured and some functions including memory are lost. It has also been shown that Aβ plaques can trigger an immunological response, causing inflammation and potentially damaging adjacent neurons [116]. Aβ aggregates can also deposit on the outside of blood vessels, causing angiopathy. Angiopathy will eventually result in haemorrhage or vascular rupture [117]. The widely accepted amyloid protein-based hypothesis suggests Aβ aggregation as the main cause driving AD pathogenesis [118]. Most of the characteristics of AD, namely the development of tau protein-containing NFTs, synaptic and neuronal damage, and neuroinflammation, are considered to be caused by an imbalance between Aβ production and elimination. However, there is still debate about whether Aβ plaques are a causal factor or an outcome of AD, but the existing evidence suggests the former is the main factor in AD pathology [119]. This ambiguity persists, especially in the context of the multiple AD medication failures, most of which were directed at key mechanisms important to the amyloid hypothesis. Although the amyloid hypothesis remains the most accepted theory, medicines aiming at decreasing Aβ levels have mainly proven unsuccessful. For instance, monoclonal antibodies against Aβ which have been approved as an anti-amyloid AD drug, such as lecanemab, only improve 27% of cognitive decline in early AD patients [120]. Additionally, aducanumab, the first to be approved by the Food and Drug Administration (FDA), was withdrawn from the market a few days ago due to poor efficacy and more side effects [121]. This strongly shows that Aβ does not cause the majority of the damage in AD. Other pathogenetic processes must therefore exist in AD, and these may account for the majority of the damage.
Structural modifications in the conformations of certain proteins or peptides, followed by their aggregation to form insoluble fibrils, have been hypothesized as the root cause of various neurodegenerative diseases, including AD [122]. These conformational changes can be established as a valuable tool for distinguishing different diseases [123]. As previously stated, mounting evidence suggests that monomeric Aβ peptides act as a basic unit and lead to the formation of amyloid fibers through several intermediary stages [124]. Aβ aggregation in AD has been reported to include a conformational transition from α-helix to expanded β-sheet in the protein [125]. The role of anti-parallel sheet structures is proposed for fibrils generated by short peptides with only one β-strand segment.
Metal dysmetabolism, involving the dysregulation of metals such as Cu, Fe, and Zn, extends its detrimental effects beyond its association with beta-amyloid in AD. Primarily, this dysmetabolism leads to oxidative stress, resulting in increased production of ROS and subsequent damage to cellular components such as lipids, proteins, and DNA. Additionally, dysregulated metal levels aggravate neuroinflammatory responses by activating microglia, which release pro-inflammatory cytokines and chemokines, eventually leading to neuronal injury and neurodegeneration. Disrupted metal homeostasis also impairs synaptic function and plasticity, thereby contributing to the cognitive deficits observed in AD [71, 126]. Moreover, mitochondrial dysfunction ensues from dysmetabolism, which diminishes energy production, elevates oxidative stress, and induces cellular dysfunction and apoptosis. Beyond beta-amyloid, metal dysmetabolism fosters protein misfolding and aggregation by binding to other proteins, such as tau, thereby exacerbating neurodegeneration. Furthermore, impaired metal ion transport disrupts essential metal-dependent processes and signaling pathways, further exacerbating cellular dysfunction and neurodegeneration. Consequently, metal dysmetabolism orchestrates multifaceted effects on neuronal function and viability, significantly contributing to the pathogenesis of AD [71, 127].
Tau Protein Aggregation
The Tau proteins on the surface of microtubules improve the packing architecture of microtubules. They interact to microtubules from the carboxy-terminal side, which contains three or four semi-homologous repeats of 31 or 32 amino acids [128]. It has been shown that Aβ plaques produced in extracellular regions activate signalling pathways within the cell that lead to the activation of kinases. Activated kinases provide a phosphate group to the tau protein, which then exits the microtubule. The phosphorylated tau proteins then aggregate and form NFTs. The absence of tau proteins weakens the microtubule, causing it to lose its signalling function, which can ultimately lead to cell death [113]. The aggregated tau protein conformation disrupts the axonal transport and contributes to microtubule destabilization [129]. When compared to healthy controls, AD patients have four times the number of aberrant or hyperphosphorylated tau proteins. Furthermore, the misfolded tau proteins lose their fundamental action of microtubule stabilization, possess increased aggregation ability, and are discovered to be potential neurotoxic agents [130, 131]. Finally, the functions of tau-microtubule are disrupted, resulting in the loss of synaptic plasticity and aberrant axonal transport, culminating in cognition related impairments.
Tau aggregates are capable of moving from one cell to other and are secreted in the extracellular space via a prion-like mechanism, resulting in the spread of tau related adverse effects in the distinct regions of the brain. These fundamental mechanisms have been used as therapeutic targets in the treatment of tau related pathologies and other neurodegenerative diseases [132, 133]. Several in-vivo investigations have provided significant evidence that establishes a synergistic relation between different pathogenic proteins, such as synthetic Aβ fibrils and tau-related pathologies [134, 135]. Besides this, these research findings showed a significant mechanistic relationship between amyloid and tau proteins and neurodegenerative disease. Tau protein has been proven to be an essential component in Aβ-induced cognitive failure, implying that the interaction between Aβ and tau is critical for the onset as well as progression of AD. As a result, investigating the possible molecular interactions between Aβ and tau pathology can be a valuable approach to understanding AD progression. The in-vitro investigations with several cell types, ranging from neuronal cell lines to cortical neurons, organotypic hippocampus, and native hippocampal cultures, revealed Aβ-mediated tau hyperphosphorylation and cytosolic as well as dendritic tau translocation [136, 137]. According to some recent findings, amyloid disease causes tau hyperphosphorylation in both the cytosol and synaptic regions. Reports have also shown that the presence of misfolded Aβ can increase the seeding properties and aggregation of human tau protein. It is also observed that the exposure of neuronal cell lines to the Aβ potentially activates the GSK-3β and thus enhances tau protein phosphorylation. This method of Aβ-mediated tau phosphorylation was subsequently tested using antisense suppression of GSK-3β and tau knockout mice, both of which failed to show Aβ-induced mortality [138, 139].
Therapeutic Strategy
For several decades, several therapeutic options have been investigated; nonetheless, there is currently no curative treatment, and the ultimate objective remains prevention. Since metallic burden is one of the causative factors for the development of AD, implementing metal restricted diet can be one of the therapeutic approaches for the disease. Further, chelating the excessive metals with a suitable chelating agent also serves the purpose of alleviating AD. Antioxidant therapy might be helpful in AD patients as oxidative stress is a primary phenomenon having an important role in the pathogenesis of AD.
Copper-Restricted Diet
A Cu-restricted diet exerts beneficial effects in AD management by minimizing oxidative damage and neuroinflammation. A metal-restricted diet reduces the production of ROS and ameliorates oxidative stress in the brain by reducing the intake of metals including Cu and Fe. This may assist in protecting neurons and slow down disease development. Furthermore, lowering metal-induced neuroinflammation may aid in cognitive preservation and neuronal function [47, 140]. Cu is recognized as an essential element vital for numerous physiological functions. Both Cu deficiency and excess can have serious health implications, including potential involvement in AD. Recent studies have highlighted alterations in Cu metabolism as a potential pathogenic mechanism in AD. Cu is primarily acquired through dietary sources, with food contributing around 75% and drinking water approximately 25% of the general population’s intake [47, 141]. This underscores the significance of dietary interventions in modulating Cu levels for AD prevention. Individuals with AD have been found to show disturbances in Cu metabolism, characterized by elevated levels of non-ceruloplasmin (non-Cp) Cu. While these elevations are less severe compared to those seen in Wilson’s disease (WD), they are still correlative with AD symptomatology and cerebrospinal fluid (CSF) markers. Postmortem analysis of brain tissue from AD patients has also revealed discriminating non-Cp Cu levels, indicating potential Cu accumulation within the brain. This accumulation may disrupt homeostatic mechanisms, thereby contributing to the activation of Cu-mediated amyloid cascades implicated in AD pathogenesis. Although AD and WD are distinct conditions, they share commonalities in Cu dysregulation mechanisms, suggesting a pathogenic overlap. Accordingly, insights gleaned from WD treatment strategies may inform preventive approaches for AD. A future preventive measure involves implementing a Cu-restricted diet, particularly for individuals with disrupted Cu metabolism [140]. Such a diet would involve reducing the intake of Cu-rich foods such as shellfish, nuts, organ meats, seeds, and certain whole grains. Furthermore, individuals may be advised to limit Cu-containing supplements and avoid high-Cu water sources. The rationale behind a Cu-restricted diet lies in the hypothesis that by reducing Cu intake, the accumulation of non-Cp Cu in the body, including the brain, could be mitigated. This, in turn, might slow down the progression of the AD. However, further research is warranted to elucidate the precise role of Cu in AD pathology and to ascertain the efficacy of dietary interventions. So, a Cu-restricted diet shows promise as a defensive strategy for AD; its application should be approached judiciously in combination with other preventive measures and therapeutic interventions [47, 141].
Chelation Therapy
The concept of metal chelation encouraged the “therapeutic chelation” approach to treat AD. According to this concept, small compounds can be potential enough to inhibit metal-induced Aβ accumulation and oxidative damage in the different regions of AD brain. Numerous in-vitro studies have been conducted over the last 20 years to characterize metal binding, its effect on amyloid accumulation, redox imbalance, and cytotoxicity [142,143,144]. Regardless of the absence of proven benefits in clinical cases, the concept of therapeutic chelation is widely exploited in preclinical studies for the treatment of AD. Metal chelation therapy involves the administration of chelators (chelating agents) into the biological system, which further binds to and removes the targeted metals [145, 146]. Only a few chelator medications are suitable for AD. Desferrioxamine (DFO), bathophenanthroline, bathocuproine (BC), trientine, penicillamine, bis (thiosemicarbazone), quinoline derivatives, and tetrathiomolybdate (TTM) are typical chelator medicines used to treat AD [147,148,149].
DFO was found to be beneficial in clinical research, including in 48 AD patients. In one of the studies, DFO reduced the aluminium level in the neocortical region of the brain in AD patients who received DFO (125 mg each injection, twice daily, 5 days a week) [150]. Although it demonstrated results for aluminium, one study claimed that, given DFO’s affinity for Fe, the result could possibly have been attributable to Fe elimination [151]. DFO has a high affinity for Cu in addition to Fe [152]. In a recent study, 1.6 mg deferasirox administration thrice weekly had no effect on the memory and motor functions as determined by contextual fear conditioning and rotarod respectively, but it decreased the content of hyperphosphorylated tau as measured by immune-reactive antibodies [153]. There are multiple plausible pathways for deferasirox’s ability to lower hyperphosphorylated tau levels. Since Feca uses aggregation of hyperphosphorylated tau, the beneficial outcomes of deferasirox can be attributed to Fe chelation [154]. Another theory is that deferasirox prevents the aggregation of tau proteins by binding directly to them. Many chemicals inhibit tau aggregation, and a few of them are structurally identical to the deferasirox [155, 156]. Other chelators, including curcumin and clioquinol (CQ), have been shown to prevent neurotoxicity or behavioural dysfunctions in transgenic APP mice model [157, 43]. Similar to deferasirox, these medicines may reduce neurodegeneration through mechanisms apart from metal chelation, such as curcumin, which has the capacity to directly prevent the formation and accumulation of Aβ plaques. Cu chelators such as penicillamine, bathophenanthroline, bathocuproine, and trientine have also been demonstrated to act against the neuropathology in AD. In one of the experiments, these drugs increased the Aβ solubility by eliminating the Cu from Cu-Aβ pair. Furthermore, bathocuproine has been constantly shown to be most effective in the AD-affected regions of the brain [149]. Moreover, bis(thiosemicarbazone) has an ability to modulate Cu concentrations in AD brain [158]. Analogues of the bis(thiosemicarbazone) metal chelator were found to be effective in the AD animal model [159]. Another study employing an APP/PS1 transgenic AD mouse model found comparable results. Bis(thiosemicarbazone) removed Cu from Cu-Aβ complex, increased soluble Aβ levels, and restored the cognitive ability [160].
A series of 8-hydroxyquinoline derivatives (VK-28, HLA-20, and MA-30) have shown promising results for the management of several neurological disorders. One of the 8-hydroxyquinoline analogues, CQ, has made its way to the phase 2 clinical trial, indicating that it improves cognitive and behavioral abilities and decreases Aβ levels in the plasma of AD patients [161, 162]. CQ is a small, lipophilic, and metal chelating agent with good bioavailability. It efficiently breaches the BBB and dissolves Aβ aggregates in the brain, most likely by excluding metal ions from the structure and redistributing them [163, 164].
Although the metal chelating drugs mentioned above have demonstrated positive results in lowering Aβ formation and accumulation in AD patients, the concerns regarding metal chelation therapy still persist. First, along with the intended effects, metal chelators may have adverse effects. The requirement for the chelating agent to penetrate the BBB necessitates a lipophilic and low molecular weight chelator. This poses a significant hurdle for the development and selection of metal chelators playing a beneficial role in AD [165, 166]. Most of the currently approved chelators are hydrophilic and designed to operate on peripheral tissues, hence they are not appropriate agents for the treatment of AD [167, 168]. One study found that the administration of divalent metal chelators, including Cu, Fe, and Zn, in severe AD patients reduced the levels of essential metals along with the targeted metal ions. As a result, critical metal loss exacerbated rather than cured AD pathogenesis [169]. Further, metal chelators have been shown to induce solubilization of Aβ deposits. However, it remains disputed whether chelating agents can only solubilize the Aβ aggregates or can degrade them to monomers, oligomers, protofibrils, short fibrils, and elongated fibrils [166, 169]. Furthermore, there are several concerns about the efficacy of specific metal-chelating candidates. A Cu–Zn chelator, CQ, is being employed in a number of therapeutic investigations. However, pre-clinical and clinical studies reveal that the efficacy of CQ is still controversial [169, 170]. In one research investigation, CQ disrupted Cu and Zn homeostasis and increased their concentrations, which contradicted the intended outcomes. CQ also resulted in adverse outcomes in the mouse model, including astrogliosis, spongiosis and brain edema [171].
Antioxidant Therapy
Various natural and synthetic polyphenolic compounds are currently employed as antioxidants; however, they have not been formally accepted as a therapeutic approach for AD. Flavonoids are an essential class of polyphenolic antioxidants and are the most common component of the human diet. Natural flavonoids are abundantly found in plants and seeds such as cocoa beans and grape seeds [172]. Several flavonoids have been shown to exert neuroprotective effects in AD cases [172, 173]. A recent investigation by Singh et al. reveals that a synthetic flavonoid derivative has a neuroprotective impact, as indicated by in-silico and in-vivo approaches in a Drosophila model of AD [174]. Moreover, flavonoids protect brain cells from oxidative damage and prevent the accumulation of Aβ fibrils in-vitro [175]. Other antioxidants exerting neuroprotective effects in the brain of AD cases include vitamins and polyphenolic compounds.
Vitamin E
Vitamin E is one of the highly potential peroxyl radical antioxidants [176]. It has the ability to target lipid-soluble membrane lipoproteins as well as low-density lipoproteins. Vitamin E can help AD patients to overcome the enhanced production of α-tocopherol transfer protein (α-TTP) in their brains [177]. A systematic review reported a decreased content of vitamin E in the blood plasma of AD patients [178]. Vitamin E and Ginkgo biloba extracts significantly improved cognitive performance in one clinical research [179]. Furthermore, a low serum vitamin E concentration has been linked to AD in another meta-analysis. Furthermore, evidence suggests vitamin E potentially inhibits tau-based neuronal damage in Drosophila [45, 180, 181]. According to one recent study, vitamin E has the potential to significantly diminish oxidative and nitrosative stress-induced toxicity in AD [182]. In-vitro and in-vivo experiments with apoE have shown that vitamin E can help in preventing neurodegenerative diseases. Further, it has been shown to be neuroprotective in apoE-deficient animals and to decrease Aβ-mediated neurotoxicity in the cultured hippocampus neurons [183, 184]. Furthermore, in-vivo investigations have shown that vitamin E has neuroprotective action by preventing Aβ-induced damage [185]. Vitamin E reversed cognitive and behavioural impairments caused by synthetic Aβ infusions into the cerebro-ventricles. Conversely, evidence from human clinical studies is less compelling as vitamin E is unable to reverse the cognitive changes in AD patients, and there is minimal improvement in living performance [186]. However, the positive effects of vitamin E on the cognition and behavior in AD cases are currently being studied.
Glutathione
Glutathione (GSH) is capable of scavenging adducts of lipid peroxidation such as acrolein, 4-hydroxy-2-nonenal (HNE) and others [187]. Further, it maintains thiol redox balance in cells, detoxes free radicals, binds metal ions, and protects against oxidative burden. It can also chelate metals, which lowers metallic burden and enables metal elimination from the tissues [187, 188]. However, the mechanism conferring the exact role of GSH in AD is still unknown. It was recently reported that cholesterol-mediated mitochondrial GSH depletion is associated with enhanced Aβ-induced redox imbalance in the mitochondria [189]. The administration of GSH-ethyl ester to the transgenic model of AD with increased sterol regulatory element-binding protein-2 (SREBP-2) expression has been found to reduce neuronal damage and inflammation [190]. Recently, a study uncovered a redox mechanism related to the antioxidant capacity of GSH, which was crucial for controlling mitochondrial functions in the axons [191].
Curcumin
Curcumin has several favourable properties to be considered for neuroprotection. It is considered a potent antioxidant, protein aggregation inhibitor, and an anti-inflammatory agent [192]. Curcumin attenuated inflammation, oxidative damage, and cognitive impairments in several rodent models of AD. Curcumin has significant free radical scavenging characteristics, in which it targets NO-derived reactive species to scavenge them, hence inhibiting lipid peroxidation [193]. Additionally, curcumin has the property to chelate metal ions, which eventually prevents Aβ aggregation and reduces oxidative damage [194]. It was also reported to restore GSH levels in the brain and decrease oxidized proteins in AD mouse models [157, 195]. However, the benefit of curcumin in AD could not be determined in one of the clinical trials; this could be due to its extremely weak pharmacokinetic and pharmacodynamic characteristics [196]. Curcumin, surprisingly, has been demonstrated to inhibit Aβ-mediated production of several inflammatory mediators in both THP-1 monocytic cells and peripheral blood monocytes [197]. The neuroprotective capacity of curcumin has been demonstrated in several rodent models of AD. When Aβ-injected rats were given curcumin, their cognitive and brain function were increased significantly in comparison to the Aβ-injected rats alone [198]. Furthermore, curcumin exerted similar type of effects in the aged Tg2576 mice [157]. The additional evidence suggests that curcumin is BBB permeable and can target and remove the accumulated Aβ plaques in animal models of AD.
Catechins
Catechins are found in tea, particularly green tea, and have several biological effects. They are classified into four types: Epicatechin (EC), Epicatechin Gallate (ECG), Epigallocatechin (EGC), and Epigallocatechin Gallate (EGCG) [199]. Catechins exert antioxidant effects by scavenging free radicals and lowering the deposition of metals, including Cu, Fe, and Zn, in the cerebral regions of AD patients by chelating them [200]. In the rat hippocampus, EGCG has been reported to decrease caspase levels, oxidative stress, and lipid peroxidation [201]. Long-term research in rats found that administration of green tea catechins (0.5% aqueous solution) prevented Aβ-mediated cognitive deficits and lowered plasma lipid peroxidation and ROS levels [202]. Catechins have been reported to have anti-inflammatory and anti-acetylcholinesterase (AChE) activity, besides antioxidant effects. Simultaneously, EGCG was discovered to directly bind with Aβ peptides and inhibit their aggregation and deposition in the brain [200, 203]. Additionally, catechins are able to cross the BBB as demonstrated in several rodent models, therefore they may serve as potential therapeutic candidates for AD [200].
Resveratrol
Resveratrol has been shown to have antioxidant capabilities by lowering concentrations of lipid peroxidation adducts and nitrite and increasing GSH levels [204]. Resveratrol was found to have an anti-amyloidogenic effect in several types of cell lines (with mutant APP695) by decreasing secreted intracellular Aβ peptide levels [199]. Resveratrol increased the intracellular antioxidant capacity as evidenced by the upregulation of enzymes, including SOD, CAT, GPx, and HO-1 while decreasing lipid peroxidation [205]. Another important action of resveratrol was to reduce free radical generation in the cerebral region by inhibiting mitochondrial membrane potential disturbance [206]. Metals bind to Aβ and NFTs and increase their aggregation, resulting in an elevation in free radicals. Resveratrol mitigates this by disrupting the metallic ion’s equilibrium [204]. In addition to antioxidant capabilities, resveratrol has the ability to produce an anti-inflammatory response, lower tau protein hyperphosphorylation and tangles formation, and increase SIRT-1 expression [206]. As a result, resveratrol is an intriguing natural antioxidant in the fight against AD pathogenesis.
Recent Investigation on Cu, Fe, and Zn Targeting Therapy
Recent therapeutic approaches include novel metal-focused therapy, metal protein attenuating compounds (MPACs), and nanotechnological interventions (Table 1). Efforts to regulate cellular metal ion homeostasis are critical to ensuring that freely available metals, such as Cu and Zn, are maintained at low levels while ensuring adequate metals are obtainable for essential metal-binding proteins.
Peptides-Based Chelators
The MPACs class includes a compound with a peptide ligand, exemplified by GSH-LD. GSH, is a vital antioxidant peptide found in human cells, comprising three main amino acids: Cys, Glu, and Gly. This peptide is renowned for its multifaceted role in addressing neurodegenerative diseases. On the other hand, l-Dopa, also recognized as Levodopa (l-3,4-dihydroxyphenylalanine), is an important amino acid naturally occurring in humans, as well as in some animals. When l-Dopa and GSH are combined, they form GSH-LD. This compound exhibits a remarkable ability to selectively eliminate excessive Cu2+ ions and partially reduce the surplus of Zn2+ ions present in Aβ peptides [214]. One commonly explored pathway is metal chelation, which is considered an indirect antioxidant mechanism. This is because metal complexation can inhibit radical reactions within the chain, delaying oxidation processes. These radicals have the potential to interact with biomolecules, leading to disruptions and damage in body tissues. It is worth noting that the relatively lower concentrations of naturally occurring bioactive peptides can contribute to pathological conditions in humans. In specific studies, fresh phaseolin and bean protein hydrolysates, obtained through enzymatic digestion with pancreatin and pepsin, have been assessed for their metal-chelating and antioxidant properties [215]. The antioxidant activity of peptides often relies on their capacity to chelate transition metals and scavenge free radicals. Residues of certain amino acids play a pivotal role in this process. Residues with nucleophilic sulfur comprising side chains, such as Methionine (Met) and Cysteine (Cys), as well as those with aromatic side chains like Tyrosine (Tyr), Tryptophan (Trp), and Phenylalanine (Phe), are adept at donating hydrogen atoms. Consequently, these residues are generally considered to possess antioxidant potential, while they can exhibit pro-oxidant effects under specific conditions [215]. Additionally, the imidazole group in Histidine (His) is known to have metal-chelating properties. Furthermore, basic and acidic amino acids may also play a vital role in chelating metal ions like Fe2+ and Cu2+ [216]. Methanobactins (MBs), also known as chalkophores, are small peptides (< 1300 Da) manufactured by the methanotrophic bacterium Methlylosinus trichosporium OB3b in its growth medium. MBs exhibit a variable binding affinity for Cu depending on the ratio of MBs to Cu ions. However, the precise details of this affinity remain unclear, as Cu (II) may undergo reduction in the presence of MBs. Notably, MBs have a preference for binding to Cu over other metals like Zn and Fe. They play a crucial role in regulating the methane oxidase system and effectively mitigating Cu toxicity, making them valuable in chelation therapy for Cu-related disorders [217, 217, 218].
Nanochelators or Nanoparticle-Based Approaches
The recent developments in nanotechnology-based strategies for AD have focused on targeting transition metals, including Cu, Fe, and Zn, which play crucial roles in the pathology of AD. The smart materials such as nanoparticles (NPs) are designed to chelate these transition metals, preventing their accumulation in the brain and thereby reducing metal-induced oxidative stress and neuroinflammation [219]. Another approach involves using NPs as carriers for metal-based therapeutics, improving drug delivery to the brain and allowing for precise modulation of transition metal levels while minimizing adverse effects on other tissues [220,221,222]. Moreover, NPs can be customized to specifically target regions of the brain rich in transition metals, such as amyloid plaques, maximizing the effectiveness of therapeutic interventions. These innovative approaches hold great promise for addressing various aspects of AD pathology and may lead to advancements in disease modification and neuroprotection, pending further optimization and validation through preclinical and clinical studies [223].
Despite their proven efficacy, metal chelators face limitations, including their restricted ability to penetrate the BBB and their inability to differentiate between toxic metals associated with Aβ aggregates and those crucial for normal biological functions. To surmount these challenges, encapsulating metal chelators within NPs presents a promising solution [218, 224]. These NPs have demonstrated a pivotal role in extracting chelator-Fe complexes from the brain, thus mitigating the toxicity of Fe ions [225]. Extensive prior research has discussed the strategic amalgamation of NPs with chelating agents to enhance their delivery to the brain for AD management. This approach has proven both safe and highly effective in reducing elevated metal concentrations in the AD-afflicted brain. Some studies have delved into the coupling of hexadentate Fe chelators with NPs for brain delivery and exhibited the ability to sequester Fe from AD brain tissues as well as from essential Fe storage proteins like ferritin [18]. Recent work by Haung et al. introduced a multifunctional treatment approach utilizing polydopamine (PDA) NPs with inherent properties for chelating metal ions and scavenging ROS. Adorned with the KLVFF peptide (PDA@K), these NPs effectively cross the pathological BBB and accumulate at lesion sites by hitchhiking on Aβ. Both in vitro and in vivo trials demonstrated that PDA@K displayed a strong affinity for Aβ and improved their BBB crossing, ultimately reducing Aβ aggregation and alleviating neuroinflammation [226]. Kowalczyk et al. investigated the potential of Prussian blue (PB), a medication included in the World Health Organization (WHO) Model List of Essential Medicines, as an AD management. Both in in-vitro and in-cellular studies demonstrate that PB-nanoparticles (PB-NPs) exhibit the ability to diminish the formation of characteristic Aβ fibers, which are identified through thioflavin T fluorescence. Furthermore, PB-NPs can reinstate the regular process of amyloid fibrillation by effectively binding or isolating Cu. This metal tends to accumulate in significant amounts within senile plaques linked to AD. PB-NPs hold promise in alleviating oxidative stress through dual mechanisms, acting as both Cu2+ ion adsorbers and scavengers for hydroxyl radicals [227].
Inspired by stimuli-responsive drug delivery systems, Shi et al. introduced a unique dual-responsive system for releasing a “caged metal chelator” based on near-infrared (NIR)-absorbing gold nanocages. In this system, the chelator CQ was enclosed within the gold nanocages, and the pore was sealed by human IgG using a thermalandredox-sensitive arylboronic ester bond. Excess H2O2 triggered the cleavage of the aryl boronic ester bond, leading to the release of CQ. Additionally, exposure to NIR light generated localized heat, further facilitating the release of CQ and dissolution of Aβ deposits, all achieved through noninvasive remote control [227]. Li et al. devised a dual-responsive approach utilizing light-responsive magnetic NPs prochelator conjugates to combat metal ions induced Aβ-aggregation. In this intricate system, they employed Fe3O4 NPs to encapsulate CQ through a photoactive o-nitrobenzyl-bromide linkage. Importantly, in its prochelator form within the conjugates, CQ did not interact with Cu2+. Upon exposure to UV radiation at 365 nm, the o-nitrobenzyl-bromide linkage undergoes photolytic cleavage, releasing the caged CQ from the NPs. This liberated CQ then effectively stops metal-induced Aβ-aggregation, reduces the presence of cellular ROS, and provides cellular protection against Aβ-related toxicity [228]. Another similar platform devised by Li et al. that hinged on the responsive release of antioxidant CeO2 NPs and the metal ion chelator CQ, triggered by H2O2 [229].
To address AD through numerous pathways, Guan et al. developed CeO2 NPs encapsulated within a functional MnMoS4 shell (CeNPs@MnMoS4-n; n = 1–5). The MnMoS4 component was capable of eliminating toxic intracellular Cu2+ through ion exchange. Furthermore, the release of Mn2+encouraged neurite outgrowth. Notably, CeNPs@MnMoS4-3, with its SOD activity, contributed to a reduction in oxidative stress [230] (Fig. 3).

Reproduced from Huang et al. [226] with the kind permission of the Copyright © holder, AAAS, 2023, an open-access article distributed under a Creative Commons Attribution License 4.0 (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium
Schematic representation of PDA@K compound mechanism: melanin-like metal ion chelators and neuroinflammation regulators designed for AD therapy. These compounds synergistically reduce Aβ burden and normalize microglia dysfunction through metal chelation and ROS scavenging.
Li et al. discovered that graphene-like carbon nitride nanosheets (g-C3N4 nanosheets) possess a remarkable ability to adsorb metal ions. They found that these nanosheets can function as chelators to effectively inhibit Cu2+-induced aggregation of Aβ, break down pre-existing Aβ-Cu2+ aggregates, diminish the levels of ROS caused by Aβ-Cu2+, and mitigate Aβ-induced toxicity [231]. Gong et al. have engineered an intelligent Aβ nanocaptor. They achieved this by affixing C3N4 nanodots onto Fe3O4@MSNs and then altering them with benzothiazole aniline to create B-FeCN. Within this nanocomposite, the C3N4 nanodots play a crucial role in capturing Cu2+, thereby preventing the formation of the Aβ-Cu2+ complex and reducing Aβ aggregation. Additionally, the presence of Fe3O4 enables localized low-temperature hyperthermia, which enhances BBB permeability and aids in dissolving Aβ plaques. Furthermore, the inclusion of BTA imparts the nanocaptor with specific targeting capabilities and the ability to fluorescently image Aβ aggregates for monitoring purposes [232]. Another intriguing approach in metal-based therapeutics for AD focuses on addressing elevated brain Fe levels. This approach involves traditional Fe chelation therapy utilizing compounds designed to pass the BBB and possesses multifunctional properties, such as antioxidant or monoamine oxidase inhibition achieved by hydroxy pyridine-coumarin hybrids. Moreover, compound hybrids exhibited protective effects against oxidative stress, leading to a substantial improvement in cognitive function in a Morris water maze model [233, 234].
Conclusions and Future Perspective
AD is the most prevalent form of dementia worldwide. The disease’s intricate mechanisms involve oxidative stress, neuroinflammation, and protein misfolding and aggregation. Oxidative stress arises from imbalances in redox-sensitive transition metals, leading to ROS generation, mitochondrial dysfunction, and neurodegeneration. Neuroinflammation exacerbates AD pathology, while protein misfolding, especially of Aβ and tau proteins, contributes to neuronal damage and cognitive decline. The traditional view of AD as a brain protein aggregation disease has shifted toward understanding its connection to metal imbalances, particularly Cu, Zn, and Fe. Research in metallobiology has revealed how metals relate to neurodegenerative disorders like AD. These transition metals play vital roles in brain functions like enzyme operations, neurotransmission, and aging. Disrupted metal balance impacts gene expression, enzyme activation, and defense against ROS, affecting brain growth, development, and function. Metal imbalances, particularly involving Cu, Fe, and Zn, disrupt brain function in AD by interacting with proteins like APP, Aβ, and tau, leading to oxidative stress, protein aggregation, and synaptic dysfunction—key elements in AD pathology. Studies suggest these imbalances and associated oxidative damage significantly contribute to AD progression. The therapeutic strategy for AD primarily focuses on prevention due to the lack of a curative treatment. It involves metal-restricted diets, chelation therapy to remove excess metals, and antioxidant therapy to counter oxidative stress. Recent investigations exploring innovative peptide-based chelators, such as GSH-LD (combining GSH and l-Dopa), show promise in selectively removing excess Cu and partially reducing surplus Zn present in Aβ peptides. Metal chelation is an indirect antioxidant mechanism that inhibits radical reactions and prevents biomolecular disruptions caused by radicals. Amino acids like Cysteine, Methionine, and Tyrosine in peptides contribute to their antioxidant potential by chelating transition metals and scavenging free radicals. NPs-based approaches address the limitations of metal chelators by enhancing brain penetration and targeting specific metal imbalances associated with AD. Examples include polydopamine NPs (PDA@K) that cross the BBB and reduce Aβ aggregation, Prussian blue NPs (PB-NPs) that bind Cu, and stimuli-responsive systems releasing chelators to dissolve Aβ deposits. Other innovations involve graphene-like carbon nitride nanosheets (g-C3N4) inhibiting Aβ-Cu2+ aggregation and nanocaptors capturing Cu2+ to prevent Aβ complex formation. Additionally, research focuses on Fe chelation therapy with compounds designed to pass the BBB, exhibiting antioxidant properties and cognitive function improvement in AD models. These multifunctional compounds offer a potential therapeutic avenue for addressing elevated brain Fe levels in AD. Metal-targeted therapies using peptides and NPs to regulate cellular metal ion balance, offering potential avenues for AD treatment. However, these approaches necessitate further validation and clinical trials to assess their effectiveness in humans.
Data Availability
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- ABAD:
-
Aβ binding alcohol dehydrogenase
- APP:
-
Amyloid precursor protein
- ATP:
-
Adenosine triphosphate
- BBB:
-
Blood-brain barrier
- CNS:
-
Central nervous system
- COX:
-
Cytochrome oxidase
- Cu:
-
Copper
- ETC:
-
Electron transport chain
- Fe:
-
Iron
- GWAS:
-
Genome-wide association studies
- GSH:
-
Glutathione
- NFTs:
-
Neurofibrillary tangles
- NMDARs:
-
N-Methyl-d-aspartic acid receptors
- ROS:
-
Reactive oxygen species
- SNP:
-
Single-nucleotide polymorphisms
- Zn:
-
Zinc
References
An L (2022) 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dementia 18(4):700–789. https://doi.org/10.1002/alz.12638
Puentes-Díaz N, Chaparro D, Morales-Morales D, Flores-Gaspar A, Alí-Torres J (2023) Role of metal cations of copper, Iron, and aluminum and multifunctional ligands in Alzheimer’s disease: experimental and computational insights. ACS omega 8(5):4508–4526. https://doi.org/10.1021/acsomega.2c06939
Wang X, Wang X, Guo Z (2018) Metal-involved theranostics: An emerging strategy for fighting Alzheimer’s disease. Coord Chem Rev 362:72–84. https://doi.org/10.1016/j.ccr.2018.03.010
Wang P, Wang Z-Y (2017) Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev 35:265–290. https://doi.org/10.1016/j.arr.2016.10.003
Eisenstein RS (2000) Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu Rev Nutr 20(1):627–662. https://doi.org/10.1146/annurev.nutr.20.1.627
Vilella A, Daini E, De Benedictis C, Grabrucker A (2020) Targeting metal homeostasis as a therapeutic strategy for Alzheimer’s disease. Exon Publications:83–98. https://doi.org/10.36255/exonpublications.alzheimersdisease.2020.ch5
DeBenedictis CA, Raab A, Ducie E, Howley S, Feldmann J, Grabrucker AM (2020) Concentrations of essential trace metals in the brain of animal species—a comparative study. Brain Sci 10(7):460. https://doi.org/10.3390/brainsci10070460
Tapiero H, Tew KD (2003) Trace elements in human physiology and pathology: zinc and metallothioneins. Biomed Pharmacother 57(9):399–411. https://doi.org/10.1016/s0753-3322(03)00081-7
Prasad AS (2008) Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp Gerontol 43(5):370–377. https://doi.org/10.1016/j.exger.2007.10.013
Peña MM, Lee J, Thiele DJ (1999) A delicate balance: homeostatic control of copper uptake and distribution. J Nutr 129(7):1251–1260. https://doi.org/10.1093/jn/129.7.1251
Zheng W, Monnot AD (2012) Regulation of brain iron and copper homeostasis by brain barrier systems: implication in neurodegenerative diseases. Pharmacol Ther 133(2):177–188. https://doi.org/10.1016/j.pharmthera.2011.10.006
Sensi SL, Granzotto A, Siotto M, Squitti R (2018) Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol Sci 39(12):1049–1063. https://doi.org/10.1016/j.tips.2018.10.001
Zhao J, Gao W, Yang Z, Li H, Gao Z (2019) Nitration of amyloid-β peptide (1–42) as a protective mechanism for the amyloid-β peptide (1–42) against copper ion toxicity. J Inorg Biochem 190:15–23. https://doi.org/10.1016/j.jinorgbio.2018.10.005
Wang L, Yin Y-L, Liu X-Z, Shen P, Zheng Y-G, Lan X-R, Lu C-B, Wang J-Z (2020) Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl Neurodegener 9(1):1–13. https://doi.org/10.1016/j.nbd.2022.105824
Myhre O, Utkilen H, Duale N, Brunborg G, Hofer T, longevity c (2013) Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases: possible impact of environmental exposures. Oxidative medicine 2013. https://doi.org/10.1155/2013/726954
Waldron KJ, Rutherford JC, Ford D, Robinson NJ (2009) Metalloproteins and metal sensing. Nature 460(7257):823–830. https://doi.org/10.1038/nature08300
Markesbery W, Lovell M (1998) Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging 19(1):33–36. https://doi.org/10.1016/s0197-4580(98)00009-8
Fasae KD, Abolaji AO, Faloye TR, Odunsi AY, Oyetayo BO, Enya JI, Rotimi JA, Akinyemi RO et al (2021) Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer’s disease: limitations, and current and future perspectives. J Trace Elem Med Biol 67:126779. https://doi.org/10.1016/j.jtemb.2021.126779
Bush AI (2000) Metals and neuroscience. Curr Opin Chem Biol 4(2):184–191. https://doi.org/10.1016/j.nbd.2015.08.012
Itoh M, Ebadi M, Swanson S (1983) The presence of zinc-binding proteins in brain. J Neurochem 41(3):823–829. https://doi.org/10.1111/j.1471-4159.1983.tb04814.x
Dreosti I (1989) Neurobiology of zinc. In: Zinc in human biology. Springer, pp 235–247. https://springerlink.bibliotecabuap.elogim.com/chapter/https://doi.org/10.1007/978-1-4471-3879-2_15
Frederickson CJ (1989) Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol 31:145–238. https://doi.org/10.1159/000109536
Duce JA, Bush AI (2010) Biological metals and Alzheimer’s disease: implications for therapeutics and diagnostics. Prog Neurobiol 92(1):1–18. https://doi.org/10.1016/j.pneurobio.2010.04.003
Thompson R, Peterson D, Mahoney W, Cramer M, Maliwal BP, Suh SW, Frederickson C, Fierke C et al (2002) Fluorescent zinc indicators for neurobiology. J Neurosci Methods 118(1):63–75. https://doi.org/10.1016/s0165-0270(02)00144-9
Squitti R, Reale G, Tondolo V, Crescenti D, Bellini S, Moci M, Caliandro P, Padua L et al (2023) Imbalance of essential metals in traumatic brain injury and its possible link with disorders of consciousness. Int J Mol Sci 24(7):6867. https://doi.org/10.3390/ijms24076867
Sarris J, Ravindran A, Yatham LN, Marx W, Rucklidge JJ, McIntyre RS, Akhondzadeh S, Benedetti F et al (2022) Clinician guidelines for the treatment of psychiatric disorders with nutraceuticals and phytoceuticals: The World Federation of Societies of Biological Psychiatry (WFSBP) and Canadian Network for Mood and Anxiety Treatments (CANMAT) Taskforce. World J Biol Psychiatry 23(6):424–455. https://doi.org/10.1080/15622975.2021.2013041
Squitti R, Pal A, Picozza M, Avan A, Ventriglia M, Rongioletti MC, Hoogenraad T (2020) Zinc Therapy in early Alzheimer’s disease: Safety and potential therapeutic efficacy. Biomolecules 10(8):1164. https://doi.org/10.3390/biom10081164
Waggoner DJ, Bartnikas TB, Gitlin JD (1999) The role of copper in neurodegenerative disease. Neurobiol Dis 6(4):221–230. https://doi.org/10.1006/nbdi.1999.0250
Hung YH, Bush AI, Cherny RA (2010) Copper in the brain and Alzheimer’s disease. J Biol Inorg Chem 15:61–76. https://doi.org/10.1007/s00775-009-0600-y
Lutsenko S, Bhattacharjee A, Hubbard AL (2010) Copper handling machinery of the brain. Metallomics 2(9):596–608. https://doi.org/10.1039/c0mt00006j
Schlief ML, Gitlin JD (2006) Copper homeostasis in the CNS: a novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol Neurobiol 33:81–90. https://doi.org/10.1385/mn:33:2:81
Matheou CJ, Younan ND, Viles JH (2015) Cu2+ accentuates distinct misfolding of Aβ (1–40) and Aβ (1–42) peptides, and potentiates membrane disruption. Biochem J 466(2):233–242. https://doi.org/10.1042/bj20141168
Sastre M, Ritchie CW, Hajji N (2015) Metal ions in Alzheimer’s disease brain. JSM Alzheimer’s Dis Relat Dement 2(1):1014
Connor JR, Menzies SL, Burdo JR, Boyer PJ (2001) Iron and iron management proteins in neurobiology. Pediatr Neurol 25(2):118–129. https://doi.org/10.1016/s0887-8994(01)00303-4
Que EL, Domaille DW, Chang CJ (2008) Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem Rev 108(5):1517–1549. https://doi.org/10.1021/cr800447y
Pfaender S, Grabrucker AM (2014) Characterization of biometal profiles in neurological disorders. Metallomics 6(5):960–977. https://doi.org/10.1039/c4mt00008k
Walsh D, Minogue A, Sala Frigerio C, Fadeeva J, Wasco W, Selkoe D (2007) The APP family of proteins: similarities and differences. Biochemical Society Transactions, vol 35. Portland Press Ltd. https://doi.org/10.1042/bst0350416
Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR et al (2003) Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain: a regulator of neuronal copper homeostasis. J Biol Chem 278(19):17401–17407. https://doi.org/10.1074/jbc.M300629200
Bush AI, Multhaup G, Moir RD, Williamson TG, Small DH, Rumble B, Pollwein P, Beyreuther K et al (1993) A novel zinc (II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J Biol Chem 268(22):16109–16112. https://doi.org/10.1016/S0021-9258(19)85394-2
Venti A, Giordano T, Eder P, Bush AI, Lahiri DK, Greig NH, Rogers JT (2004) The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5′-untranslated region. Ann N Y Acad Sci 1035(1):34–48. https://doi.org/10.1196/annals.1332.003
Lovell M, Robertson J, Teesdale W, Campbell J, Markesbery W (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158(1):47–52. https://doi.org/10.1016/j.pneurobio.2011.05.001
Minicozzi V, Stellato F, Comai M, Dalla Serra M, Potrich C, Meyer-Klaucke W, Morante S (2008) Identifying the minimal copper-and zinc-binding site sequence in amyloid-β peptides. J Biol Chem 283(16):10784–10792. https://doi.org/10.1074/jbc.m707109200
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I et al (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30(3):665–676. https://doi.org/10.1016/s0896-6273(01)00317-8
Telling ND, Everett J, Collingwood JF, Dobson J, van der Laan G, Gallagher JJ, Wang J et al (2017) Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer’s disease. Cell Chem Biol 24(10):1205-1215.e1203. https://doi.org/10.1016/j.chembiol.2017.07.014
Dong Y, Chen X, Liu Y, Shu Y, Chen T, Xu L, Li M, Guan X (2018) Do low-serum vitamin E levels increase the risk of Alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. Int J Geriatr Psychiatry 33(2):e257–e263. https://doi.org/10.1002/gps.4780
Chapleau M, La Joie R, Yong K, Agosta F, Allen IE, Apostolova L, Best J, Boon BD et al (2024) Demographic, clinical, biomarker, and neuropathological correlates of posterior cortical atrophy: an international cohort study and individual participant data meta-analysis. Lancet Neurol 23(2):168–177. https://doi.org/10.1016/S1474-4422(23)00414-3
Squitti R, Polimanti R, Bucossi S, Ventriglia M, Mariani S, Manfellotto D, Vernieri F, Cassetta E et al (2013) Linkage disequilibrium and haplotype analysis of the ATP7B gene in Alzheimer’s disease. Rejuvenation Res 16(1):3–10. https://doi.org/10.1089/rej.2012.1357
Nerattini M, Jett S, Andy C, Carlton C, Zarate C, Boneu C, Battista M, Pahlajani S et al (2023) Systematic review and meta-analysis of the effects of menopause hormone therapy on risk of Alzheimer’s disease and dementia. Front Aging Neurosci 15:1260427. https://doi.org/10.3389/fnagi.2023.1260427
Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM (2011) Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog Neurobiol 94(3):296–306. https://doi.org/10.1016/j.pneurobio.2011.05.001
Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang X, Atwood CS, Beyreuther K, Tanzi RE et al (1999) Aqueous dissolution of Alzheimer’s disease Aβ amyloid deposits by biometal depletion. J Biol Chem 274(33):23223–23228. https://doi.org/10.1074/jbc.274.33.23223
Perry G, Taddeo MA, Petersen RB, Castellani RJ, Harris PL, Siedlak SL, Cash AD, Liu Q et al (2003) Adventiously-bound redox active iron and copper are at the center of oxidative damage in Alzheimer disease. Biometals 16:77–81. https://doi.org/10.1023/a:1020731021276
Noy D, Solomonov I, Sinkevich O, Arad T, Kjaer K, Sagi I (2008) Zinc-amyloid β interactions on a millisecond time-scale stabilize non-fibrillar Alzheimer-related species. J Am Chem Soc 130(4):1376–1383. https://doi.org/10.1021/ja076282l
Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ (2010) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9(1):119–128. https://doi.org/10.1016/S1474-4422(09)70299-6
Squitti R, Ventriglia M, Simonelli I, Bonvicini C, Costa A, Perini G, Binetti G, Benussi L et al (2021) Copper imbalance in Alzheimer’s disease: meta-analysis of serum, plasma, and brain specimens, and replication study evaluating ATP7B gene variants. Biomolecules 11(7):960. https://doi.org/10.3390/biom11070960
Squitti R, Faller P, Hureau C, Granzotto A, White AR, Kepp KP (2021) Copper imbalance in Alzheimer’s disease and its link with the amyloid hypothesis: towards a combined clinical, chemical, and genetic etiology. J Alzheimers Dis 83(1):23–41. https://doi.org/10.3233/JAD-201556
Gong L, Sun J, Cong S (2023) Levels of iron and iron-related proteins in Alzheimer’s disease: a systematic review and meta-analysis. Journal of Trace Elements in Medicine Biology 127304. https://doi.org/10.1016/j.jtemb.2023.127304
Ayton S, Portbury S, Kalinowski P, Agarwal P, Diouf I, Schneider JA, Morris MC, Bush AI (2021) Regional brain iron associated with deterioration in Alzheimer’s disease: a large cohort study and theoretical significance. Alzheimer Dementia 17(7):1244–1256. https://doi.org/10.1002/alz.12282
Ventriglia M, Brewer GJ, Simonelli I, Mariani S, Siotto M, Bucossi S, Squitti R (2015) Zinc in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis 46(1):75–87. https://doi.org/10.3233/JAD-141296
Panghal A (1868) Flora SJS (2024) Nanotechnology in the diagnostic and therapy for Alzheimer’s disease. Biochim Biophys Acta 3:130559. https://doi.org/10.1016/j.bbagen.2024.130559
Abubakar MB, Sanusi KO, Ugusman A, Mohamed W, Kamal H, Ibrahim NH, Khoo CS, Kumar J (2022) Alzheimer’s disease: an update and insights into pathophysiology. Front Aging Neurosci 14:742408. https://doi.org/10.3389/fnagi.2022.742408
Jellinger KA (2010) Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med 14(3):457–487. https://doi.org/10.1111/j.1582-4934.2010.01010.x
Butterfield DA, Halliwell B (2019) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20(3):148–160. https://doi.org/10.1038/s41583-019-0132-6
Chen Z, Zhong C (2014) Oxidative stress in Alzheimer’s disease. Neurosci Bull 30(2):271–281. https://doi.org/10.1007/s12264-013-1423-y
Bai R, Guo J, Ye XY, Xie Y, Xie T (2022) Oxidative stress: the core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res Rev 77:101619. https://doi.org/10.1016/j.arr.2022.101619
Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, Boonpraman N, Yi SS et al (2021) NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol 41:101947. https://doi.org/10.1016/j.redox.2021.101947
Takeuchi M, Yamagishi S (2008) Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr Pharm Des 14(10):973–978. https://doi.org/10.2174/138161208784139693
Gella A, Durany N (2009) Oxidative stress in Alzheimer disease. Cell Adh Migr 3(1):88–93. https://doi.org/10.4161/cam.3.1.7402
Peña-Bautista C, Baquero M, Vento M, Cháfer-Pericás C (2019) Free radicals in Alzheimer’s disease: lipid peroxidation biomarkers. Clin Chim Acta 491:85–90. https://doi.org/10.1016/j.cca.2019.01.021
Butterfield DA, Hensley K, Cole P, Subramaniam R, Aksenov M, Aksenova M, Bummer PM, Haley BE et al (1997) Oxidatively induced structural alteration of glutamine synthetase assessed by analysis of spin label incorporation kinetics: relevance to Alzheimer’s disease. J Neurochem 68(6):2451–2457. https://doi.org/10.1046/j.1471-4159.1997.68062451.x
He W, Wang C, Chen Y, He Y, Cai Z (2017) Berberine attenuates cognitive impairment and ameliorates tau hyperphosphorylation by limiting the self-perpetuating pathogenic cycle between NF-κB signaling, oxidative stress and neuroinflammation. Pharmacol Rep : PR 69(6):1341–1348. https://doi.org/10.1016/j.pharep.2017.06.006
Aaseth J, Skalny AV, Roos PM, Alexander J, Aschner M, Tinkov AA (2021) Copper, iron, selenium and lipo-glycemic dysmetabolism in Alzheimer’s disease. Int J Mol Sci 22:17. https://doi.org/10.3390/ijms22179461
Rogers JT, Bush AI, Cho HH, Smith DH, Thomson AM, Friedlich AL, Lahiri DK, Leedman PJ et al (2008) Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: riboregulation against neural oxidative damage in Alzheimer’s disease. Biochem Soc Trans 36(Pt 6):1282–1287. https://doi.org/10.1042/bst0361282
Mezzaroba L, Alfieri DF, Colado Simão AN, Vissoci Reiche EM (2019) The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 74:230–241. https://doi.org/10.1016/j.neuro.2019.07.007
Miura T, Suzuki K, Kohata N, Takeuchi H (2000) Metal binding modes of Alzheimer’s amyloid beta-peptide in insoluble aggregates and soluble complexes. Biochemistry 39(23):7024–7031. https://doi.org/10.1021/bi0002479
Butterfield DA (2002) Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. Rev Free Radic Res 36(12):1307–1313. https://doi.org/10.1080/1071576021000049890
Li W, Zhang J, Su Y, Wang J, Qin M, Wang W (2007) Effects of zinc binding on the conformational distribution of the amyloid-beta peptide based on molecular dynamics simulations. J Phys Chem B 111(49):13814–13821. https://doi.org/10.1021/jp076213t
Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA (2000) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J Neurochem 74(1):270–279. https://doi.org/10.1046/j.1471-4159.2000.0740270.x
Ashraf A, Jeandriens J, Parkes HG, So PW (2020) Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: evidence of ferroptosis. Redox Biol 32:101494. https://doi.org/10.1016/j.redox.2020.101494
Hofer T, Perry G (2016) Nucleic acid oxidative damage in Alzheimer’s disease-explained by the hepcidin-ferroportin neuronal iron overload hypothesis? J Trace Elem Med Biol 38:1–9. https://doi.org/10.1016/j.jtemb.2016.06.005
Bhatia V, Sharma S (2021) Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J Neurol Sci 421:117253. https://doi.org/10.1016/j.jns.2020.117253
Grimm A, Eckert A (2017) Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem 143(4):418–431. https://doi.org/10.1111/jnc.14037
Swerdlow RH, Burns JM (1842) Khan SM (2014) The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochem Biophys Acta 8:1219–1231. https://doi.org/10.1016/j.bbadis.2013.09.010
Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14:450–464. https://doi.org/10.1016/j.redox.2017.10.014
Desler C, Lillenes MS, Tønjum T, Rasmussen LJ (2018) The role of mitochondrial dysfunction in the progression of Alzheimer’s disease. Curr Med Chem 25(40):5578–5587. https://doi.org/10.2174/0929867324666170616110111
Sen A, Nelson TJ, Alkon DL, Hongpaisan J (2018) Loss in PKC Epsilon causes downregulation of MnSOD and BDNF expression in neurons of Alzheimer’s disease hippocampus. J Alzheim Dis : JAD 63(3):1173–1189. https://doi.org/10.3233/jad-171008
Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK (2006) Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am J Pathol 168(5):1608–1618. https://doi.org/10.2353/ajpath.2006.051223
Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, Guo L, Chen D et al (2011) Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J Neurosci 31(6):2313–2320. https://doi.org/10.1523/jneurosci.4717-10.2011
Echtay KS (2007) Mitochondrial uncoupling proteins—what is their physiological role? Free Radical Biol Med 43(10):1351–1371. https://doi.org/10.1016/j.freeradbiomed.2007.08.011
de la Monte SM, Wands JR (2006) Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheim Dis : JAD 9(2):167–181. https://doi.org/10.3233/jad-2006-9209
Wu Z, Zhang J, Zhao B (2009) Superoxide anion regulates the mitochondrial free Ca2+ through uncoupling proteins. Antioxid Redox Signal 11(8):1805–1818. https://doi.org/10.1089/ars.2009.2427
Frost GR, Jonas LA, Li YM (2019) Friend, foe or both? Immune activity in Alzheimer’s disease. Front Aging Neurosc 11:337. https://doi.org/10.3389/fnagi.2019.00337
Leng F, Edison P (2021) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17(3):157–172. https://doi.org/10.1038/s41582-020-00435-y
Villa V, Thellung S, Bajetto A, Gatta E, Robello M, Novelli F, Tasso B, Tonelli M et al (2016) Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90–231 or lipopolysaccharide. Pharmacol Res 113(Pt A):500–514. https://doi.org/10.1016/j.phrs.2016.09.010
Brosseron F, Krauthausen M, Kummer M, Heneka MT (2014) Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol 50(2):534–544. https://doi.org/10.1007/s12035-014-8657-1
Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, Cotman CW (2012) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation 9:179. https://doi.org/10.1186/1742-2094-9-179
Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP (2006) Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation 3:27. https://doi.org/10.1186/1742-2094-3-27
Martínez Leo EE, Segura Campos MR (2019) Systemic oxidative stress: a key point in neurodegeneration—a review. J Nutr Health Aging 23(8):694–699. https://doi.org/10.1007/s12603-019-1240-8
Ashour NH, El-Tanbouly DM, El Sayed NS, Khattab MM (2021) Roflumilast ameliorates cognitive deficits in a mouse model of amyloidogenesis and tauopathy: involvement of nitric oxide status, Aβ extrusion transporter ABCB1, and reversal by PKA inhibitor H89. Prog Neuropsychopharmacol Biol Psychiatry 111:110366. https://doi.org/10.1016/j.pnpbp.2021.110366
Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D (2014) Neuroinflammation: the role and consequences. Neurosci Res 79:1–12. https://doi.org/10.1016/j.neures.2013.10.004
Mishra A, Kim HJ, Shin AH, Thayer SA (2012) Synapse loss induced by interleukin-1β requires pre- and post-synaptic mechanisms. J Neuroimmune Pharmacol 7(3):571–578. https://doi.org/10.1007/s11481-012-9342-7
Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114(2):181–190. https://doi.org/10.1016/s0092-8674(03)00521-x
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Sci (New York, NY) 352(6286):712–716. https://doi.org/10.1126/science.aad8373
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheim Dementia 12(6):719–732. https://doi.org/10.1016/j.jalz.2016.02.010
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T et al (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153(3):707–720. https://doi.org/10.1016/j.cell.2013.03.030
Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S et al (2013) CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci 16(7):848–850. https://doi.org/10.1038/nn.3435
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH et al (2013) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78(4):631–643. https://doi.org/10.1016/j.neuron.2013.04.014
Lee HJ, Korshavn KJ, Kochi A, Derrick JS, Lim MH (2014) Cholesterol and metal ions in Alzheimer’s disease. Chem Soc Rev 43(19):6672–6682. https://doi.org/10.1039/C4CS00005F
Xu JJ, Guo S, Xue R, Xiao L, Kou JN, Liu YQ, Han JY, Fu JJ et al (2021) Adalimumab ameliorates memory impairments and neuroinflammation in chronic cerebral hypoperfusion rats. Aging 13(10):14001–14014. https://doi.org/10.18632/aging.203009
Zhang Y, Zhao Y, Zhang J, Yang G (2020) Mechanisms of NLRP3 inflammasome activation: its role in the treatment of Alzheimer’s disease. Neurochem Res 45(11):2560–2572. https://doi.org/10.1007/s11064-020-03121-z
Hanslik KL, Ulland TK (2020) The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol 11:570711. https://doi.org/10.3389/fneur.2020.570711
Chiu YJ, Lin CH, Lee MC, Hsieh-Li HM, Chen CM, Wu YR, Chang KH, Lee-Chen GJ (2021) Formulated Chinese medicine Shaoyao Gancao Tang reduces NLRP1 and NLRP3 in Alzheimer’s disease cell and mouse models for neuroprotection and cognitive improvement. Aging 13(11):15620–15637. https://doi.org/10.18632/aging.203125
Fox LM, Yamamoto A (2015) Chapter 7 - macroautophagy of aggregation-prone proteins in neurodegenerative disease. In: Hayat MA (ed) Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Academic Press, Amsterdam, pp 117–137. https://doi.org/10.1016/B978-0-12-801043-3.00007-8
Ashrafian H, Zadeh EH, Khan RH (2021) Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int J Biol Macromol 167:382–394. https://doi.org/10.1016/j.ijbiomac.2020.11.192
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA et al (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14(8):837–842. https://doi.org/10.1038/nm1782
Hu NW, Smith IM, Walsh DM, Rowan MJ (2008) Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain 131(Pt 9):2414–2424. https://doi.org/10.1093/brain/awn174
Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B et al (2004) An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-beta derivatives. J Neurosci 24(28):6277–6282. https://doi.org/10.1523/jneurosci.1344-04.2004
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118(1):103–113. https://doi.org/10.1007/s00401-009-0522-3
Barage SH, Sonawane KD (2015) Amyloid cascade hypothesis: pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 52:1–18. https://doi.org/10.1016/j.npep.2015.06.008
Paroni G, Bisceglia P, Seripa D (2019) Understanding the amyloid hypothesis in Alzheimer’s disease. J Alzheim Dis : JAD 68(2):493–510. https://doi.org/10.3233/jad-180802
van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D et al (2023) Lecanemab in Early Alzheimer’s Disease. N Engl J Med 388(1):9–21. https://doi.org/10.1056/NEJMoa2212948
Carma Hassan JC (2024) Biogen discontinues Alzheimer’s medication Aduhelm. Accessed March 18 2024
Celej MS, Sarroukh R, Goormaghtigh E, Fidelio GD, Ruysschaert JM, Raussens V (2012) Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem J 443(3):719–726. https://doi.org/10.1042/bj20111924
Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6(1):11–22. https://doi.org/10.1038/nrn1587
Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, Shea JE (2006) Amyloid beta-protein monomer structure: a computational and experimental study. Protein Sci 15(3):420–428. https://doi.org/10.1110/ps.051762406
Ding F, Borreguero JM, Buldyrey SV, Stanley HE, Dokholyan NV (2003) Mechanism for the alpha-helix to beta-hairpin transition. Proteins 53(2):220–228. https://doi.org/10.1002/prot.10468
Bruno MA, Leon WC, Fragoso G, Mushynski WE, Almazan G, Cuello AC (2009) Amyloid β-induced nerve growth factor dysmetabolism in Alzheimer disease. J Neuropathol Exp Neurol 68(8):857–869. https://doi.org/10.1097/NEN.0b013e3181aed9e6
Sharp FR, DeCarli CS, Jin L-W, Zhan X (2023) White matter injury, cholesterol dysmetabolism, and APP/Abeta dysmetabolism interact to produce Alzheimer’s disease (AD) neuropathology: a hypothesis and review. Front Aging Neurosci 15:1096206. https://doi.org/10.3389/fnagi.2023.1096206
Boutajangout A, Wisniewski T (2014) Tau-based therapeutic approaches for Alzheimer’s disease—a mini-review. Gerontology 60(5):381–385. https://doi.org/10.1159/000358875
Muralidar S, Ambi SV, Sekaran S, Thirumalai D, Palaniappan B (2020) Role of tau protein in Alzheimer’s disease: the prime pathological player. Int J Biol Macromol 163:1599–1617. https://doi.org/10.1016/j.ijbiomac.2020.07.327
Mamun AA, Uddin MS, Mathew B, Ashraf GM (2020) Toxic tau: structural origins of tau aggregation in Alzheimer’s disease. Neural Regen Res 15(8):1417–1420. https://doi.org/10.4103/1673-5374.274329
Xia Y, Sorrentino ZA, Kim JD, Strang KH, Riffe CJ, Giasson BI (2019) Impaired tau-microtubule interactions are prevalent among pathogenic tau variants arising from missense mutations. J Biol Chem 294(48):18488–18503. https://doi.org/10.1074/jbc.RA119.010178
Brunello CA, Merezhko M, Uronen RL, Huttunen HJ (2020) Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci 77(9):1721–1744. https://doi.org/10.1007/s00018-019-03349-1
Jucker M, Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501(7465):45–51. https://doi.org/10.1038/nature12481
Götz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Sci (New York, NY) 293(5534):1491–1495. https://doi.org/10.1126/science.1062097
Chabrier MA, Blurton-Jones M, Agazaryan AA, Nerhus JL, Martinez-Coria H, LaFerla FM (2012) Soluble aβ promotes wild-type tau pathology in vivo. J Neurosci 32(48):17345–17350. https://doi.org/10.1523/jneurosci.0172-12.2012
DeVos SL, Corjuc BT, Commins C, Dujardin S, Bannon RN, Corjuc D, Moore BD, Bennett RE et al (2018) Tau reduction in the presence of amyloid-β prevents tau pathology and neuronal death in vivo. Brain 141(7):2194–2212. https://doi.org/10.1093/brain/awy117
Blurton-Jones M, Laferla FM (2006) Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res 3(5):437–448. https://doi.org/10.2174/156720506779025242
Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H (1998) Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res 31(4):317–323. https://doi.org/10.1016/s0168-0102(98)00061-3
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99(9):6364–6369. https://doi.org/10.1073/pnas.092136199
Kaler SG (2011) ATP7A-related copper transport diseases—emerging concepts and future trends. Nat Rev Neurol 7(1):15–29. https://doi.org/10.1038/nrneurol.2010.180
Squitti R, Siotto M, Polimanti R (2014) Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol Aging 35:S40–S50. https://doi.org/10.1016/j.neurobiolaging.2014.02.031
Guo JP, Pan JX, Xiong L, Xia WF, Cui S, Xiong WC (2015) Iron Chelation Inhibits Osteoclastic Differentiation In Vitro and in Tg2576 Mouse Model of Alzheimer’s Disease. PLoS ONE 10(11):e0139395. https://doi.org/10.1371/journal.pone.0139395
Yang X, Cai P, Liu Q, Wu J, Yin Y, Wang X, Kong L (2018) Novel 8-hydroxyquinoline derivatives targeting β-amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorg Med Chem 26(12):3191–3201. https://doi.org/10.1016/j.bmc.2018.04.043
Pavlidis N, Kofinas A, Papanikolaou MG, Miras HN, Drouza C, Kalampounias AG, Kabanos TA, Konstandi M et al (2021) Synthesis, characterization and pharmacological evaluation of quinoline derivatives and their complexes with copper(ΙΙ) in in vitro cell models of Alzheimer’s disease. J Inorg Biochem 217:111393. https://doi.org/10.1016/j.jinorgbio.2021.111393
Flora SJ, Pachauri V (2010) Chelation in metal intoxication. Int J Environ Res Public Health 7(7):2745–2788. https://doi.org/10.3390/ijerph7072745
Flora SJS, Jain K, Panghal A, Patwa J (2022) Chemistry, pharmacology, and toxicology of monoisoamyl dimercaptosuccinic acid: a chelating agent for chronic metal poisoning. Chem Res Toxicol 35(10):1701–1719. https://doi.org/10.1021/acs.chemrestox.2c00129
Squitti R, Zito G (2009) Anti-copper therapies in Alzheimer’s disease: new concepts. Recent Pat CNS Drug Discovery 4(3):209–219. https://doi.org/10.2174/157488909789104802
Huang W, Wei W, Shen Z (2014) Drug-like chelating agents: a potential lead for Alzheimer’s disease. RSC Adv 4(94):52088–52099. https://doi.org/10.1039/C4RA09193K
Cherny RA, Barnham KJ, Lynch T, Volitakis I, Li QX, McLean CA, Multhaup G, Beyreuther K et al (2000) Chelation and intercalation: complementary properties in a compound for the treatment of Alzheimer’s disease. J Struct Biol 130(2–3):209–216. https://doi.org/10.1006/jsbi.2000.4285
McLachlan DR, Smith WL, Kruck TP (1993) Desferrioxamine and Alzheimer's disease: video home behavior assessment of clinical course and measures of brain aluminum. Therapeutic drug monitoring 15 (6):602–607. https://cir.nii.ac.jp/crid/1571135650836546304
Liu G, Garrett MR, Men P, Zhu X, Perry G, Smith MA (2005) Nanoparticle and other metal chelation therapeutics in Alzheimer disease. Biochem Biophys Acta 1741(3):246–252. https://doi.org/10.1016/j.bbadis.2005.06.006
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet (London, England) 337(8753):1304–1308. https://doi.org/10.1016/0140-6736(91)92978-b
Kwan P, Ho A, Baum L (2022) Effects of Deferasirox in Alzheimer’s Disease and Tauopathy Animal Models. Biomolecules 12:3. https://doi.org/10.3390/biom12030365
Yamamoto A, Shin RW, Hasegawa K, Naiki H, Sato H, Yoshimasu F, Kitamoto T (2002) Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J Neurochem 82(5):1137–1147. https://doi.org/10.1046/j.1471-4159.2002.t01-1-01061.x
Brunden KR, Trojanowski JQ, Lee VM (2009) Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discovery 8(10):783–793. https://doi.org/10.1038/nrd2959
Bulic B, Pickhardt M, Schmidt B, Mandelkow EM, Waldmann H, Mandelkow E (2009) Development of tau aggregation inhibitors for Alzheimer’s disease. Angew Chem Int Ed Engl 48(10):1740–1752. https://doi.org/10.1002/anie.200802621
Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM (2001) The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci 21(21):8370–8377. https://doi.org/10.1523/jneurosci.21-21-08370.2001
Lambert C, Beraldo H, Lievre N, Garnier-Suillerot A, Dorlet P, Salerno M (2013) Bis(thiosemicarbazone) copper complexes: mechanism of intracellular accumulation. J Biol Inorg Chem 18(1):59–69. https://doi.org/10.1007/s00775-012-0949-1
McKenzie-Nickson S, Bush AI, Barnham KJ (2016) Bis(thiosemicarbazone) Metal complexes as therapeutics for neurodegenerative diseases. Curr Top Med Chem 16(27):3058–3068. https://doi.org/10.2174/1568026616666160216155746
Esmieu C, Guettas D, Conte-Daban A, Sabater L, Faller P, Hureau C (2019) Copper-targeting approaches in Alzheimer’s disease: how to improve the fallouts obtained from in vitro studies. Inorg Chem 58(20):13509–13527. https://doi.org/10.1021/acs.inorgchem.9b00995
Zheng H, Youdim MB, Weiner LM, Fridkin M (2005) Synthesis and evaluation of peptidic metal chelators for neuroprotection in neurodegenerative diseases. J Peptide Res 66(4):190–203. https://doi.org/10.1111/j.1399-3011.2005.00289.x
Deraeve C, Pitié M, Mazarguil H, Meunier B (2007) Bis-8-hydroxyquinoline ligands as potential anti-Alzheimer agents. New J Chem 31(2):193–195. https://doi.org/10.1039/B616085A
Bush AI (2002) Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol Aging 23(6):1031–1038. https://doi.org/10.1016/s0197-4580(02)00120-3
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R et al (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 60(12):1685–1691. https://doi.org/10.1001/archneur.60.12.1685
Bolognin S, Drago D, Messori L, Zatta P (2009) Chelation therapy for neurodegenerative diseases. Med Res Rev 29(4):547–570. https://doi.org/10.1002/med.20148
Budimir A (2011) Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm (Zagreb, Croatia) 61(1):1–14. https://doi.org/10.2478/v10007-011-0006-6
Yang GJ, Liu H, Ma DL, Leung CH (2019) Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem 24(8):1159–1170. https://doi.org/10.1007/s00775-019-01712-y
Kenche VB, Barnham KJ (2011) Alzheimer’s disease & metals: therapeutic opportunities. Br J Pharmacol 163(2):211–219. https://doi.org/10.1111/j.1476-5381.2011.01221.x
Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K et al (2009) Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheim Dis : JAD 17(3):457–468. https://doi.org/10.3233/jad-2009-1068
Kavirajan H, Schneider LS (2007) Efficacy and adverse effects of cholinesterase inhibitors and memantine in vascular dementia: a meta-analysis of randomised controlled trials. Lancet Neurol 6(9):782–792. https://doi.org/10.1016/s1474-4422(07)70195-3
Zatta P, Raso M, Zambenedetti P, Wittkowski W, Messori L, Piccioli F, Mauri PL, Beltramini M (2005) Copper and zinc dismetabolism in the mouse brain upon chronic cuprizone treatment. Cell Mol Life Sci 62(13):1502–1513. https://doi.org/10.1007/s00018-005-5073-8
Pritam P, Deka R, Bhardwaj A, Srivastava R, Kumar D, Jha AK, Jha NK, Villa C et al (2022) Antioxidants in Alzheimer’s disease: current therapeutic significance and future prospects. Biology 11:2. https://doi.org/10.3390/biology11020212
Baptista FI, Henriques AG, Silva AM, Wiltfang J, da Cruz e Silva OA (2014) Flavonoids as therapeutic compounds targeting key proteins involved in Alzheimer's disease. ACS Chem Neurosc 5 (2):83-92 https://doi.org/10.1021/cn400213r
Singh SK, Gaur R, Kumar A, Fatima R, Mishra L, Srikrishna S (2014) The flavonoid derivative 2-(4’ Benzyloxyphenyl)-3-hydroxy-chromen-4-one protects against Aβ42-induced neurodegeneration in transgenic Drosophila: insights from in silico and in vivo studies. Neurotox Res 26(4):331–350. https://doi.org/10.1007/s12640-014-9466-z
Ishige K, Schubert D, Sagara Y (2001) Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radical Biol Med 30(4):433–446. https://doi.org/10.1016/s0891-5849(00)00498-6
Lloret A, Esteve D, Monllor P, Cervera-Ferri A, Lloret A (2019) The Effectiveness of vitamin E treatment in Alzheimer’s disease. Int J Mol Sci 20:4. https://doi.org/10.3390/ijms20040879
Zhang SM, Hernán MA, Chen H, Spiegelman D, Willett WC, Ascherio A (2002) Intakes of vitamins E and C, carotenoids, vitamin supplements, and PD risk. Neurology 59(8):1161–1169. https://doi.org/10.1212/01.wnl.0000028688.75881.12
La Fata G, Weber P, Mohajeri MH (2014) Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 6(12):5453–5472. https://doi.org/10.3390/nu6125453
Kandiah N, Ong PA, Yuda T, Ng LL, Mamun K, Merchant RA, Chen C, Dominguez J et al (2019) Treatment of dementia and mild cognitive impairment with or without cerebrovascular disease: expert consensus on the use of Ginkgo biloba extract, EGb 761(®). CNS Neurosci Ther 25(2):288–298. https://doi.org/10.1111/cns.13095
Cowan CM, Sealey MA, Mudher A (2021) Suppression of tau-induced phenotypes by vitamin E demonstrates the dissociation of oxidative stress and phosphorylation in mechanisms of tau toxicity. J Neurochem 157(3):684–694. https://doi.org/10.1111/jnc.15253
Iijima-Ando K, Iijima K (2010) Transgenic Drosophila models of Alzheimer’s disease and tauopathies. Brain Struct Funct 214(2–3):245–262. https://doi.org/10.1007/s00429-009-0234-4
Casati M, Boccardi V, Ferri E, Bertagnoli L, Bastiani P, Ciccone S, Mansi M, Scamosci M et al (2020) Vitamin E and Alzheimer’s disease: the mediating role of cellular aging. Aging Clin Exp Res 32(3):459–464. https://doi.org/10.1007/s40520-019-01209-3
Veinbergs I, Van Uden E, Mallory M, Alford M, McGiffert C, DeTeresa R, Orlando R, Masliah E (2001) Role of apolipoprotein E receptors in regulating the differential in vivo neurotrophic effects of apolipoprotein E. Exp Neurol 170(1):15–26. https://doi.org/10.1006/exnr.2001.7684
Butterfield DA, Koppal T, Subramaniam R, Yatin S (1999) Vitamin E as an antioxidant/free radical scavenger against amyloid beta-peptide-induced oxidative stress in neocortical synaptosomal membranes and hippocampal neurons in culture: insights into Alzheimer’s disease. Rev Neurosci 10(2):141–149. https://doi.org/10.1515/revneuro.1999.10.2.141
Yamada K, Tanaka T, Han D, Senzaki K, Kameyama T, Nabeshima T (1999) Protective effects of idebenone and alpha-tocopherol on beta-amyloid-(1–42)-induced learning and memory deficits in rats: implication of oxidative stress in beta-amyloid-induced neurotoxicity in vivo. Eur J Neurosci 11(1):83–90. https://doi.org/10.1046/j.1460-9568.1999.00408.x
Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J et al (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. New Engl J Med 336(17):1216–1222. https://doi.org/10.1056/nejm199704243361704
Butterfield D, Castegna A, Pocernich C, Drake J, Scapagnini G, Calabrese V (2002) Nutritional approaches to combat oxidative stress in Alzheimer’s disease. J Nutr Biochem 13(8):444. https://doi.org/10.1016/s0955-2863(02)00205-x
Dringen R, Brandmann M, Hohnholt MC, Blumrich EM (2015) Glutathione-dependent detoxification processes in astrocytes. Neurochem Res 40(12):2570–2582. https://doi.org/10.1007/s11064-014-1481-1
Marí M, de Gregorio E, de Dios C, Roca-Agujetas V, Cucarull B, Tutusaus A, Morales A, Colell A (2020) Mitochondrial Glutathione: Recent Insights and Role in Disease. Antioxid (Basel, Switzerland) 9:10. https://doi.org/10.3390/antiox9100909
Barbero-Camps E, Fernández A, Martínez L, Fernández-Checa JC, Colell A (2013) APP/PS1 mice overexpressing SREBP-2 exhibit combined Aβ accumulation and tau pathology underlying Alzheimer’s disease. Hum Mol Genet 22(17):3460–3476. https://doi.org/10.1093/hmg/ddt201
Allen M, Zou F, Chai HS, Younkin CS, Miles R, Nair AA, Crook JE, Pankratz VS et al (2012) Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: an association study with mechanistic implications. Mol Neurodegener 7:13. https://doi.org/10.1186/1750-1326-7-13
Naqvi S, Panghal A, Flora SJS (2020) Nanotechnology: a promising approach for delivery of neuroprotective drugs. Front Neurosci 14:494. https://doi.org/10.3389/fnins.2020.00494
Wei QY, Chen WF, Zhou B, Yang L, Liu ZL (2006) Inhibition of lipid peroxidation and protein oxidation in rat liver mitochondria by curcumin and its analogues. Biochem Biophys Acta 1760(1):70–77. https://doi.org/10.1016/j.bbagen.2005.09.008
Baum L, Ng A (2004) Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J Alzheim Dis 6(4):367–377. https://doi.org/10.3233/jad-2004-6403. (discussion 443-369)
Nishinaka T, Ichijo Y, Ito M, Kimura M, Katsuyama M, Iwata K, Miura T, Terada T et al (2007) Curcumin activates human glutathione S-transferase P1 expression through antioxidant response element. Toxicol Lett 170(3):238–247. https://doi.org/10.1016/j.toxlet.2007.03.011
Serafini MM, Catanzaro M, Rosini M, Racchi M, Lanni C (2017) Curcumin in Alzheimer’s disease: Can we think to new strategies and perspectives for this molecule? Pharmacol Res 124:146–155. https://doi.org/10.1016/j.phrs.2017.08.004
Giri RK, Rajagopal V, Kalra VK (2004) Curcumin, the active constituent of turmeric, inhibits amyloid peptide-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulating early growth response-1 transcription factor. J Neurochem 91(5):1199–1210. https://doi.org/10.1111/j.1471-4159.2004.02800.x
Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM (2001) Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol Aging 22(6):993–1005. https://doi.org/10.1016/s0197-4580(01)00300-1
Singh SK, Srikrishna S, Castellani RJ, Perry G (2017) Antioxidants in the Prevention and Treatment of Alzheimer’s Disease. In: Al-Gubory KH, Laher I (eds) Nutritional antioxidant therapies: treatments and perspectives. Springer International Publishing, Cham, pp 523–553. https://doi.org/10.1007/978-3-319-67625-8_20
Ide K, Matsuoka N, Yamada H, Furushima D, Kawakami K (2018) Effects of tea catechins on Alzheimer’s disease: recent updates and perspectives. Mol (Basel, Switzerland) 23:9. https://doi.org/10.3390/molecules23092357
Choi YT, Jung CH, Lee SR, Bae JH, Baek WK, Suh MH, Park J, Park CW et al (2001) The green tea polyphenol (-)-epigallocatechin gallate attenuates beta-amyloid-induced neurotoxicity in cultured hippocampal neurons. Life Sci 70(5):603–614. https://doi.org/10.1016/s0024-3205(01)01438-2
Haque AM, Hashimoto M, Katakura M, Hara Y, Shido O (2008) Green tea catechins prevent cognitive deficits caused by Abeta1-40 in rats. J Nutr Biochem 19(9):619–626. https://doi.org/10.1016/j.jnutbio.2007.08.008
Kaur T, Pathak CM, Pandhi P, Khanduja KL (2008) Effects of green tea extract on learning, memory, behavior and acetylcholinesterase activity in young and old male rats. Brain Cogn 67(1):25–30. https://doi.org/10.1016/j.bandc.2007.10.003
Gomes BAQ, Silva JPB, Romeiro CFR, Dos Santos SM, Rodrigues CA, Gonçalves PR, Sakai JT, Mendes PFS et al (2018) Neuroprotective mechanisms of Resveratrol in Alzheimer’s disease: role of SIRT1. Oxid Med Cell Longev 2018:8152373. https://doi.org/10.1155/2018/8152373
Kong D, Yan Y, He XY, Yang H, Liang B, Wang J, He Y, Ding Y et al (2019) Effects of resveratrol on the mechanisms of antioxidants and estrogen in Alzheimer’s disease. Biomed Res Int 2019:8983752. https://doi.org/10.1155/2019/8983752
Arbo BD, André-Miral C, Nasre-Nasser RG, Schimith LE, Santos MG, Costa-Silva D, Muccillo-Baisch AL, Hort MA (2020) Resveratrol derivatives as potential treatments for Alzheimer’s and Parkinson’s disease. Front Aging Neurosci 12:103. https://doi.org/10.3389/fnagi.2020.00103
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R et al (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Archives of neurology 60 (12):1685–1691. https://www.doi.org/https://doi.org/10.1001/archneur.60.12.1685
Wang C-Y, Xie J-W, Xu Y, Wang T, Cai J-H, Wang X, Zhao B-L, An L et al (2013) Trientine reduces BACE1 activity and mitigates amyloidosis via the AGE/RAGE/NF-κB pathway in a transgenic mouse model of Alzheimer’s disease. Antioxid Redox Signal 19(17):2024–2039. https://doi.org/10.1089/ars.2012.5158
Xu W, Xu Q, Cheng H, Tan X (2017) The efficacy and pharmacological mechanism of Zn7MT3 to protect against Alzheimer’s disease. Sci Rep 7(1):13763. https://doi.org/10.1038/s41598-017-12800-x
Dedeoglu A, Cormier K, Payton S, Tseitlin KA, Kremsky JN, Lai L, Li X, Moir RD et al (2004) Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp Gerontol 39(11–12):1641–1649. https://doi.org/10.1016/j.exger.2004.08.016
McLachlan DC, Kruck T, Kalow W, Andrews D, Dalton A, Bell M, Smith W (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. The Lancet 337(8753):1304–1308. https://doi.org/10.1016/0140-6736(91)92978-B
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L et al (2010) PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J Alzheim Dis 20(2):509–516. https://doi.org/10.3233/JAD-2010-1390
Lee J-Y, Friedman JE, Angel I, Kozak A, Koh J-Y (2004) The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human β-amyloid precursor protein transgenic mice. Neurobiol Aging 25(10):1315–1321. https://doi.org/10.1016/j.neurobiolaging.2004.01.005
Cacciatore I, Marinelli L, Di Stefano A, Di Marco V, Orlando G, Gabriele M, Gatta DMP, Ferrone A et al (2018) Chelating and antioxidant properties of l-Dopa containing tetrapeptide for the treatment of neurodegenerative diseases. Neuropeptides 71:11–20. https://doi.org/10.1016/j.npep.2018.06.002
Carrasco-Castilla J, Hernández-Álvarez AJ, Jiménez-Martínez C, Jacinto-Hernández C, Alaiz M, Girón-Calle J, Vioque J, Dávila-Ortiz G (2012) Antioxidant and metal chelating activities of peptide fractions from phaseolin and bean protein hydrolysates. Food Chem 135(3):1789–1795. https://doi.org/10.1016/j.foodchem.2012.06.016
Chen H-M, Muramoto K, Yamauchi F, Nokihara K (1996) Antioxidant activity of designed peptides based on the antioxidative peptide isolated from digests of a soybean protein. J Agric 44(9):2619–2623. https://doi.org/10.1021/jf950833m
El Ghazouani A, Basle A, Firbank SJ, Knapp CW, Gray J, Graham DW, Dennison C (2011) Copper-binding properties and structures of methanobactins from Methylosinus trichosporium OB3b. Inorg Chem 50(4):1378–1391. https://doi.org/10.1021/ic101965j
McCabe JW, Vangala R, Angel LA (2017) Binding selectivity of methanobactin from Methylosinus trichosporium OB3b for copper (I), silver (I), zinc (II), nickel (II), cobalt (II), manganese (II), lead (II), and iron (II). J Am Soc Mass Spectrom 28(12):2588–2601. https://doi.org/10.1007/s13361-017-1778-9
Jadhav K, Jhilta A, Singh R, Ray E, Sharma N, Shukla R, Singh AK, Verma RK (2023) Clofazimine nanoclusters show high efficacy in experimental TB with amelioration in paradoxical lung inflammation. Biomaterials Advances 154:213594. https://doi.org/10.1016/j.bioadv.2023.213594
Singh R, Jadhav K, Vaghasiya K, Ray E, Shukla R, Verma RK (2023) New generation smart drug delivery systems for rheumatoid arthritis. Curr Pharm Des 29(13):984–1001. https://doi.org/10.2174/1381612829666230406102935
Jadhav K, Ray E, Vaghasiya K, Verma RK (2022) Neurodegenerative disorders due to inhalation of various small particles. In: Nanomedical Drug Delivery for Neurodegenerative Diseases. Elsevier, pp 41–54. https://doi.org/10.1016/B978-0-323-85544-0.00010-1
Shukla R, Thok K, Alam I, Singh R (2020) Nanophytomedicine market: global opportunity analysis and industry forecast. Nanophytomedicine: Concept to Clinic:19–31. https://springerlink.bibliotecabuap.elogim.com/chapter/https://doi.org/10.1007/978-981-15-4909-0_2
Vaghasiya K, Ray E, Singh R, Jadhav K, Sharma A, Khan R, Katare OP, Verma RK (2021) Efficient, enzyme responsive and tumor receptor targeting gelatin nanoparticles decorated with concanavalin-A for site-specific and controlled drug delivery for cancer therapy. Mater Sci Eng: C 123:112027. https://doi.org/10.1016/j.msec.2021.112027
Huang Y, Chang Y, Liu L, Wang J (2021) Nanomaterials for modulating the aggregation of β-amyloid peptides. Mol (Basel, Switzerland) 26(14):4301. https://doi.org/10.3390/molecules26144301
Liu G, Men P, Harris PL, Rolston RK, Perry G, Smith MA (2006) Nanoparticle iron chelators: a new therapeutic approach in Alzheimer disease and other neurologic disorders associated with trace metal imbalance. Neurosci Lett 406(3):189–193. https://doi.org/10.1016/j.neulet.2006.07.020
Huang Q, Jiang C, Xia X, Wang Y, Yan C, Wang X, Lei T, Yang X et al (2023) Pathological BBB crossing melanin-like nanoparticles as metal-ion chelator and neuroinflammation regulator against Alzheimer’s disease. Research. 6:0180. https://doi.org/10.34133/research.0180
Kowalczyk J, Grapsi E, Espargaro A, Caballero AB, Juárez-Jiménez J, Busquets MA, Gamez P, Sabate R et al (2021) Dual effect of prussian blue nanoparticles on Aβ40 aggregation: β-Sheet fibril reduction and copper dyshomeostasis regulation. Biomacromol 22(2):430–440. https://doi.org/10.1021/acs.biomac.0c01290
Li M, Liu Z, Ren J, Qu X (2012) Inhibition of metal-induced amyloid aggregation using light-responsive magnetic nanoparticle prochelator conjugates. Chem Sci 3(3):868–873. https://doi.org/10.1039/C1SC00631B
Li M, Shi P, Xu C, Ren J, Qu X (2013) Cerium oxide caged metal chelator: anti-aggregation and anti-oxidation integrated H2O2-responsive controlled drug release for potential Alzheimer’s disease treatment. Chem Sci 4(6):2536–2542. https://doi.org/10.1039/C3SC50697E
Guan Y, Gao N, Ren J, Qu X (2016) Rationally designed CeNP@ MnMoS4 core-shell nanoparticles for modulating multiple facets of Alzheimer’s disease. Chem - A Eur J 22(41):14523–14526. https://doi.org/10.1002/chem.201603233
Li M, Guan Y, Ding C, Chen Z, Ren J, Qu X (2016) An ultrathin graphitic carbon nitride nanosheet: a novel inhibitor of metal-induced amyloid aggregation associated with Alzheimer’s disease. J Mater Chem B Chem Sci 4(23):4072–4075. https://doi.org/10.1039/C6TB01215A
Gong L, Zhang X, Ge K, Yin Y, Machuki JOA, Yang Y, Shi H, Geng D et al (2021) Carbon nitride-based nanocaptor: an intelligent nanosystem with metal ions chelating effect for enhanced magnetic targeting phototherapy of Alzheimer’s disease. Biomaterials 267:120483. https://doi.org/10.1016/j.biomaterials.2020.120483
Zhang C, Yang K, Yu S, Su J, Yuan S, Han J, Chen Y, Gu J et al (2019) Design, synthesis and biological evaluation of hydroxypyridinone-coumarin hybrids as multimodal monoamine oxidase B inhibitors and iron chelates against Alzheimer’s disease. Eur J Med Chem 180:367–382. https://doi.org/10.1016/j.ejmech.2019.07.031
Weinreb O, Amit T, Bar-Am O, Youdim MB (2016) Neuroprotective effects of multifaceted hybrid agents targeting MAO, cholinesterase, iron and β-amyloid in ageing and Alzheimer’s disease. British J Pharmacol 173(13):2080–2094. https://doi.org/10.1111/bph.13318
Acknowledgements
The authors are also thankful to Chembiodraw for providing a platform for drawing illustrations.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Archna Panghal: writing—original draft. Raghuraj Singh: writing—original draft. Krishna Jadhav: writing—original draft. Ashima Thakur: writing—original draft. Rahul Kumar Verma— review and editing. Charan Singh: writing—review and editing. Manoj Goyal: writing—review and editing. Jayant Kumar: conceptualization and writing—review and editing. Ajay G. Namdeo: review and editing.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Singh, R., Panghal, A., Jadhav, K. et al. Recent Advances in Targeting Transition Metals (Copper, Iron, and Zinc) in Alzheimer’s Disease. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04256-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04256-8