
Abstract
Pyrroline-5-carboxylate reductase (PYCR), the last enzyme in proline synthesis that converts P5C into proline, was found promoting cancer growth and inhibiting apoptosis through multiple approaches, including regulating cell cycle and redox homeostasis, and promoting growth signaling pathways. Proline is abnormally up-regulated in multiple cancers and becomes one of the critical players in the reprogramming of cancer metabolism. As the last key enzymes in proline generation, PYCRs have been the subject of many investigations, and have been demonstrated to play an indispensable role in promoting tumorigenesis and cancer progression. In this article, we will thoroughly review the recent investigations on PYCRs in cancer development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
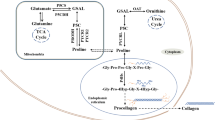
Proline is a special non-essential amino acid, as its side chain forms a pyrrolidine loop with nitrogen, making it the only proteinogenic amino acid. The cyclic structure contained in proline causes its extraordinary rigidity and influences the nearby secondary structure of proteins, making it a sequence-recognition motif (Phang 2019). Proline is one of the most predominant amino acids in the cell microenvironment, and it forms collagen via the intermediate hydroxyproline. Unlike other amino acids, proline has its own metabolic features with an exclusive family of metabolic enzymes. There are two distinct pathways involved in proline biosynthesis due to different origins of resources, namely glutamate, and ornithine (Tanner et al. 2018). However, all roads lead to Δ1-pyrroline-5-carboxylate (P5C), the precursor of proline, and pyrroline-5-carboxylate reductases (PYCRs/P5CRs) are the last enzymes that convert P5C into proline through NAD(P)H oxidation (Christensen et al. 2017). This process is highly conserved and is exhibited in almost all species, and indispensable for proline production (Adams and Frank 1980).
Three homologous genes have been discovered to encode PYCRs, including PYCR1, PYCR2, and PYCR3/PYCRL. PYCR1 and PYCR2 are both located in the mitochondria and share 84% structure homology, functioning similarly in the last step of the glutamate-P5C-proline pathway with a preference for NADH as the cofactor. However, PYCR3 lacks 40 amino acids in the C terminal compared to the others and is mainly located in the cytoplasm. In addition, PYCR3 prefers to catalyze proline production from ornithine, using NADPH as the cofactor (De Ingeniis et al. 2012). The crystal structure of human PYCR revealed a decameric architecture consisting of five basic homodimer subunits and ten catalytic sites. The ring-shaped holoenzyme contains a circular groove that serves as the binding sites for substrates and cofactors (Nocek et al. 2005; Meng et al. 2006).
PYCRs are linked to many human diseases. The missense mutation in PYCR1 was found to cause autosomal-recessive cutis laxa type 2 (ARCL2), a multisystem disorder including premature aging, wrinkled and lax skin, joint laxity, and developmental delay (Guernsey et al. 2009). Two homozygous mutations in PYCR2 were reported to cause microcephaly and hypomyelination, suggesting a crucial role of PYCR2 in human nervous system development (Nakayama et al. 2015). Since the reprogramming of metabolism emerges as a vital hallmark of cancer, an increasing number of metabolic pathways have been deeply investigated (Hanahan and Weinberg 2011; Pavlova and Thompson 2016), including the proline cycle, which found proline to be abnormally up-regulated in multiple cancers (Sun et al. 2019; Tanner et al. 2018; Tang et al. 2018). PYCRs, as the last key enzyme in proline generation, have attracted many investigations, and have been demonstrated to play an indispensable role in promoting tumorigenesis and cancer progression. As proline metabolism became evident in the metabolic reprogramming of cancer, a few reviews have summarized the aberrant proline cycle in tumors and the important enzymes involved in the process (Tanner et al. 2018; Phang 2019; Burke et al. 2020; D'Aniello et al. 2020). However, there is still short of a global description for PYCR in tumor initiation and development. This review, therefore, summarized the distinct role and its mechanisms of PYCR in cancer progression, including those unrelated to proline production.
Up-regulated in multiple cancers
Through immunohistochemical staining, PYCRs have been found to be up-regulated in different cancer tissues, including lung cancer (Cai et al. 2018; Gao et al. 2020; Guo et al. 2019; She et al. 2019), liver cancer (Zhuang et al. 2019), colorectal cancer (Yan et al. 2019), gastric cancer (Xiao et al. 2020), breast cancer (Ding et al. 2017; Craze et al. 2018), prostate cancer (Zeng et al. 2017), bladder cancer (Song et al. 2021; Du et al. 2021) and esophageal squamous cell carcinoma (Sun et al. 2019). High expression of PYCRs is positively correlated with poor cancer prognoses. Over-expression of PYCRs in different tumors was also demonstrated by analysis from different databases such as The Cancer Genome Atlas (TCGA) (Sang et al. 2019; Hollinshead et al. 2018; Cheng et al. 2020) (Table 1). Increased expression of PYCRs resulted in an elevation of proline concentration, which was recognized as a metabolic addiction of cancer cells (Ding et al. 2020), making proline a trustworthy therapeutical target (Loayza-Puch et al. 2016; Sun et al. 2019).
Aside from the abnormal expression levels, Hong et al. reported that PYCR1 had novel splicing variants in non-small cell lung cancer. They found that exon 2 or 3 of PYCR1 is skipped in cancer tissues, indicating that PYCR1 might gain a novel function in cancer via alternative splicing (Hong et al. 2016). Above all, up-regulating the expression level of PYCR was shown to be a potent strategy of cancer cells, making it a novel oncogene and a promising target to block tumor progression and improve survival.
Promote proliferation and inhibit apoptosis
Many studies have demonstrated that PYCR1 plays a crucial role in tumor growth and progression. Knockdown of PYCR1 significantly suppressed tumor growth and induced apoptosis in various cancers. Specifically, reduction of PYCR1 significantly down-regulated the expression level of Bcl-2 and Bcl-xl, two key anti-apoptotic proteins, and promoted the cleavage of caspase-3. Meanwhile, Bax, a pro-apoptotic protein and tumor suppressor, was increased after the silencing of PYCR1 (Ye et al. 2018; Cai et al. 2018; Wang et al. 2019). Reduction of PYCR3/PYCRL in a subset of cancer cells that are independent of exogenous proline inhibited colony formation and cell proliferation following proline starvation (Sahu et al. 2016).
Regulate cell cycle
Cell cycle transition is a complicated but accurate process controlled by a number of cyclin-dependent kinases, and each cycle is associated with a unique cyclin-CDK activity. Loss of PYCR1 induces cell cycle arrest in the G1 phase and subsequent apoptosis in non-small cell lung cancer cells. Mechanically, the expression of cyclin D1, which functions in promoting cell cycle entering S phase from the G1 phase, decreased dramatically after the silencing of PYCR1 (Tetsu and McCormick 1999; Cai et al. 2018). The cyclin B1/CDK1 complex is activated and accumulated at the centrosome during interphase, which controls both centrosome separation and mitotic spindle assembly (Nigg 2001). In prostate cancer cells, ablation of PYCR1 caused cell cycle arrested at the G2/M phase via down-regulating the expression levels of CDK1, CDK2, CDK4 and Cyclin B1 (Zeng et al. 2017). In hepatocellular carcinoma, Ding et al. performed an RNA-sequencing analysis after knockdown PYCR and found a strong correlation between PYCR1 and cell cycle-related genes, which was not observed in PYCR2 knockdown cells. The silencing of PYCR1 significantly repressed the expression of crucial cell cycle regulators, including cyclins, CDKs, E2F and MYC (Ding et al. 2020). However, Liu et al. found that impaired proline biogenesis by down-regulation of PYCR had no effect on cell cycle in lymphoma and lung cancer cells (Liu et al. 2015). The possible explanation is that these groups chose different types of cancer cell lines to carry out the experiments for the cell cycle. The PC9 cell chosen by Liu et al. was shown a rather low expression level of PYCR among lung cancer cells by other group (Cai et al. 2018), which might partially make a case for why PYCR had different roles in cell cycle regulation in different cancer cells.
Regulate redox homeostasis
Proline was shown a potent anti-oxidative stress reagent that capable to protect cells from various reactive oxygen species (ROS) inducers such as H2O2, tert-butyl hydroperoxide, and carcinogenic oxidative stress. In the presence of H2O2, the expression level of PYCR increased, resulted in more proline production, and consequently increased cell survival (Krishnan et al. 2008). In addition, PARK7/DJ-1 could directly enhance the enzymatic activity of PYCR1 and promote proline production. Knockdown either PYCR1 or DJ-1 inhibited cell resistance to H2O2, however, deletion both DJ-1 and PYCR1 did not significantly enhance the protection, indicating that PYCR1 and DJ-1 might work together in the same pathway in anti-oxidative protection (Yasuda et al. 2013). Ribonucleotide-diphosphate reductase subunit M2 B (RRM2B) plays a vital role in protecting cells from oxidative stress through its intrinsic catalase activity that reduces hydrogen peroxide into water. PYCR1 and PYCR2 can interact with RRM2B. Silencing of PYCR1 and PYCR2 not only directly causes fragmentation of mitochondria, sensitizing cells to oxidative stress, but also eliminates the resistance to hydrogen peroxide achieved by over-expression of RRM2B, indicating that this complex functioned together to protect cells from oxidative stress (Kuo et al. 2016). Isocitrate dehydrogenase 1 (IDH1) was frequently mutated in various cancers, especially in gliomas and glioblastoma. One of the oncogenic effects of this mutation is breaking the redox homeostasis due to the acquirement of the NADPH-coupled reducing activity. In IDH1 mutated cancers, the cellular NADPH: NADP+ ratio is altered, which makes it more sensitive to oxidative stress. PYCR1 was found up-regulated in IDH1-mutated gliomas, along with an increase in proline synthesis and proline concentration, serving as an endogenous antioxidant. In addition, the enhanced PYCR1 activity consumed the sparing oxygen by oxidizing NAD(P)H, resulting in partially uncoupling respiration from TCA cycle activity, and sustaining cellular anabolism (Hollinshead et al. 2018). Although it still remained elusive that whether anti-oxidation could prevent or promote cancer, overloaded ROS was harmful to cancer cell survival, due to the ruinous oxidation of DNA, protein and lipids. PYCR could directly respond to excessive ROS level such as H2O2, and promote redox homeostasis via multiple strategies, including proline biogenesis, up-regulation of RRM2B and balancing NADPH: NADP+ ratio.
Regulate cell growth and proliferation signaling pathways
STAT3 and NF-κB are two distinct transcription factors that can affect the activity of each other, and both play crucial roles in tumorigenesis (Johnson et al. 2018). STAT3 is able to activate NF-κB via binding and recruiting its acetyltransferase p300 and enhancing the nuclear retention of NF-κB (He and Karin 2011). Yan et al. found that PYCR1 directly interacts with STAT3, and overexpression of PYCR1 elevates the protein levels of STAT3. Silencing of PYCR1 decreased the phosphorylation level of NF-κB p65 and p38 MAPK, which could be reversed by over-expression of STAT3. This suggested that PYCR1 is involved in the STAT3-MAPK/NF-κB axis and regulates the progression of cancer (Yan et al. 2019). Jun N-terminal kinases (JNKs) are members of the MAPKs superfamily that function in regulating cell proliferation, differentiation, and apoptosis. Upon phosphorylation by upstream MAP2Ks, JNK translocates and activates c-Jun, which consequently increases the transcription of its downstream target genes (Dhanasekaran and Reddy 2008). A gene expression array was performed to investigate the difference between shPYCR1 and control hepatocellular carcinoma (HCC), which revealed that silencing PYCR1 significantly altered the JNK signaling pathway. In addition, an obvious reduction of mRNA level of c-Jun was detected after PYCR1 knockdown, indicating that PYCR1 knockdown could inhibit cell proliferation in HCC through regulating JNK pathway (Zhuang et al. 2019).
Knockdown PYCR1 significantly impaired the phosphorylation levels of the serine/threonine kinase AKT and its target p70, and this inhibitory effect could be reversed by additional stimulation with insulin-like growth factor 1 (IGF-1), an activator of T308 phosphorylation site of AKT. IGF-1 also rescued the proliferation inhibition in PYCR1 knockdown cells, demonstrating that PYCR1 promoted tumor growth via the AKT pathway (Ye et al. 2018). Wang et al. also reported that not only AKT but also the phosphorylation level of mTOR, was obviously down-regulated following reduction of PYCR1 (Wang and Liu 2019). In addition, Du et al. discovered that, in bladder cancer cells, PYCR1 knockdown impeded tumor growth and invasion via diminishing the phosphorylation levels of AKT, and GSK-3β which is the down-stream target of AKT. Reduction of PYCR1 also decreased the active level of β-catenin, whereas over-expression of PYCR1 had the opposite effect. More importantly, re-activation of β-catenin was capable to reverse the inhibition effect caused by PYCR1 knockdown, which demonstrated that PYCR1 promoted AKT/GSK-3β/β-catenin signaling in bladder cancer progression (Du et al. 2021). The ERK pathway is a highly evolutionarily conserved signaling cascade composed of several crucial components including Ras, Raf, MEK, and ERK (Samatar and Poulikakos 2014). Reduction of PYCR1 decreased both the protein level and phosphorylation level of ERK in breast cancer cells, and thus significantly impaired cell growth. However, there was no obvious change in either the ERK pathway or cell growth after PYCR2 knockdown, suggesting a unique role of PYCR1 in regulating the ERK signaling pathway (Ding et al. 2017).
In summary, down-regulation of PYCR impaired various proliferative signaling such as STAT3, AKT, and mTOR pathways. However, PYCR is not an inherent component or mediator in these pathways, which means that PYCR regulated growth signaling through an indirect mechanism, which has not been elucidated yet. Since ablation of PYCR decreased proline level, and supplement with proline could activate mTOR signaling in embryonic stem (ES) cells, the inhibitory effect on mTOR after knockdown PYCR in cancer cells could be put down to the shortage of proline. The redox imbalance due to PYCR silencing could be the explanation for NF-κB and STAT3 activation, as ROS is an intrinsic messenger molecule which can activate multiple signaling such as NF-κB pathway (Fig. 1).

The role of PYCRs in tumorigenesis and progression
Promote cancer invasion and migration
Epithelial-mesenchymal transition (EMT) is an orderly reversible process that is crucial for epithelial cancer progression and metastasis. During EMT, the interaction between cancer cells and the cell-extracellular matrix is remodeled, facilitating cancer cells to detach from the basement for migrating (Dongre and Weinberg 2019). Multiple groups have demonstrated that reduction of PYCR1 significantly impaired the migration and invasion capability of cancer cells (Cheng et al. 2020; Gao et al. 2020; Song et al. 2021) via down-regulating the EMT markers including N-cadherin, Vimentin, Snail and Slug, whereas promoting the expression of E-cadherin that hindered the EMT process (Sang et al. 2019; Ding et al. 2017). Mitochondrial Lon is a stress response protein that functions in maintaining mitochondrial proteostasis. Lon also plays an important role in promoting EMT via increasing cellular ROS generation and inducing chronic inflammation in a tumor microenvironment (Pinti et al. 2015). PYCR1 bound to Lon and functioned as an ROS regulator. The over-expression of PYCR1 could increase ROS generation. Under oxidative stress, the interaction between PYCR1 and Lon increased, which resulted in EMT activation and secretion of pro-inflammation cytokines. Besides, the induction of EMT by Lon could be obviously inhibited by PYCR1 knockdown. This finding suggests that PYCR1 can promote EMT via an indirect mechanism (Kuo et al. 2020). The extracellular matrix (ECM) of the tumor microenvironment plays a vital role in the development of cancer, such as gaining invasiveness, undergoing the EMT process, and metastasis. Aberrant ECM remodeling including proteolysis and crosslinking contributes to the signaling transduction and angiogenesis, which in turn promotes cancer cell invasion (Yuzhalin et al. 2018). Collagen is the most abundant component of ECM, and around 25% amino acids in collagen are proline and hydroxylated-proline (Phang 2019). Since up-regulation of PYCR in cancer cells promoted proline biogenesis, it is reasonable to speculate that PYCR1 might promote matrix protein production and thus contribute to ECM remodeling. Although there is no direct evidence, several recent works already gave some hints (Chen et al. 2021; Liang et al. 2019). For example, transforming growth factor-beta (TGF-β) is a potent inducer of EMT, which was shown to elevate the expression levels of all enzymes participated in proline biogenesis including PYCR (Schworer et al. 2020). Therefore, it is safe to hypothesize that PYCR plays an important role in tumor migration induced by collagen production and ECM remodeling.
Induce drug resistance and cancer relapse
Chemoresistance and cancer relapse have become an increasingly major challenge for cancer therapy and patient prognosis. Neoadjuvant treatment (NAT) is an administration of therapy agents or radiation before the surgery, aimed to shrink large tumors and obtain prognostic information about drug sensitivity (Trimble et al. 1993). Shenoy et al. performed LC–MS/MS proteomic analysis of breast cancer tissue before and after neoadjuvant treatment and found that the expression level of PYCR1 was tightly associated with drug resistance. Specifically, PYCR1/2 were highly expressed in cancer tissue before and after treatment, which was correlated with poorer prognoses and higher tumor recurrence (Shenoy et al. 2020). In vivo and in vitro functional experiments further demonstrated that PYCR1 deletion increased glutamine flux to the TCA cycle, which restricted the proliferation and invasion capability of cancer cells and increased drug sensitivity (Shenoy et al. 2020). Knockdown PYCR1 also diminished IC50 for 5-FU in colorectal cancer cells, and overexpression of STAT3 could reverse the effect. This finding suggests that PYCR1 promotes drug resistance by activating the STAT3 signaling pathway (Yan et al. 2019). Cancer stem-like cells (CSLC) are a population of cancer cells inside the tumor mass and possess the ability of self-renewal, which are believed capable to induce drug resistance and relapse. Proline was shown that it is able to induce embryonic-stem-cell-to-mesenchymal-like transition (esMT) in ES cells, and PYCR1 was shown to be regulated by TAp73 in CSLC. These evidences all suggested an important role of proline metabolism in cancer stem-like cells, which calls for more investigations.
Involved in Virus-related cancer
About 15–20% of cancers are associated with parasitic infection, of which the most common occurrence is through viruses. Oncovirus infection could produce oncogenic effects through multiple mechanisms such as induction of DNA damage and chronic inflammation (Tashiro and Brenner 2017). Kaposi’s sarcoma-associated herpesvirus (KSHV) infection increases levels of nonessential amino acids, especially proline. KSHV K1 oncoprotein directly binds with host PYCR1 and PYCR2 via its C terminus, and this interaction enhances PYCR enzymatic activity, resulting in increased proline synthesis. Additionally, PYCR activity is also crucial for regulating ROS production in infected cells and indispensable for K1-mediated transformation and tumor growth (Choi et al. 2020). A systematic proteogenomic analysis of Hepatitis B virus (HBV)-related HCC tissues revealed an abnormal activation of key signaling pathways and liver-related metabolic reprogramming, compared with paired adjacent normal tissues. PYCR2 expression was found to be significantly different, indicating the crucial role of PYCR2 in metabolic reprogramming. Further immunostaining results also demonstrated that PYCR2 is tightly correlated with patient survival, suggesting that PYCR2 might be a robust prognostic marker in HBV-related HCC (Gao et al. 2019).
The regulation of PYCR
As a crucial enzyme of proline metabolism, extracellular proline is the natural regulator of PYCR activity (Lorans and Phang 1981). A measurement of ribosome profiling for detecting restricted amino acids found a limitation of available proline for kidney cancer. The deficiency of proline and its precursor resulted in up-regulation and activation of PYCR1, indicating the importance of its regulation in tumor growth (Loayza-Puch et al. 2016).
Transcription
PI3K/AKT was proposed to be the upstream regulator of PYCR1 by GSEA analysis, and there was a positive correlation of mRNA expression levels between PIK3CB, AKT1, and PYCR1 in GC cohort from TCGA database. In addition, inhibition of PI3K significantly decreased the mRNA and protein levels of PYCR1 (Xiao et al. 2020). Using chromatin immunoprecipitation (ChIP) assay, PYCR1 was discovered as a target for androgen receptor (AR) in prostate cancer. AR occupancy was enriched at the PYCR1 locus, and the expression of PYCR1 responded to DHT treatment (Jariwala et al. 2007). PYCR1 was also found as a potential downstream target of the oncogene c-MYC. Liu et al. reported that c-MYC could directly increase the expression level of PYCR1 to facilitate proline production from glutamate while silencing c-MYC inversely downregulated PYCR1 (Liu et al. 2012). The positive correlation between c-MYC and PYCR1 mRNA level was also confirmed in a study by Craze et al. (Craze et al. 2018). The transcription factor MZF1 promoted PYCR1 expression as well, enhancing proline production to support neuroblastoma progression (Fang et al. 2019). TAp73, a tumor suppressor, was recently found to regulate PYCR1 in cancer stem-like cells. Mechanically, TAp73 knockdown significantly inhibited the expression levels of PYCR1 mRNA and protein, leading to a metabolic shift from proline and glutamate synthesis to the urea cycle, which caused a deficiency in proline concentration, limiting cell growth (Sharif et al. 2019).
Post-transcription
The expression of PYCR1 was negatively regulated by miR-488, a microRNA that was found down-regulated in both non-small-cell lung cancer cell lines and patient tissues, which resulted in highly elevated PYCR1 expression. Furthermore, re-expression of PYCR1 could rescue the inhibitory effect in cell proliferation caused by miR-488 over-expression on NSCLC cells, suggesting a potential therapeutic role of miR-488/PYCR1 axis in lung cancer (Wang et al. 2019). Another microRNA, miR-328-3p was recently shown to directly target PYCR1 and repress its expression in lung adenocarcinoma, and thus inhibit cell viability and migration of cancer cells (Lu et al. 2021). Alpha-ketoglutarate-dependent dioxygenase (FTO) is a vital RNA demethylase that mediates the demethylation of the 6th nitrogen within the adenosine base (m6A) and regulates multiple biological activities (Chen et al. 2019b). Song et al. found that PYCR1 mRNA was a novel substrate of FTO, as knockdown of FTO significantly increased the m6A level of PYCR1 mRNA. FTO promotes the stability of PYCR1 mRNA by preventing its degradation and causes the abnormally high expression of PYCR1 in bladder cancer. This discovery demonstrates an important role of FTO/PYCR1 signaling in bladder cancer development (Song et al. 2021).
Post-translation
It was recently found that PYCR1 can be regulated by acetylation, which was its first reported post-translational modification (PTM). PYCR1 is acetylated by acetyltransferase CREB-binding protein (CBP) at the 228th lysine residue (K228), which is inversely removed by histone deacetylase Sirtuin 3 (SIRT3). The hyperacetylation level of K228 significantly impaired the formation of the decamer, thus inhibiting its enzymatic activity. The stable re-construction of wild-type PYCR1 and its different acetylation level mimics were generated from PYCR1 knockout cell lines and revealed that the acetylation of PYCR1K228 obviously impeded cancer cell proliferation. This discovery demonstrated that PTM is an indispensable regulator of PYCRs (Chen et al. 2019a). DJ-1, encoded by the PARK7 gene, also regulated the enzymatic activity of PYCR1 and enhanced its anti-oxidative stress function via a direct interaction (Yasuda et al. 2013). Kindlin-2 is a widely expressed protein that regulates the adhesion between cells and the extracellular matrix and is also involved in signaling transduction. Kindlin-2 was reported to co-localize and interact with PYCR1. Ablation of Kindlin-2 in lung cancer cells caused the obvious reduction in PYCR1 protein level but not mRNA level, which indicated that the interaction with Kindlin-2 prevented the protein degradation of PYCR1. Moreover, this interaction can be regulated by ECM stiffening, suggesting that the mechano-environment is also a regulator for PYCR (Guo et al. 2019, 2020). They also found that PINCH-1, a focal adhesion protein, was able to promote the association between PYCR1 and Kindlin-2 via affecting the mitochondrial translocation of Kindlin-2. Furthermore, ablation of PINCH-1 obviously impaired proline biogenesis, thus inhibiting tumor growth in lung adenocarcinoma (Guo et al. 2020).
In general, several oncogenes like c-MYC has been identified as the transcription factor of PYCR, which underlined the special demand for proline metabolism in oncogenic transformation and partially explained the high-expression of PYCR in various cancer cells. Nevertheless, more transcription factors are still in need to be discovered, since not all cancer cells possess the activation of c-MYC. Besides, as the posttranslational modification was recognized as the crucial regulation of enzymes in mitochondrial, the new modifications such as succinylation and phosphorylation should be further investigated on PYCR to further elucidate the complicated regulation network (Fig. 2).

The regulation of PYCRs at different levels
Inhibitors of PYCR as an anti-cancer strategy
Since a great amount of evidence has suggested the important function of PYCR in tumorigenesis and tumor progression, searching for targeted inhibitors thereof has become a viable strategy for cancer treatment. Milne et al. screened a commercially available compound library against PYCR1 and found pargyline as a fragment-like hit, whose IC50 was 198 µM. Based on the structure and activity relationship (SAR), a series of derived compounds were designed. One of effect with an IC50 of 8.8 µM, and its anti-cancer activity was demonstrated and proven in breast cancer cell lines (Milne et al. 2019). Christensen et al. performed another screen for proline analogs via X-ray crystallography and discovered five other potent inhibitors. The co-crystallization of PYCR1 with these inhibitors were all resolved. Among them, N-formyl L-proline (NFLP) had the smallest competitive constant at 100 µM and demonstrated a robust anticancer activity in breast cancer cells (Christensen et al. 2020).
Perspective
PYCR, the last enzyme in proline synthesis that converts P5C into proline, was found to be abnormally up-regulated in various cancers. PYCR promoted cancer growth and inhibited apoptosis through multiple approaches, including regulating the cell cycle, redox homeostasis and promoting growth signaling pathways. Elevating PYCR levels were also positively correlated with drug resistance and cancer relapse. However, the molecular mechanisms are not fully elucidated yet, especially those indirectly associated with its enzymatic activity. For example, how PYCR promotes the phosphorylation level of AKT or ERK, and whether PYCR is a component of some key signaling pathways or simply an indirect factor, require further investigation. Several works found that loss of PYCR changed the expression levels of multiple proteins such as EMT markers and CDKs, suggesting an important role of PYCR in regulating gene expression. Nevertheless, the explanation to this phenomenon is unclear. It could be attributed to the elevated ROS levels or endoplasmic reticulum (ER) stress caused by the ablation of PYCR, which then might activate crucial transcription factors. Numerous studies have investigated the complicated regulation network of PYCR and found that dysregulation of PYCR is tightly connected with cancer progression. Besides acetylation, there are a few other post-translational modifications like succinylation, phosphorylation and ubiquitination predicted within PYCR, which still remain to be further investigated. A few specific inhibitors were discovered to block the enzymatic activity of PYCR and showed potent anti-cancer effects, but these tests were temporarily limited to only cancer cell lines. Therefore, more data about safety and efficiency from animal models are urgently needed, to validate the therapeutic value of inhibiting PYCR in cancers.
References
Adams E, Frank L (1980) Metabolism of proline and the hydroxyprolines. Annu Rev Biochem 49:1005–1061. https://doi.org/10.1146/annurev.bi.49.070180.005041
Burke L, Guterman I, Palacios Gallego R, Britton RG, Burschowsky D, Tufarelli C, Rufini A (2020) The Janus-like role of proline metabolism in cancer. Cell Death Discov 6:104. https://doi.org/10.1038/s41420-020-00341-8
Cai F, Miao Y, Liu C, Wu T, Shen S, Su X, Shi Y (2018) Pyrroline-5-carboxylate reductase 1 promotes proliferation and inhibits apoptosis in non-small cell lung cancer. Oncol Lett 15(1):731–740. https://doi.org/10.3892/ol.2017.7400
Chen S, Yang X, Yu M, Wang Z, Liu B, Liu M, Liu L, Ren M, Qi H, Zou J, Vucenik I, Zhu WG, Luo J (2019a) SIRT3 regulates cancer cell proliferation through deacetylation of PYCR1 in proline metabolism. Neoplasia 21(7):665–675. https://doi.org/10.1016/j.neo.2019.04.008
Chen XY, Zhang J, Zhu JS (2019b) The role of m(6)A RNA methylation in human cancer. Mol Cancer 18(1):103. https://doi.org/10.1186/s12943-019-1033-z
Chen K, Guo L, Wu C (2021) How signaling pathways link extracellular mechano-environment to proline biosynthesis: a hypothesis. BioEssays. https://doi.org/10.1002/bies.202100116
Cheng C, Song D, Wu Y, Liu B (2020) RAC3 promotes proliferation, migration and invasion via PYCR1/JAK/STAT signaling in bladder cancer. Front Mol Biosci 7:218. https://doi.org/10.3389/fmolb.2020.00218
Choi UY, Lee JJ, Park A, Zhu W, Lee HR, Choi YJ, Yoo JS, Yu C, Feng P, Gao SJ, Chen S, Eoh H, Jung JU (2020) Oncogenic human herpesvirus hijacks proline metabolism for tumorigenesis. Proc Natl Acad Sci U S A 117(14):8083–8093. https://doi.org/10.1073/pnas.1918607117
Christensen EM, Patel SM, Korasick DA, Campbell AC, Krause KL, Becker DF, Tanner JJ (2017) Resolving the cofactor-binding site in the proline biosynthetic enzyme human pyrroline-5-carboxylate reductase 1. J Biol Chem 292(17):7233–7243. https://doi.org/10.1074/jbc.M117.780288
Christensen EM, Bogner AN, Vandekeere A, Tam GS, Patel SM, Becker DF, Fendt SM, Tanner JJ (2020) In crystallo screening for proline analog inhibitors of the proline cycle enzyme PYCR1. J Biol Chem. https://doi.org/10.1074/jbc.RA120.016106
Craze ML, Cheung H, Jewa N, Coimbra NDM, Soria D, El-Ansari R, Aleskandarany MA, Wai Cheng K, Diez-Rodriguez M, Nolan CC, Ellis IO, Rakha EA, Green AR (2018) MYC regulation of glutamine-proline regulatory axis is key in luminal B breast cancer. Br J Cancer 118(2):258–265. https://doi.org/10.1038/bjc.2017.387
D’Aniello C, Patriarca EJ, Phang JM, Minchiotti G (2020) Proline metabolism in tumor growth and metastatic progression. Front Oncol 10:776. https://doi.org/10.3389/fonc.2020.00776
De Ingeniis J, Ratnikov B, Richardson AD, Scott DA, Aza-Blanc P, De SK, Kazanov M, Pellecchia M, Ronai Z, Osterman AL, Smith JW (2012) Functional specialization in proline biosynthesis of melanoma. PLoS ONE 7(9):e45190. https://doi.org/10.1371/journal.pone.0045190
Dhanasekaran DN, Reddy EP (2008) JNK signaling in apoptosis. Oncogene 27(48):6245–6251. https://doi.org/10.1038/onc.2008.301
Ding J, Kuo ML, Su L, Xue L, Luh F, Zhang H, Wang J, Lin TG, Zhang K, Chu P, Zheng S, Liu X, Yen Y (2017) Human mitochondrial pyrroline-5-carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis 38(5):519–531. https://doi.org/10.1093/carcin/bgx022
Ding Z, Ericksen RE, Escande-Beillard N, Lee QY, Loh A, Denil S, Steckel M, Haegebarth A, Wai Ho TS, Chow P, Toh HC, Reversade B, Gruenewald S, Han W (2020) Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J Hepatol 72(4):725–735. https://doi.org/10.1016/j.jhep.2019.10.026
Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20(2):69–84. https://doi.org/10.1038/s41580-018-0080-4
Du S, Sui Y, Ren W, Zhou J, Du C (2021) PYCR1 promotes bladder cancer by affecting the Akt/Wnt/beta-catenin signaling. J Bioenerg Biomembr 53(2):247–258. https://doi.org/10.1007/s10863-021-09887-3
Fang E, Wang X, Yang F, Hu A, Wang J, Li D, Song H, Hong M, Guo Y, Liu Y, Li H, Huang K, Zheng L, Tong Q (2019) Therapeutic targeting of MZF1-AS1/PARP1/E2F1 axis inhibits proline synthesis and neuroblastoma progression. Adv Sci (weinh) 6(19):1900581. https://doi.org/10.1002/advs.201900581
Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, Huang C, Li J, Dong X, Zhou Y, Liu Q, Ma L, Wang X, Zhou J, Liu Y, Boja E, Robles AI, Ma W, Wang P, Li Y, Ding L, Wen B, Zhang B, Rodriguez H, Gao D, Zhou H, Fan J (2019) Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell 179 (2):561–577 e522. https://doi.org/10.1016/j.cell.2019.08.052
Gao Y, Luo L, Xie Y, Zhao Y, Yao J, Liu X (2020) PYCR1 knockdown inhibits the proliferation, migration, and invasion by affecting JAK/STAT signaling pathway in lung adenocarcinoma. Mol Carcinog 59(5):503–511. https://doi.org/10.1002/mc.23174
Guernsey DL, Jiang H, Evans SC, Ferguson M, Matsuoka M, Nightingale M, Rideout AL, Provost S, Bedard K, Orr A, Dubé M-P, Ludman M, Samuels ME (2009) Mutation in pyrroline-5-carboxylate reductase 1 gene in families with cutis laxa type 2. Am J Hum Genet 85(1):120–129. https://doi.org/10.1016/j.ajhg.2009.06.008
Guo L, Cui C, Zhang K, Wang J, Wang Y, Lu Y, Chen K, Yuan J, Xiao G, Tang B, Sun Y, Wu C (2019) Kindlin-2 links mechano-environment to proline synthesis and tumor growth. Nat Commun 10(1):845. https://doi.org/10.1038/s41467-019-08772-3
Guo L, Cui C, Wang J, Yuan J, Yang Q, Zhang P, Su W, Bao R, Ran J, Wu C (2020) PINCH-1 regulates mitochondrial dynamics to promote proline synthesis and tumor growth. Nat Commun 11(1):4913. https://doi.org/10.1038/s41467-020-18753-6
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
He G, Karin M (2011) NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res 21(1):159–168. https://doi.org/10.1038/cr.2010.183
Hollinshead KER, Munford H, Eales KL, Bardella C, Li C, Escribano-Gonzalez C, Thakker A, Nonnenmacher Y, Kluckova K, Jeeves M, Murren R, Cuozzo F, Ye D, Laurenti G, Zhu W, Hiller K, Hodson DJ, Hua W, Tomlinson IP, Ludwig C, Mao Y, Tennant DA (2018) Oncogenic IDH1 mutations promote enhanced proline synthesis through PYCR1 to support the maintenance of mitochondrial redox homeostasis. Cell Rep 22(12):3107–3114. https://doi.org/10.1016/j.celrep.2018.02.084
Hong Y, Kim WJ, Bang CY, Lee JC, Oh YM (2016) Identification of alternative splicing and fusion transcripts in non-small cell lung cancer by RNA sequencing. Tuberc Respir Dis (seoul) 79(2):85–90. https://doi.org/10.4046/trd.2016.79.2.85
Jariwala U, Prescott J, Jia L, Barski A, Pregizer S, Cogan JP, Arasheben A, Tilley WD, Scher HI, Gerald WL, Buchanan G, Coetzee GA, Frenkel B (2007) Identification of novel androgen receptor target genes in prostate cancer. Mol Cancer 6:39. https://doi.org/10.1186/1476-4598-6-39
Johnson DE, O’Keefe RA, Grandis JR (2018) Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 15(4):234–248. https://doi.org/10.1038/nrclinonc.2018.8
Krishnan N, Dickman MB, Becker DF (2008) Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic Biol Med 44(4):671–681. https://doi.org/10.1016/j.freeradbiomed.2007.10.054
Kuo ML, Lee MB, Tang M, den Besten W, Hu S, Sweredoski MJ, Hess S, Chou CM, Changou CA, Su M, Jia W, Su L, Yen Y (2016) PYCR1 and pycr2 interact and collaborate with RRM2B to protect cells from overt oxidative stress. Sci Rep 6:18846. https://doi.org/10.1038/srep18846
Kuo CL, Chou HY, Chiu YC, Cheng AN, Fan CC, Chang YN, Chen CH, Jiang SS, Chen NJ, Lee AY (2020) Mitochondrial oxidative stress by Lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett 474:138–150. https://doi.org/10.1016/j.canlet.2020.01.019
Liang ST, Audira G, Juniardi S, Chen JR, Lai YH, Du ZC, Lin DS, Hsiao CD (2019) Zebrafish carrying pycr1 gene deficiency display aging and multiple behavioral abnormalities. Cells. https://doi.org/10.3390/cells8050453
Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U S A 109(23):8983–8988. https://doi.org/10.1073/pnas.1203244109
Liu W, Hancock CN, Fischer JW, Harman M, Phang JM (2015) Proline biosynthesis augments tumor cell growth and aerobic glycolysis: involvement of pyridine nucleotides. Sci Rep 5:17206. https://doi.org/10.1038/srep17206
Loayza-Puch F, Rooijers K, Buil LC, Zijlstra J, Oude Vrielink JF, Lopes R, Ugalde AP, van Breugel P, Hofland I, Wesseling J, van Tellingen O, Bex A, Agami R (2016) Tumour-specific proline vulnerability uncovered by differential ribosome codon reading. Nature 530(7591):490–494. https://doi.org/10.1038/nature16982
Lorans G, Phang JM (1981) Proline synthesis and redox regulation: Differential functions of pyrroline-5-carboxylate reductase in human lymphoblastoid cell lines. Biochem Biophys Res Commun 101(3):1018–1025. https://doi.org/10.1016/0006-291X(81)91850-7
Lu J, Lin J, Zhou Y, Ye K, Fang C (2021) MiR-328-3p inhibits lung adenocarcinoma-genesis by downregulation PYCR1. Biochem Biophys Res Commun 550:99–106. https://doi.org/10.1016/j.bbrc.2021.02.029
Meng Z, Lou Z, Liu Z, Li M, Zhao X, Bartlam M, Rao Z (2006) Crystal structure of human pyrroline-5-carboxylate reductase. J Mol Biol 359(5):1364–1377. https://doi.org/10.1016/j.jmb.2006.04.053
Milne K, Sun J, Zaal EA, Mowat J, Celie PHN, Fish A, Berkers CR, Forlani G, Loayza-Puch F, Jamieson C, Agami R (2019) A fragment-like approach to PYCR1 inhibition. Bioorg Med Chem Lett 29(18):2626–2631. https://doi.org/10.1016/j.bmcl.2019.07.047
Nakayama T, Al-Maawali A, El-Quessny M, Rajab A, Khalil S, Stoler JM, Tan WH, Nasir R, Schmitz-Abe K, Hill RS, Partlow JN, Al-Saffar M, Servattalab S, LaCoursiere CM, Tambunan DE, Coulter ME, Elhosary PC, Gorski G, Barkovich AJ, Markianos K, Poduri A, Mochida GH (2015) Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am J Hum Genet 96(5):709–719. https://doi.org/10.1016/j.ajhg.2015.03.003
Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2(1):21–32. https://doi.org/10.1038/35048096
Nocek B, Chang C, Li H, Lezondra L, Holzle D, Collart F, Joachimiak A (2005) Crystal structures of delta1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J Mol Biol 354(1):91–106. https://doi.org/10.1016/j.jmb.2005.08.036
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23(1):27–47. https://doi.org/10.1016/j.cmet.2015.12.006
Phang JM (2019) Proline metabolism in cell regulation and cancer biology: recent advances and hypotheses. Antioxid Redox Signal 30(4):635–649. https://doi.org/10.1089/ars.2017.7350
Pinti M, Gibellini L, Liu Y, Xu S, Lu B, Cossarizza A (2015) Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell Mol Life Sci 72(24):4807–4824. https://doi.org/10.1007/s00018-015-2039-3
Sahu N, Dela Cruz D, Gao M, Sandoval W, Haverty PM, Liu J, Stephan JP, Haley B, Classon M, Hatzivassiliou G, Settleman J (2016) Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. Cell Metab 24(5):753–761. https://doi.org/10.1016/j.cmet.2016.08.008
Samatar AA, Poulikakos PI (2014) Targeting RAS–ERK signalling in cancer: promises and challenges. Nat Rev Drug Discovery 13(12):928–942. https://doi.org/10.1038/nrd4281
Sang S, Zhang C, Shan J (2019) Pyrroline-5-Carboxylate reductase 1 accelerates the migration and invasion of nonsmall cell lung cancer in vitro. Cancer Biother Radiopharm 34(6):380–387. https://doi.org/10.1089/cbr.2019.2782
Schworer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, Cross JR, Thompson CB (2020) Proline biosynthesis is a vent for TGFbeta-induced mitochondrial redox stress. EMBO J 39(8):e103334. https://doi.org/10.15252/embj.2019103334
Sharif T, Dai C, Martell E, Ghassemi-Rad MS, Hanes MR, Murphy PJ, Kennedy BE, Venugopal C, Subapanditha M, Giacomantonio CA, Marcato P, Singh SK, Gujar S (2019) TAp73 modifies metabolism and positively regulates growth of cancer stem-like cells in a Redox-sensitive manner. Clin Cancer Res 25(6):2001–2017. https://doi.org/10.1158/1078-0432.CCR-17-3177
She Y, Mao A, Li F, Wei X (2019) P5CR1 protein expression and the effect of gene-silencing on lung adenocarcinoma. PeerJ 7:e6934. https://doi.org/10.7717/peerj.6934
Shenoy A, Belugali Nataraj N, Perry G, Loayza Puch F, Nagel R, Marin I, Balint N, Bossel N, Pavlovsky A, Barshack I, Kaufman B, Agami R, Yarden Y, Dadiani M, Geiger T (2020) Proteomic patterns associated with response to breast cancer neoadjuvant treatment. Mol Syst Biol 16(9):e9443. https://doi.org/10.15252/msb.20209443
Song W, Yang K, Luo J, Gao Z, Gao Y (2021) Dysregulation of USP18/FTO/PYCR1 signaling network promotes bladder cancer development and progression. Aging (albany NY). https://doi.org/10.18632/aging.202359
Sun C, Li T, Song X, Huang L, Zang Q, Xu J, Bi N, Jiao G, Hao Y, Chen Y, Zhang R, Luo Z, Li X, Wang L, Wang Z, Song Y, He J, Abliz Z (2019) Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc Natl Acad Sci U S A 116(1):52–57. https://doi.org/10.1073/pnas.1808950116
Tang L, Zeng J, Geng P, Fang C, Wang Y, Sun M, Wang C, Wang J, Yin P, Hu C, Guo L, Yu J, Gao P, Li E, Zhuang Z, Xu G, Liu Y (2018) Global metabolic profiling identifies a pivotal role of proline and hydroxyproline metabolism in supporting hypoxic response in hepatocellular carcinoma. Clin Cancer Res 24(2):474–485. https://doi.org/10.1158/1078-0432.CCR-17-1707
Tanner JJ, Fendt SM, Becker DF (2018) The proline cycle as a potential cancer therapy target. Biochemistry 57(25):3433–3444. https://doi.org/10.1021/acs.biochem.8b00215
Tashiro H, Brenner MK (2017) Immunotherapy against cancer-related viruses. Cell Res 27(1):59–73. https://doi.org/10.1038/cr.2016.153
Tetsu O, McCormick F (1999) β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398(6726):422–426. https://doi.org/10.1038/18884
Trimble EL, Ungerleider RS, Abrams JA, Kaplan RS, Feigal EG, Smith MA, Carter CL, Friedman MA (1993) Neoadjuvant therapy in cancer treatment. Cancer 72(11 Suppl):3515–3524. https://doi.org/10.1002/1097-0142(19931201)72:11+%3c3515::aid-cncr2820721619%3e3.0.co;2-a
Wang QL, Liu L (2019) PYCR1 is Associated with papillary renal cell carcinoma progression. Open Med (wars) 14:586–592. https://doi.org/10.1515/med-2019-0066
Wang D, Wang L, Zhang Y, Yan Z, Liu L, Chen G (2019) PYCR1 promotes the progression of non-small-cell lung cancer under the negative regulation of miR-488. Biomed Pharmacother 111:588–595. https://doi.org/10.1016/j.biopha.2018.12.089
Xiao S, Li S, Yuan Z, Zhou L (2020) Pyrroline-5-carboxylate reductase 1 (PYCR1) upregulation contributes to gastric cancer progression and indicates poor survival outcome. Ann Transl Med 8(15):937. https://doi.org/10.21037/atm-19-4402
Yan K, Xu X, Wu T, Li J, Cao G, Li Y, Ji Z (2019) Knockdown of PYCR1 inhibits proliferation, drug resistance and EMT in colorectal cancer cells by regulating STAT3-Mediated p38 MAPK and NF-kappaB signalling pathway. Biochem Biophys Res Commun 520(2):486–491. https://doi.org/10.1016/j.bbrc.2019.10.059
Yasuda T, Kaji Y, Agatsuma T, Niki T, Arisawa M, Shuto S, Ariga H, Iguchi-Ariga SM (2013) DJ-1 cooperates with PYCR1 in cell protection against oxidative stress. Biochem Biophys Res Commun 436(2):289–294. https://doi.org/10.1016/j.bbrc.2013.05.095
Ye Y, Wu Y, Wang J (2018) Pyrroline-5-carboxylate reductase 1 promotes cell proliferation via inhibiting apoptosis in human malignant melanoma. Cancer Manag Res 10:6399–6407. https://doi.org/10.2147/CMAR.S166711
Yuzhalin AE, Lim SY, Kutikhin AG, Gordon-Weeks AN (2018) Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim Biophys Acta Rev Cancer 1870(2):207–228. https://doi.org/10.1016/j.bbcan.2018.09.002
Zeng T, Zhu L, Liao M, Zhuo W, Yang S, Wu W, Wang D (2017) Knockdown of PYCR1 inhibits cell proliferation and colony formation via cell cycle arrest and apoptosis in prostate cancer. Med Oncol 34(2):27. https://doi.org/10.1007/s12032-016-0870-5
Zhuang J, Song Y, Ye Y, He S, Ma X, Zhang M, Ni J, Wang J, Xia W (2019) PYCR1 interference inhibits cell growth and survival via c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathway in hepatocellular cancer. J Transl Med 17(1):343. https://doi.org/10.1186/s12967-019-2091-0
Acknowledgements
We thank Thomas Luo for editorial assistance. This work was supported by the National Natural Science Foundation of China (81874147, 81671389).
Author information
Authors and Affiliations
Contributions
Supervision, review, and editing: JL; literature search and writing: YTL; draft revising: JTB, CS, MHL. All authors have read and agreed to publish this version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors of this manuscript have no competing interests.
Informed consent
Written informed consents were obtained from all participants.
Additional information
Handling editor: J. M. Phang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, Y., Bie, J., Song, C. et al. PYCR, a key enzyme in proline metabolism, functions in tumorigenesis. Amino Acids 53, 1841–1850 (2021). https://doi.org/10.1007/s00726-021-03047-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-021-03047-y