
Abstract
Most cases of frontotemporal lobar degeneration (FTLD) are characterized by abnormal intracellular accumulation of either tau or TDP-43 protein. However, in ~10% of cases, composed of a heterogenous collection of uncommon disorders, the molecular basis remains to be uncertain. We recently discovered that the pathological changes in several tau/TDP-43-negative FTLD subtypes are immunoreactive (ir) for the fused in sarcoma (FUS) protein. In this study, we directly compared the pattern of FUS-ir pathology in cases of atypical FTLD-U (aFTLD-U, N = 10), neuronal intermediate filament inclusion disease (NIFID, N = 5) and basophilic inclusion body disease (BIBD, N = 8), to determine whether these are discrete entities or represent a pathological continuum. All cases had FUS-ir pathology in the cerebral neocortex, hippocampus and a similar wide range of subcortical regions. Although there was significant overlap, each group showed specific differences that distinguished them from the others. Cases of aFTLD-U consistently had less pathology in subcortical regions. In addition, the neuronal inclusions in aFTLD-U usually had a uniform, round shape, whereas NIFID and BIBD were characterized by a variety of inclusion morphologies. In all cases of aFTLD-U and NIFID, vermiform neuronal intranuclear inclusions (NII) were readily identified in the hippocampus and neocortex. In contrast, only two cases of BIBD had very rare NII in a single subcortical region. These findings support aFTLD-U, NIFID and BIBD as representing closely related, but distinct entities that share a common molecular pathogenesis. Although cases with overlapping pathology may exist, we recommend retaining the terms aFTLD-U, NIFID and BIBD for specific FTLD-FUS subtypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The clinical syndrome of frontotemporal dementia (FTD) is associated with heterogenous neuropathology [42]. Relatively selective degeneration of the frontal and temporal cerebral lobes is a consistent feature (frontotemporal lobar degeneration FTLD). In addition, most cases are characterized by abnormal intracellular accumulation of protein, forming inclusion bodies, in neurons and glia. A system of nomenclature and nosology was recently introduced that groups the FTLD subtypes into broad categories, based on the molecular defect that is currently felt to be most characteristic (FTLD-protein) [31]. In the majority of cases, the pathological protein is either the microtubule-associated protein tau or the transactive response DNA-binding protein with M r 43 kDa, TDP-43 (FTLD-tau and FTLD-TDP, respectively). However, there remain ~10% of cases, composed of a heterogenous collection of uncommon disorders, for which the molecular basis remains to be uncertain [21, 30, 42, 47]. We recently discovered that the pathological changes in several of these tau/TDP-43-negative FTLD subtypes are immunoreactive for the fused in sarcoma protein (FUS) [38–40], thus defining a new molecular category (FTLD-FUS) [32].
Most cases of tau-negative FTLD were previously defined by the presence of neuronal inclusions that could only be demonstrated with immunohistochemistry (IHC) for ubiquitin (FTLD-U) [20, 34]. In the majority of these cases, the ubiquitinated pathological protein was subsequently discovered to be TDP-43 (FTLD-TDP) [2, 10, 41]. However, 10–20% of FTLD-U does not show evidence of abnormal TDP-43 [21, 30, 47]. We previously reported that these cases of “atypical” FTLD-U (aFTLD-U) have an unusual and highly consistent clinical phenotype characterized by sporadic, early onset, severe and rapidly progressive psychobehavioral abnormalities, in the absence of significant language or motor deficits [30, 47]. The neuropathology is also atypical. In addition to ubiquitin-positive, tau/TDP-43-negative neuronal cytoplasmic inclusions (NCI), cases of aFTLD-U have unusual neuronal intranuclear inclusions (NII) that appear as long thick filaments that may be straight, curved or twisted (vermiform). In a recent study, we found the NCI and NII in aFTLD-U to be consistently immunoreactive (ir) with antibodies against FUS [40]. Moreover, FUS-IHC demonstrated additional types of NCI, as well as glial cytoplasmic inclusions (GCI) that had not previously been appreciated with ubiquitin IHC. Biochemical analysis of postmortem aFTLD-U brain tissue demonstrated a relative shift of FUS protein to the insoluble fraction, but no evidence of abnormal molecular species. None of the cases in which genetic analysis was performed was found to harbor mutations in the FUS gene.
Basophilic inclusion body disease (BIBD) is a term that has been used for a small number of clinically and pathologically heterogeneous cases, in which the common finding is NCI that are basophilic with hematoxylin and eosin stain (basophilic inclusions, BI). The clinical phenotypes include sporadic ALS [24, 25], familial ALS [53], ALS with dementia [15, 18, 61] and pure FTD [37, 61]. Although cases of BIBD with clinical FTD show chronic degeneration of the frontotemporal neocortex, the BI tend to be most numerous in subcortical regions, such as the basal ganglia and brainstem tegmentum [37, 61]. We recently reported that these BI are also consistently immunoreactive for FUS [38]. In addition, FUS-IHC in our cases of BIBD demonstrated abundant neuronal and glial pathology that had not previously been recognized, including in the frontotemporal neocortex and hippocampus.
Neuronal intermediate filament inclusion disease (NIFID) is an uncommon neurological disorder [54] with pathology characterized by NCI that are immunoreactive for all of the class IV neuronal intermediate filaments (IF), which include light, medium and heavy weight neurofilament (NF) subunits and α-internexin [7, 9]. The typical clinical presentation is early onset, sporadic FTD, associated with a pyramidal and/or extrapyramidal movement disorder [5, 7, 8, 19, 22, 29, 36, 46, 61]. Although immunoreactivity for IF was initially described as the defining pathological feature of NIFID, all cases show a wide range of NCI morphologies with only a small proportion being IF-ir. Our recent study of NIFID found that (1) FUS-IHC demonstrated many more NCI than IF-IHC, (2) several morphological types of inclusions (including vermiform NII and GCI) were only FUS-ir and (3) all neurons that contained abnormal IF accumulation also contained a FUS-ir inclusion [40]. We interpreted these results as suggesting that FUS plays a more central role in the pathogenesis of NIFID and that the abnormal accumulation of IF in a proportion of inclusions is likely a secondary phenomenon.
Based on these findings, aFTLD-U, BIBD and NIFID have now been grouped together under the broad designation of FTLD-FUS [32]. Although it is now clear that these entities are more closely related than previously thought, the degree of overlap remains uncertain. In the present study, we directly compared the pattern of FUS-ir pathology in cases of aFTLD-U, BIBD and NIFID, to help in determining whether these are discrete entities or represent a pathological continuum.
Materials and methods
Cases
Cases were selected that had originally received a neuropathological diagnosis of aFTLD-U (N = 10), BIBD (N = 8) or NIFID (N = 5). Detailed clinical and pathological description of each case had been published previously (Table 1). Significant tau, TDP-43 and α-synuclein pathology had already been excluded. In each case, sections stained with hematoxylin and eosin (HE), ubiquitin and IF-IHC, were re-evaluated and a final classification made according to current criteria [42]. A diagnosis of BIBD was based on the presence of numerous round BI, in multiple neuroanatomical regions as seen on HE-stained sections. Cases of NIFID had significant numbers of IF-ir NCI in the neocortex and hippocampus. aFTLD-U was characterized by cortical NCI that were ubiquitin-ir, but did not label for other neurodegenerative proteins (including, tau, α-synuclein, TDP-43 and IF) and were not visible with histochemical techniques, such as HE stain or silver impregnation methods.
Immunohistochemistry
Immunohistochemistry was performed on 5-μm thick sections of formalin-fixed, paraffin-embedded tissue using the Ventana BenchMark® XT automated staining system (Ventana, Tuscon, AZ) and developed with aminoethylcarbizole (AEC). The primary antibodies employed recognized FUS (Sigma-Aldrich anti-FUS; 1:25–1:200 and Bethyl Laboratories A300-302A; 1:500, both with initial overnight incubation at room temperature, following microwave antigen retrieval), ubiquitin (DAKO anti-ubiquitin; 1:500, following microwave antigen retrieval), α-internexin (Zymed anti-α-internexin; 1:500 with 3-h initial incubation at room temperature, following microwave antigen retrieval), nonphosphorylated neurofilament (NF) (DAKO anti-neurofilament protein; 1:2,000, following protease digestion), phosphorylated neurofilament (pNF) (Sternberger SMI 31; 1:8,000, following protease digestion).
Based on the amount of normal physiological staining, it was apparent that the anti-FUS sensitivity was greatly influenced by the degree of tissue fixation and that this was only partially reversed by antigen retrieval. Therefore, the dilution of the primary antibody was adjusted in each case to allow for faint physiological staining that ensured sensitivity (internal positive control) but did not compromise visualization of the pathology. Most of the quantitation was based on the sections stained using the FUS antibody from Sigma, with the Bethyl antibody being used primarily for confirmation.
Semiquantitative evaluation of FUS-ir pathology
The number of FUS-ir inclusions was scored in different anatomical regions using a semiquantitative grading system, in which NCI and NII were each rated as being absent (−), rare (+), occasional (++), moderate (+++) or numerous (++++) [39]. Inclusions were considered “rare” if only a few examples could be found in the entire region examined, “occasional” if they were relatively easy to find, but not present in every medium power microscopic field, “moderate” if at least a few examples were present in most microscopic fields, and “numerous” when many were present in every microscopic field. The different morphological subtypes of NCI were also recorded in each region as being common or infrequent.
Statistical analysis
Owing to the small size of the groups and the fact that the scores were not normally distributed, a non-parametric method (Kruskal–Wallis one-way analysis of variance) was performed to compare the inclusion scores among the diagnostic groups, in each anatomical region. A result with p < 0.05 indicated that at least one group was significantly different from the other two. Student–Neuman–Keuls at p < 0.05 was used as a post hoc test to determine which group(s) differed from which others.
Results
Core diagnostic features
Each case fulfilled only one set of neuropathological criteria and, in every case, the original diagnosis was confirmed (Table 2). All cases had ub-ir NCI in the cerebral cortex and hippocampus. These were numerous in all cases of NIFID, more variable but usually numerous in aFTLD-U and moderate-to-numerous in cases of BIBD, where the immunoreactivity was often less intense. All cases originally diagnosed as NIFID had moderate-to-numerous IF-ir NCI, of variable morphology, in the cerebral neocortex and limbic regions. IF-ir NCI were also present in some cases of aFTLD-U and BIBD, however, these were usually rare, restricted to a few hippocampal pyramidal neurons, and had a tangle-like morphology. All cases originally diagnosed as BIBD had numerous BI in a wide range of neuroanatomical locations. BI were also found in most cases of aFTLD-U and NIFID, but were few in number (rare or occasional) and restricted to the midbrain periaqueductal gray matter or substantia nigra. In all cases of aFTLD-U, NCI in the neocortex and hippocampus could only be appreciated with ubiquitin IHC. In contrast, cortical NCI were easily identified on HE sections of NIFID and BIBD.
FUS-ir NCI and NII
aFTLD-U
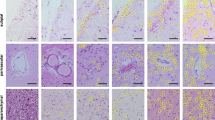
In all cases, FUS-ir NCI were present in a wide range of neuroanatomical regions (Table 3). There was significant variation among cases, but the neocortex, dentate fascia of the hippocampus, striatum and periaqueductal gray matter tended to be most affected (Fig. 1a–c). Other brainstem nuclei, thalamus, globus pallidus and pyramidal layer of hippocampus were also consistently involved, but usually to a lesser extent. In the pyramidal layer of the hippocampus, NCI were only common in CA3 and CA4 regions, however, most cases showed a near complete loss of neurons from CA1 and the subiculum (hippocampal sclerosis). In all cases in which the spinal cord was available, a small number of lower motor neurons (LMN) were found to harbor inclusions. Rare NCI were present in the cerebellar dentate nucleus in a minority of cases and the cerebellar cortex was never affected. In all anatomical regions, the predominant NCI morphology was small, compact, round, oval or kidney-shaped inclusions, approximately the size of the nucleus or smaller (Fig. 1a–c; Table 4). Crescentic NCI were only common in the striatum and non-compact aggregates of FUS-ir coarse granules made up a significant proportion of the NCI in pyramidal neurons of the hippocampus and in the periaqueductal gray matter. Filamentous, often vermiform NII were present in the hippocampus in all cases, being more numerous in the dentate granule cells than in pyramidal neurons (Fig. 1d; Table 5). Small numbers of NII were also present in the cerebral neocortex, striatum, thalamus, pontine nuclei and inferior olivary nucleus in the majority of cases and were less consistently found in all other regions examined.

FUS-immunoreactive pathology in aFTLD-U. Small, round, compact neuronal cytoplasmic inclusions were the most common pathological change, and were most abundant in the neocortex (a, b) and dentate fascia of the hippocampus (c). Vermiform and filamentous neuronal intranuclear inclusions were present in all cases, being most numerous in the dentate granule cells of the hippocampus (d), but also present in hippocampal pyramidal neurons, neocortex and many subcortical regions. FUS immunohistochemistry. Scale bar 40 µm (a), 20 µm (b, c) and 8 µm (d)
NIFID
In all cases, FUS-ir NCI were numerous, in virtually all regions evaluated (Table 3). The only area examined that showed significant variation (from rare to numerous) was the hypoglossal nucleus, but even it was affected in all cases. The cerebellar cortex was the only region to be spared. In most anatomical areas, several different NCI morphologies were common. Small round or oval compact inclusions were present in all regions (Fig. 2a). Crescentic, annular and tangle-like NCI were also common in the cerebral neocortex, hippocampus and basal ganglia (Fig. 2b–d). NCI were present in all sections of the hippocampal pyramidal layer, but were particularly numerous in CA1 and the subiculum, where they were often flame-shaped, sometimes resembling tangles while others were compact (Fig. 2e). Aggregates of coarse granules were common in the globus pallidus, thalamus, most brainstem nuclei, cerebellar dentate nucleus and LMN (Fig. 2f). In several subcortical regions, a minority of the NCI had a complex morphology, being composed of a non-compact collection of thick filaments and globular structures (Fig. 2g). These were thought to represent the hyaline conglomerate inclusions seen on HE stain. Variable numbers of vermiform NII were present in the hippocampus (Fig. 2h) in all cases, but they were rare elsewhere (Table 5).

FUS-immunoreactive pathology in NIFID. Neuronal cytoplasmic inclusions (NCI) were numerous in most regions examined. Small, round compact NCI were present in all areas involved, and were the predominant morphology in the hippocampal dentate granule cells (a). Crescentic, annular and tangle-like NCI were also common in the cerebral neocortex (b, c) and basal ganglia (d). Flame-shaped NCI were numerous in hippocampal CA1 region and subiculum (e). Aggregates of coarse cytoplasmic granules were common in many subcortical regions, including spinal cord motor neurons (f). NCI with a complex morphology were thought to correspond to hyaline conglomerate inclusions (g). Variable numbers of vermiform neuronal intranuclear inclusions were present in the hippocampus (arrows h), but rare elsewhere. FUS immunohistochemistry. Scale bar 20 µm (a, c, e), 40 µm (b, d), 10 µm (f, g) and 8 µm (h)
BIBD
Neuronal cytoplasmic inclusions were scored as either moderate or numerous in all cases in the cerebral neocortex, hippocampal pyramidal layer (sections CA3 and CA4), globus pallidus, thalamus, midbrain, pons, inferior olivary nucleus and LMN of the spinal cord (Table 3). The hippocampal dentate granule cells, striatum, hypoglossal nucleus and cerebellar dentate nucleus were also consistently affected, but to a more variable degree. A single round NCI was present in a cerebellar Purkinje cell in one case. Multiple NCI morphologies were common in most anatomical regions (Table 4). Round compact NCI were present in all areas, and were usually one of the most frequent types. Particularly large examples, present in larger neurons of subcortical regions, likely corresponded to BI (Fig. 3a, b). Crescentic, annular and tangle-like inclusions were also common in the cerebral neocortex, hippocampus and basal ganglia (Fig. 3c–e). Non-compact collections of coarse granules were also common in the thalamus, many brainstem nuclei and cerebellar dentate nucleus. NCI with complex morphology (see above) were common in the inferior olivary nucleus and infrequently found in other brainstem nuclei and the thalamus. NII were only identified in two cases (Table 5). In one, rare vermiform NII were present in a focal area of the thalamus and in the other case, two neurons with nuclear inclusions were identified in the basis pontis (Fig. 3f). NII were never identified in the hippocampus or neocortex.

FUS-immunoreactive pathology in BIBD. Large, round, neuronal cytoplasmic inclusions in large subcortical neurons likely correspond to basophilic inclusions (a, b). Small round, crescentic, annular and tangle-like NCI were common in the cerebral neocortex (c), hippocampus (d) and basal ganglia (e). NII were identified in only two cases; one with two neurons with nuclear inclusions in the basis pontis and the other had rare NII in the thalamus (f). FUS immunohistochemistry. Scale bar 30 µm (a), 15 µm (b), 40 µm (c), 20 µm (d, e) and 8 µm (f)
Other FUS-ir pathology
In addition to the neuronal inclusions described above, affected areas of all cases demonstrated variable numbers of small round or crescentic FUS-ir GCI and short cell processes. Some of the processes were thick enough to be identified as dystrophic neurites, while smaller thread-like structures could have been of either neuronal or glial origin. The glial and neuritic pathology was difficult to characterize and quantify, but roughly paralleled the number of NCI and did not offer any independent discriminatory value (data not shown).
Group comparisons
Cases from all three groups were found to have FUS-ir pathology in the cerebral neocortex, hippocampus and a similar wide range of subcortical regions (Table 3). Although there was significant overlap, each group showed specific features that allowed it to be distinguished from the others.
Cases of aFTLD-U had the most distinctive pattern of FUS-ir pathology. They consistently had the fewest NCI in subcortical regions and in many areas this difference was discriminatory (both specific and sensitive), as there was no overlap in the range of severity among the aFTLD-U cases when compared with that of the other two groups (aFTLD-U = +/++ vs. NIFID and BIBD = ++++) (Table 3). In addition, the vast majority of NCI in aFTLD-U had a uniform, round or oval shape and in only a few regions were other NCI morphologies common (Table 4). This contrasted with NIFID and BIBD, both of which were characterized by a wide range of inclusion morphologies. Finally, all cases of aFTLD-U had significant numbers of vermiform NII (Table 5). The presence of NII in neocortex and hippocampus was a feature that absolutely distinguished aFTLD-U from BIBD cases, whereas the broader anatomical distribution of NII in aFTLD-U showed relative difference when compared with NIFID.
Cases of NIFID and BIBD demonstrated greater similarity. Numerous NCI was a more consistent finding in most neuroanatomical regions in NIFID compared with BIBD cases, which showed greater variability; however, this difference only reached statistical significance in the striatum and globus pallidus and there was always overlap in the range of scores found in the two groups (Table 3). The variety of NCI morphologies was also similar between these groups (Table 4). The pattern of involvement of the hippocampal pyramidal layer in cases of NIFID was consistent and unique, with numerous flame-shaped NCI in the CA1 section and subiculum; this differed from both aFTLD-U and BIBD, where sections CA3 and CA4 were preferentially involved. The only finding on FUS-IHC that absolutely distinguished cases of NIFID from BIBD was the presence of at least some vermiform NII in hippocampal dentate granule cells and pyramidal neurons in all cases of NIFID, but none of the BIBD group (Table 5).
Discussion
Fused in sarcoma is a ubiquitously expressed [1], multifunctional, DNA/RNA-binding protein that shares striking functional homology with TDP-43 [27, 60]. It continually shuttles between the nucleus and cytoplasm [62], but in neurons and glia, it has a predominantly nuclear localization [1]. The normal physiological role of FUS in the brain in not completely understood, but it may be involved in neuronal plasticity and the maintenance of dendritic integrity [11, 12].
The FUS gene, on chromosome 16, was originally identified as a component of fusion oncogenes in a variety of human cancers [44]. More recently, missense mutations in FUS were discovered to be the cause of familial ALS (FALS) type 6 [26, 56]. Studies from many different centers have now confirmed that FUS mutations are responsible for ~4% of FALS and <1% of sporadic ALS [33]. A small number of these studies have included a description of the neuropathology which consistently shows cytoplasmic inclusions that are TDP-43-negative, but FUS-ir [14, 16, 26, 43, 45, 56, 58]. The role of FUS mutations in causing FTLD is less certain. A few patients have been reported with FALS due to FUS mutations, who also developed clinical FTD [6, 52, 59] and one study identified a novel (M254V) FUS mutation in a patient with sporadic, adult-onset FTD without evidence of ALS [57]. Unfortunately, none of these reports included neuropathological evaluation. All of the cases used in the present study were sporadic and none in which genetic analysis was performed were found to have FUS mutations [39, 40].
It was the description of TDP-43-negative, FUS-ir pathology in the original reports of FALS with FUS mutations [26, 56] that prompted us to investigate the possible role of FUS in tau/TDP-43-negative forms of FTLD. Since our initial publications [38–40], several other studies have evaluated FUS-ir pathology in subtypes of FTLD. A group from the Netherlands identified four cases of FTLD-FUS (5%) in a collection of 74 autopsy specimens from patients with clinical FTD [49]. The phenotype and neuropathology matched previous descriptions of aFTLD-U [30, 47] and the findings on FUS-IHC were compatible with ours. A similar frequency of FTLD-FUS was reported in a study from the UK, in which 5 (5%) of 100 FTLD autopsy cases were found to have FUS pathology [48]. Four were diagnosed as aFTLD-U and the other as NIFID; however, detailed neuropathological descriptions were not provided. Interestingly, one of the patients with aFTLD-U was from a family with autosomal dominant dementia and, although the mother was also found to have FTLD-FUS, no FUS mutation was identified. Screening of the Sydney Brain Bank for patients matching the clinical phenotype of aFTLD-U also identified one case with FUS pathology [28]. Finally, a large international study, that intentionally excluded BIBD and NIFID found FUS pathology in 34/37 (92%) cases of tau/TDP-43-negative FTLD [55]. The material originated from 12 different centers and included the 15 cases used in our original study [39] as well as 22 that had not previously been evaluated for FUS. The clinical phenotype was remarkably consistent and typical of aFTLD-U and no pathogenic FUS mutations were identified. Description of the neuropathology was brief, but all were reported to have FUS-ir NCI and NII.
The only additional reports of FUS in BIBD have been in patients with a pure ALS clinical phenotype. These have included sporadic cases with juvenile [3, 17] and adult-onset [13, 35] and familial adult-onset cases [23, 51]. FUS mutations were identified in most [3, 17, 23, 50, 51] of the cases. Although none of these cases had clinical FTD, all of the neuropathology reports described widespread FUS-ir pathology involving many neuroanatomical regions [3, 13, 17, 23, 35, 51].
The diagnosis of aFTLD-U, NIFID and BIBD are each based on the finding of a specific type of NCI [7, 30, 37]. However, none of these conditions is common and very few previous studies have directly compared their neuropathological features [61]. The finding of abundant FUS pathology in all cases examined to date indicates that aFTLD-U, BIBD and NIFID share a common aberrant molecular pathway and are more closely related than previously recognized. To determine the degree of overlap, we performed the first direct comparison of the FUS-ir pathology among these FTLD subtypes. Cases of aFTLD-U had the most distinct pattern of FUS pathology, characterized by more modest involvement of subcortical regions, less variation in NCI morphology and the most numerous and anatomically widespread NII. Cases of NIFID and BIBD showed greater similarity, but the complete absence of NII in hippocampus and neocortex was unique to the BIBD cases. Therefore, the cases showed many similarities, but there were sufficient differences on FUS-IHC to allow each group to be distinguished from the other two, with some findings being absolutely group specific. This is similar to the results of our re-evaluation of the core diagnostic features; BI and IF-ir NCI were a common finding in cases of all diagnostic groups, but their number and anatomical distribution allowed for definitive classification of each case (Table 2). Therefore, our findings on FUS-IHC provide validation of the classical diagnostic criteria and suggest that aFTLD-U, NIFID and BIBD are relatively distinct entities rather than a pathological continuum.
Similar to their pathology, aFTLD-U, NIFID and BIBD show overlapping but divergent clinical features. Although we did not have sufficient information to allow evaluation of individual cases, our findings on FUS-IHC appear to show some general correlations with the characteristic clinical phenotype of each group. Cases of NIFID usually present with a combination of FTD, pyramidal and extrapyramidal movement disorder [5, 7, 8, 19, 22, 29, 36, 46, 61] and were found to have abundant FUS-ir pathology in both cerebral cortex and many subcortical regions. BIBD often has a more limited phenotype (i.e. FTD or ALS only) [24, 25, 37, 53, 61] and had more variable involvement of different neuroanatomical regions. In aFTLD-U, the relative sparing of subcortical regions may explain the absence of significant movement disorder [30, 47, 55].
In summary, this study provides the first direct comparison of the FUS pathology that characterizes the three major FTLD-FUS subtypes. Our findings validate the specificity of the classical neuropathological diagnostic criteria for aFTLD-U, NIFID and BIBD, and support these as representing closely related but distinct entities that share a common molecular pathogenesis. This relationship is perhaps analogous to that of progressive supranuclear palsy and corticobasal degeneration, in which the neuropathology is not as unique as Pick’s disease, but less of a continuum than is seen in the Lewy body disorders (Parkinson’s disease and dementia with Lewy bodies). Therefore, although FTLD-FUS cases with overlapping pathology may exist, we recommend retaining the terms aFTLD-U, NIFID and BIBD for specific FTLD-FUS subtypes. Diagnosis should continue to be based on the original neuropathological criteria for each condition; however, the results of FUS-IHC should also be taken into account. Additional studies are needed to determine where cases of FTLD and ALS with FUS mutations fall within this spectrum.
References
Andersson MK, Stahlberg A, Arvidsson Y et al (2008) The multifunctional FUS, EWS, and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 9:37
Arai T, Hasegawa M, Akiyama H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Baumer D, Hilton D, Paine AML et al (2010) Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 75:611–618
Behring B, Beuche W, Kretzschmar HA (1998) Progressive dementia with parkinsonism in corticobasal degeneration and brainstem degeneration with neuronal inclusions. Neurology 51:285–288
Bigio EH, Lipton AM, White CL, Dickson DW, Hirano A (2003) Frontotemporal and motor neurone degeneration with neurofilament inclusion bodies: additional evidence for overlap between FTD and ALS. Neuropathol Appl Neurobiol 29:239–253
Blair IP, Williams KL, Warrich ST et al (2010) FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry 81:639–645
Cairns NJ, Grossman M, Arnold SE et al (2004) Clinical and neuropathological variation in neuronal intermediate filament inclusion disease. Neurology 63:1376–1384
Cairns NJ, Perry RH, Jaros E et al (2003) Patients with a novel neurofilamentopathy: dementia with neurofilament inclusions. Neurosci Lett 341:177–180
Cairns NJ, Zhukareva V, Uryu K et al (2004) α-Internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol 164:2153–2161
Davidson Y, Kelley T, Mackenzie IRA et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113:521–533
Fujii R, Okabe S, Urushido T et al (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15:587–593
Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118:5755–5765
Fujita Y, Fujita S, Takatama M, Ikeda M, Okamoto K (2010) Numerous FUS-positive inclusions in an elderly woman with motor neuron disease. Neuropathology. doi:10.1111/j.1440-1789.2010.01146.x
Groen EJN, van ES MA, van Vught PWJ et al (2010) FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch Neurol 67:224–230
Hamada K, Fukazawa T, Yanagihara T et al (1995) Dementia with ALS features and diffuse Pick body-like inclusions (atypical Pick’s disease?). Clin Neuropathol 14:1–6
Hewitt C, Kirby J, Highley R et al (2010) Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 67:455–461
Huang EJ, Zhang J, Geser F et al (2010) Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol 20:1069–1076
Ishihara K, Araki S, Ihori N et al (2006) An autopsy case of frontotemporal dementia with severe dysarthria and motor neuron disease showing numerous basophilic inclusions. Neuopathology 26:447–454
Josephs KA, Holton JL, Rossor MN et al (2003) Neurofilament inclusion body disease: a new proteinopathy? Brain 126:2291–2303
Josephs KA, Holton JL, Rossor MN et al (2004) Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol 30:369–373
Josephs KA, Lin WL, Ahmed Z, Stroh DA, Graff-Radford NR, Dickson DW (2008) Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116:159–167
Josephs KA, Uchikado H, McComb RD et al (2005) Extending the clinicopathological spectrum of neurofilament inclusion disease. Acta Neuropathol 109:427–432
Kobayashi Z, Tsuchiya K, Arai T et al (2010) Occurence of basophilic inclusions and FUS-immunoreactive neuronal and glial inclusions in a case of familial amyotrophic lateral sclerosis. J Neurol Sci 293:6–11
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665
Kusaka H, Matsumoto S, Imai T (1993) Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuopathol 12:215–218
Kwiatkowski TJ, Bosco DA, LeClerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136:1001–1004
Loy CT, McCusker E, Kril JJ et al (2010) Very early-onset frontotemporal dementia with no family history predicts underlying fused in sarcoma pathology. Brain. doi:10.1093/brain/awq186
Mackenzie IR, Feldman H (2004) Neurofilament inclusion body disease with early onset frontotemporal dementia and primary lateral sclerosis. Clin Neuropathol 23:183–193
Mackenzie IRA, Foti D, Woulfe J, Hurwitz TA (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293
Mackenzie IR, Neumann M, Bigio EH et al (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18
Mackenzie IRA, Neumann M, Bigio EH et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4
Mackenzie IRA, Rademakers R, Neumann M (2010) TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9:995–1007
Mackenzie IRA, Shi J, Shaw CL et al (2006) Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol 112:551–559
Matsuoka T, Fujii N, Kondo A et al (2010) An autopsied case of sporadic adult-onset amyotrophic lateral sclerosis with FUS-positive basophilic inclusions. Neuropathology. doi:10.1111/j.1440-1789.2010.01129.x
Molina-Porcel L, Llado A, Rey MJ et al (2008) Clinical and pathological heterogeneity of neuronal intermediate filament inclusion disease. Arch Neurol 65:272–275
Munoz-Garcia D, Ludwin SK (1984) Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 16:467–480
Munoz DG, Neumann M, Kusaka H et al (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA (2009) Frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IRA (2009) Abundant FUS pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Neumann M, Tolnay M, Mackenzie IRA (2009) The molecular basis of frontotemporal dementia. Exp Rev Mol Med 11:e23
Rademakers R, Stewart H, DeJesus-Hernandez M et al (2010) FUS gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42:170–176
Riggi N, Cironi L, Suva ML, Stamenkovic I (2007) Sarcomas: genetics, signalling, and cellular origins. Part I: The fellowship of TET. J Pathol 213:4–20
Robertson J, Bilbao J, Zinman L et al (2010) A novel double mutation in FUS gene causing sporadic ALS. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2010.05.015
Roeber S, Bazner H, Hennerici M, Porstmann R, Kretzschmar HA (2006) Neurodegeneration with features of NIFID and ALS-extended clinical and neuropathological spectrum. Brain Pathol 16:228–234
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Rohrer JD, Lashley T, Holton J et al (2010) The clinical and neuroanatomical phenotype of FUS associated frontotemporal lobar degeneration. J Neurol Neursurg Psychiatry. doi:10.1136/jnnp.2010.214437
Seelar H, Klijnsma KY, de Koning I et al (2010) Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol 257:747–753
Suzuki N, Aoki M, Warita H et al (2010) FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusions. J Hum Genet 55:252–254
Tateishi T, Hokonohara T, Yamasaki R et al (2010) Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol 119:255–364
Ticozzi N, Silani V, LeClerc AL et al (2009) Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73:1180–1185
Tsuchiya K, Matsunaga T, Aoki M et al (2001) Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol 20:53–59
Uchikado H, Shaw G, Wang DS, Dickson DW (2005) Screening for neurofilament inclusion disease using alpha-internexin immunohistochemistry. Neurology 64:1658–1659
Urwin H, Josephs KA, Rohrer JD et al (2010) FUS pathology defines the majority of tau-and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 120:33–41
Vance C, Rogelj B, Hortobagyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Van Langenhove T, van der Zee J, Sleegers K et al (2010) Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74:366–371
Yamamoto-Watanabe Y, Watanabe M, Okamoto K et al (2010) A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: a clinical, pathological and genetic report. J Neurol Sci 296:59–63
Yan J, Deng HX, Siddique N et al (2010) Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75:807–814
Yang S, Warraich ST, Nicholson GA, Blair IP (2010) Fused in sarcoma/translocated in liposarcoma: a multifunctional DNA/RNA binding protein. Int J Biochem Cell Biol 42:1408–1411
Yokota O, Tsuchiya K, Terada S et al (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115:561–575
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110:1741–1750
Acknowledgments
We thank Margaret Luk and Mareike Schroff for their excellent technical assistance. This work was supported by grants from Canadian Institutes of Health Research (grant number 74580, IM); the Pacific Alzheimer Research Foundation (IM); the German Federal Ministry of Education and Research (grant number 01GI0704, MN); the Stavros-Niarchos Foundation (MN); the Synapsis Foundation (MN); and the German Brain Bank “BrainNet” (HK).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mackenzie, I.R.A., Munoz, D.G., Kusaka, H. et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 121, 207–218 (2011). https://doi.org/10.1007/s00401-010-0764-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0764-0