
Abstract
Frontotemporal lobar degeneration (FTLD) can be pathologically subdivided into tau-positive and tau-negative types. The most common tau-negative variant is FTLD with ubiquitin-immunoreactive lesions (FTLD-U). Recently, the TAR DNA binding protein 43 (TDP-43) was identified in neuronal inclusions in FTLD-U. After applying TDP-43 immunohistochemistry to a series of 44 cases of FTLD-U with no secondary pathology, three cases (7%) were identified with ubiquitin- and p62-positive neuronal cytoplasmic inclusions (NCI) that were negative for TDP-43. All the three cases had marked brain atrophy with striking atrophy of the striatum. Cases 1 and 2 presented at ages 43 and 38, respectively, as behavioral variant frontotemporal dementia (1 with positive family history) and had ubiquitin- and p62-positive NCI in frontotemporal neocortex and dentate granule cells of the hippocampus. Case 3 presented with the corticobasal syndrome. Unlike the other two cases, ubiquitin- and p62-positive NCI were also visible on hematoxylin and eosin stain. There were no neuronal intranuclear inclusions. Electron microscopic examination of the NCI in cases 2 and 3 revealed granulofilamentous inclusions. These cases confirm the existence of TDP-43-negative FTLD-U and extend the clinical and pathological spectrum of this disorder. The findings raise the possibly of an as yet identified protein that may play a pathogenic role in tau-negative FTLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Frontotemporal lobar degeneration (FTLD) can be pathologically subdivided into cases with tau-positive and tau-negative immunoreactive lesions; the latter group being more common [14, 20]. Recently, the TAR DNA binding protein 43 (TDP-43) was identified in neuronal inclusions in tau-negative FTLD with ubiquitin-immunoreactive lesions (FTLD-U) and has been suggested to be a sensitive and specific marker for those disorders [2, 26]. On the other hand, TDP-43 immunoreactivity has also been reported in amyotrophic lateral sclerosis (ALS) [2, 26], Guam Parkinson dementia complex [9, 11], Parkinson dementia [25], as well as many cases of hippocampal sclerosis and some cases of Alzheimer’s disease [1]. The latter findings raise questions about the specificity of TDP-43 for FTLD-U.
Recently it has been proposed to classify FTLD into subtypes based upon the distribution and morphology of TDP-43 immunoreactive lesions in frontotemporal neocortices and dentate granule cells of the hippocampus [3, 21]. In addition, there is a small subset of cases of FTLD-U that do not show TDP-43 immunoreactivity [3, 22]. In this report, we describe the frequency and clinicopathologic characteristics of cases of FTLD with ubiquitin-positive neuronal cytoplasmic inclusions (NCI) that show no evidence of TDP-43 immunoreactivity in a collection of 44 cases of FTLD-U without co-morbid pathologic processes. Three cases were identified and studied with light and electron microscopic immunohistochemistry.
Materials and methods
Subject selection
The neuropathological database at the Mayo Clinic, Jacksonville, FL, was queried to identify all cases of FTLD that had been histologically analyzed by one neuropathologist (DWD) between October 1998 and July 2007, and that also had stored paraffin blocks and a complete set of slides. A total of 68 cases were identified that met these criteria. Those with a secondary neurodegenerative diagnosis were excluded: Alzheimer’s disease (Braak neurofibrillary tangle stage ≥ IV; N = 5), Lewy body disease (N = 5), argyrophilic grain disease (N = 11), neurofilament inclusion body disease (N = 2), and one case of pseudodementia. A total of 44 cases met inclusion and exclusion criteria and underwent immunostaining with TDP-43 (rabbit polyclonal: 1:3,000; ProteinTech Group, Chicago, IL). TDP-43 immunostaining was performed on brain tissue sections from ten different regions, including frontal, temporal and parietal neocortices, hippocampus, amygdala, striatum, midbrain, pons, medulla and cerebellum. Of the 44 cases, 41 showed abnormal TDP-43 immunoreactivity. In the remaining three cases, there was no abnormal TDP-43 immunoreactivity in any of the ten sections.
Pathological methods
All three cases without abnormal TDP-43 immunoreactivity underwent further histologic and ultrastructural evaluation. Tissue sections were stained with hematoxylin and eosin (H&E), Luxol fast blue-periodic acid Schiff (LFB-PAS) and Bielschowsky silver stains. Immunohistochemical staining was performed using standard methods. The deparaffinized and rehydrated sections were steamed in distilled water for 30 min and immunostained in batches to assure consistency with a DAKO Autostainer (DAKO, Carpinteria, CA) using 3, 3′-diaminobenzidine as the chromogen. After immunostaining, the sections were lightly counterstained with hematoxylin. The following antibodies were used: phosphorylated neurofilament (SMI-31, 1:20,000; Covance, Berkeley, CA); ubiquitin (mouse monoclonal Ubi-1, 1:40,000; Encor Biotechnology, Alachua, FL; rabbit polyclonal UBQ [6], 1:500 and rabbit polyclonal UH-19 [18], 1:2,500); phospho-tau (CP13, 1:100; Peter Davies, Albert Einstein College of Medicine, Bronx, NY); α-synuclein (NACP, 1:3,000 [10]); p62 (guinea pig polyclonal; 1;2500; Progen Biotechnik GmbH, Heidelberg, Germany) and α-internexin (1:100; EnCor Biotechnology, Alachua, FL). The presence or absence of motor neuron disease was assessed and defined as previously described, including stains for activated microglia [15].
Electron microscopy
Small pieces of tissue (hippocampus from cases 1 and 3, frontal cortex of case 2) from formalin-fixed brains were post-fixed in 2.5% glutaraldehyde, 0.1 M cacodylate buffer, pH 7.0, and then in aqueous 2% OsO4 and 1% uranyl acetate, 50% ethanol. After dehydration in an ascending series of alcohols and propylene oxide, they were infiltrated and embedded in Epon 812 (Polysciences, Warrington, PA). Thin sections were stained with uranyl acetate and lead citrate.
For immunoelectron microscopy, tissues were dehydrated in 30, 50, 70, and 90% ethanol for 10 min each infiltrated with 1:1 and 1:2 ratios of 90% ethanol: LR White for 20 and 40 min, respectively, followed by pure LR White for 1 h and overnight at room temperature. The tissues were embedded in BEEM® capsules, capped, and polymerized in a vacuum oven at 50°C for 2 days. Immunogold labeling was performed according to previous published methods [19] with two polyclonal antibodies to ubiquitin [6, 18] used at 1:20, which gave comparable results.
Results
Clinical histories
Case 1 was a 53-year-old man with 10-year duration of illness who presented with personality change at the age of 43. He would no longer repair broken items around the house, something that he had enjoyed doing prior. He began watching a significant amount of television. He had difficulty maintaining concentration. He became socially disinhibited and would say inappropriate things in public. He had significant difficulty sleeping and had hallucinations at night where he would see animals and people. He also had a veracious appetite and especially for sweets. Family history was negative for any neurodegenerative diseases. When first examined at age 49 his Mini-Mental State Examination (MMSE) [7] was 25 out of 30. There was no evidence of motor neuron disease or Parkinsonism. An MRI of the brain demonstrated evidence of hydrocephalus. An 18F-fluoro-deoxy-glucose positron emission tomography (FDG-PET) study demonstrated bifrontal hypometabolism. His neurodegenerative syndrome was most consistent with a clinical diagnosis of behavioral variant frontotemporal dementia (bvFTD).
Case 2 was a 42-year-old man with 4-year duration of illness who presented with personality change and hypersexuality at the age of 38. It was reported that he stopped attending to tasks that he would previously complete. He had a change in his appetite and ate whatever was available, especially sweets. He was socially disinhibited. Family history was significant for an uncle who was clinically diagnosed with progressive supranuclear palsy. On examination at age 39 his MMSE was 29 out of 30. Frontal release signs were present. There was no evidence of motor neuron disease or Parkinsonism. An MRI of the brain demonstrated frontal and temporal lobe atrophy. His neurodegenerative syndrome was most consistent with a clinical diagnosis of bvFTD.
Case 3 was a 68-year-old man with 5-year duration of illness who presented with language difficulty and trouble with arm movements. His language problems were characterized by difficulty with expression as well as trouble finding the correct word. His comprehension was relatively intact. He also had become socially isolated, being previously a more outgoing person. He had impaired coordination and had difficulty writing, cutting and eating his food, using tools and opening bottles. His episodic memory was preserved, and he had good insight into his problems. Family history was negative for any neurodegenerative disease. When first examined at age 65 his MMSE score was 23 out of 30. He had a partial Gerstmann syndrome. He had features of Parkinsonism, including reduced arm swing. A mild pronator drift was noted. He had evidence of apraxia. There was no evidence of motor neuron disease. An MRI showed left greater than right parietal atrophy as well as some left temporal lobe atrophy. His neurodegenerative syndrome was most consistent with a clinical diagnosis of corticobasal syndrome.
Neuropathological findings
Macroscopic findings
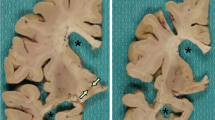
Neuropathological features are summarized in Table 1. The fixed brain weights, based upon doubling the weight of the available hemibrain, ranged from 920 to 1,240 g. The sulci and gyri revealed varying degrees of frontal, temporal and parietal lobe atrophy. In case 1, the atrophy was most marked in frontal and medial temporal structures (Fig. 1), while in case 2 atrophy was concentrated in the frontal lobe, including the precentral gyrus (Fig. 2). In case 3, atrophy was more diffuse, affecting all but the occipital lobe (Fig. 3). There was severe atrophy of basal ganglia nuclei with marked flattening of the caudate nuclei in all cases (Figs. 1, 2, 3).

Case 1: grossly there is frontal atrophy, sparing peri-rolandic sulci. Coronal sections show massive ventricular enlargement, severe atrophy of the striatum and atrophy of the frontal and temporal lobes with attenuation of the cerebral white matter. Inclusions in the dentate fascia are not visible on H&E (b), but are immunopositive for ubiquitin (c) and p62/sequestosome (d) and negative for TDP-43 (e). b–e ×400

Case 2: grossly there is frontal atrophy that encroaches on precentral gyrus. Coronal sections show ventricular enlargement, severe atrophy of the striatum and depigmentation of the substantia nigra. Inclusions in the dentate fascia are not visible on H&E (b), but are immunopositive for ubiquitin (c) and p62/sequestosome (d) and negative for TDP-43 (e). b–e ×400

Case 3: grossly there is global atrophy that is very severe in inferior parietal and posterior temporal lobes. Coronal sections show ventricular enlargement, severe atrophy of the striatum, severe medial temporal atrophy and depigmentation of the substantia nigra. Inclusions in the dentate fascia, which resemble Pick bodies, are readily apparent on H&E (b) and are positive for ubiquitin (c) and p62/sequestosome (not shown), but they are negative on Bielschowsky (d) and Gallyas (e) stains and for tau (not shown). They are also negative for α-internexin (f) and TDP-43 (g). b–g ×400
Microscopic findings
The neocortex showed marked thinning of the cortical ribbon most severe in temporal lobe and less so in frontal lobe in case 1; frontal, temporal and parietal lobe in case 3; and frontal lobe in case 2. With thioflavin-S fluorescent microscopy no senile plaques or neurofibrillary tangles were detected in any of the cases in all cortical sections. Tau, α-synuclein, and α-internexin failed to reveal neuronal or glial inclusions in gray or white matter. There were ubiquitin immunoreactive NCI in neocortex in cases 2 and 3. Some of the NCI in case 3 were visible on H&E in cortex, basal ganglia and hippocampus.
The hippocampus had extensive neuronal loss in Sommer’s sector, with more severe neuronal loss and gliosis in the subiculum than CA1 in all three cases. Only a few neurofibrillary tangles were noted in the CA2 region of case 3. The dentate fascia showed mild to moderate neuronal loss and gliosis, most marked in case 1. On H&E, there were well-circumscribed amphophilic-to-slightly basophilic inclusions in the perinuclear cytoplasm in case 3, only (Fig. 3). They were moderate in number in the dentate fascia and insular cortex, scant in cingular cortex and absent in the amygdala. No similar inclusions were noted in cases 1 and 2 on H&E. In all cases there were ubiquitin immunoreactive NCI in the dentate fascia (Figs. 1c, 2c, 3c). In cases 2 and 3 these inclusions were round, and well defined, while the inclusions in case 1 were smaller and less well defined (Fig. 1). In addition, these ubiquitin-positive inclusions were scant (<10 inclusion) in case 1, moderate-frequent in number in case 2, and moderate in case 3.
In all three cases, the NCI showed immunoreactivity to p62/sequestosome (Figs. 1d, 2d), but were negative to TDP-43 (Figs. 1e, 2e, 3g), tau, α-synuclein, neurofilament and α-internexin (Fig. 3f). There were no neuronal intranuclear inclusions in any of the three cases.
The basal nucleus of Meynert had no significant neuronal loss in all three cases, with only a few neurofibrillary tangles in case 3. The basal ganglia had no senile plaques or neurofibrillary tangles, but showed severe neuronal loss and gliosis in all three cases, particularly in the head of the caudate nucleus. Ubiquitin immunohistochemistry revealed NCI that were moderate in density in case 3, sparse in case 2 and rare in case 1. There were a moderate number of axonal spheroids in case 1, while they were sparse in case 2 and absent in case 3. The thalamus showed variable neuronal loss and gliosis, mostly in anterior and medial nuclei (Table 1). The subthalamus was unremarkable in all three cases. The substantia nigra had moderate to marked neuronal loss with extraneuronal neuromelanin and gliosis (Table 1). No Lewy bodies or neurofibrillary tangles were present in any of the cases. The medial cerebral peduncle (frontobulbar tract) was attenuated, extending to the middle third (corticospinal tract) in case 3. The motor neurons in the medulla were spared, as were the Betz cells in the motor cortex of all three cases.
Electron microscopy
The NCI were detected in the frontal cortex in case 2 and the granule cells of dentate fascia in case 3 (Fig. 4). They were not membrane-bound and contained randomly oriented filaments with focal coating by electron dense granular material (Fig. 4b). In coated regions the filaments had a diameter of 15–20 nm, but in segments without coating, the filaments had a diameter of 10–15 nm, and appeared to be straight. Filaments were mostly short in length and not densely packed. In addition, small vesicles of various densities were present among the filaments (Fig. 4c). Some inclusion also contained interspersed mitochondria and occasional laminated bodies. In other inclusions mitochondria, lipofuscin and rough endoplasmic reticulum were displaced by the inclusion to the periphery of the perikarya (Fig. 4c). In case 1, cytoplasmic inclusions were sparse and could not be detected at the electron microscopic level. Immunoelectron microscopy with gold conjugated secondary antibodies confirmed ubiquitin labeling of the inclusions in both coated and uncoated regions of the filaments (not shown).

a Neuronal cytoplasmic inclusions (asterisk) in a hippocampal granule cell of case 2. (Bar 1 μm). b Higher magnification shows coated (double arrow) and uncoated (arrowheads) filaments in random orientations, as well as small vesicles containing dense materials (arrows). Mitochondria (M) are located at the periphery of the inclusion. Bar 0.3 μm. c Neuronal cytoplasmic inclusions (asterisk) in a cortical neuron of case 3. The filamentous inclusions displace cytoplasmic organelles. Arrows point to mitochondria. Lf Lipofuscin, N nucleus. Note deep nuclear indentation (arrowheads). Bar 1 μm
Discussion
In this study, we identified three cases of FTLD-U that had neuronal inclusions that were negative for TDP-43. Although the inclusions in case 3 were visible on H&E, suggesting that case 3 may be different from cases 1 and 2, all three cases shared the same degree of striking atrophy of the striatum and the same immunohistochemical profile. In addition, the electron microscopic appearance of the NCI was similar in cases 2 and 3. These findings suggest that the three cases may represent a single type of FTLD-U.
The term dementia lacking distinctive histology was first described by Knopman et al. [16] and was initially thought to be the most common type of FTLD. With the advent of improved methods for ubiquitin immunohistochemistry, it was demonstrated that almost all cases of dementia lacking distinctive histology had inclusions in frontotemporal neocortex or dentate granule cell layer of the hippocampus that were immunoreactive for ubiquitin, with or without motor neuron disease (MND) consistent with FTLD-U or FTLD-MND [13, 14, 20, 21]. Most recently, it has been demonstrated that the ubiquitinated inclusions are immunoreactive for TDP-43 [26]. It has been suggested that TDP-43 is a more specific marker of FTLD-U than ubiquitin because ubiquitin immunoreactivity is also present in a wide range of neuronal inclusions as well as age-related structures [5]. On the other hand, recent studies indicate that not all FTLD-U have TDP-43 immunoreactivity, including cases with α-internexin-immunoreactive NCI [4] and so-called basophilic inclusion body disease [29].
The three cases described in this series had evidence of frontal and temporal lobe atrophy, with variable involvement of parietal lobe, on gross histology and had neuronal loss and gliosis in frontotemporal neocortices with ubiquitin-positive NCI. Hence, all three cases meet the criteria for FTLD-U. Unlike the majority of the 44 FTLD-U cases in this series, these 3 cases were unique, since in none of them did the NCI stain positive with TDP-43. Cases 1 and 2 were similar to those recently reported by Mackenzie et al. [22]. Similar to that series of six cases, our cases 1 and 2 also had a young age at onset and clinical presentation with bvFTD with disinhibition. Pathologically, cases 1 and 2 were also similar, in that NCI were found in frontotemporal neocortices and hippocampal dentate granule cells and were immunoreactive to ubiquitin and p62/sequestosome, yet negative to TDP-43. Unlike the series of six patients reported by Mackenzie et al. [22], one of our cases (case 2) has a positive family history and none had neuronal intranuclear inclusions. Furthermore, we cannot confirm a female predominance in this disease as all of our cases were male.
Case 3 in our series had a different clinical presentation, presenting with the corticobasal syndrome and at an older age of 63. Unlike cases 1 and 2, NCI were visible on H&E in neocortex and in the granule cells of the hippocampus dentate gyrus. The NCI had slightly basophilic tinctorial properties on H&E compared to the adjacent cytoplasm and were similar to NCI described in basophilic inclusion body disease, an entity initially associated with ALS [23] and later thought to be a variant (“generalized variant”) of Pick’s disease [24]. Subsequent case reports and series have shown an association between basophilic inclusion body disease and dementia or ALS [17, 23, 27, 29]. It is of note that our case 3 had corticospinal tract degeneration. In addition, the corticobasal syndrome in case 3 is similar to presentations of some patients with basophilic inclusion body disease reported by Yokota et al. [29]. The presence of ubiquitin immunoreactivity in basophilic inclusion body disease is inconsistent, with some studies reporting ubiquitin immunoreactivity as in our case 3 [27], while others do not [29]. This may be due to variations in the sensitivity of ubiquitin immunohistochemistry or due to a lack of consensus on how this entity is defined pathologically.
At the electron microscopic level, the inclusions in cases 2 and 3 appeared similar to each other and similar to those reported previously in basophilic inclusion body disease [17, 24] and for FTLD-U with TDP-43 immunoreactivity (unpublished observations). They all contained 10–15 nm diameter straight filaments that were focally coated by electron dense granular material. They were also immunoreactive for ubiquitin. Therefore, we cannot exclude the possibility that all three cases are similar. Another striking features of all three cases was the severe degree of basal ganglia atrophy, with flattening of the caudate head and nucleus accumbens, additionally suggesting that they may share a common etiopathogenesis.
An FTLD with a similar immunohistochemical profile to our cases has been associated with mutations in the charged multivesicular body protein 2b (CHMP2b) [28]. Similar to our cases, FTLD associated with CHMP2B has ubiquitin and p62 positive inclusions that are TDP-43 negative [12]. It is unlikely, however, that our cases are associated with a mutation in CHMP2B as this genetic defect has only been described in a single Danish family and two of the three cases did not have a positive family history. Moreover, this is a rare mutation and has not been found in screens of over 140 FTLD families [8].
In summary, the present study confirms the existence of atypical FTLD-U without TDP-43 immunoreactivity [22] and suggests that there is likely to be another protein(s) that plays a pathogenic role in tau-negative FTLD.
References
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R et al (2007) TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol 61:435–445. doi:10.1002/ana.21154
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. doi:10.1016/j.bbrc.2006.10.093
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22. doi:10.1007/s00401-007-0237-2
Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH et al (2004) Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 63:1376–1384
Dickson DW, Wertkin A, Kress Y, Ksiezak-Reding H, Yen SH (1990) Ubiquitin immunoreactive structures in normal human brains. Distribution and developmental aspects. Lab Invest 63:87–99
Dickson DW, Wertkin A, Mattiace LA, Fier E, Kress Y, Davies P et al (1990) Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer’s disease. Acta Neuropathol 79:486–493. doi:10.1007/BF00296107
Folstein MF, Folstein SE, McHugh PR (1975) Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12:189–198. doi:10.1016/0022-3956(75)90026-6
Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J et al (2006) Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15:2988–3001. doi:10.1093/hmg/ddl241
Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM et al (2008) Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol 115:133–145. doi:10.1007/s00401-007-0257-y
Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH et al (2000) Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol 99:663–672. doi:10.1007/s004010051177
Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T et al (2007) TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 130:1386–1394. doi:10.1093/brain/awm065
Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 66:884–891. doi:10.1097/nen.0b013e3181567f02
Josephs KA, Dickson DW (2007) Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging 28:1718–1722. doi:10.1016/j.neurobiolaging.2006.07.010
Josephs KA, Holton JL, Rossor MN, Godbolt AK, Ozawa T, Strand K et al (2004) Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol 30:369–373. doi:10.1111/j.1365-2990.2003.00545.x
Josephs KA, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW (2006) Clinically undetected motor neuron disease in pathologically proven frontotemporal lobar degeneration with motor neuron disease. Arch Neurol 63:506–512. doi:10.1001/archneur.63.4.506
Knopman DS, Mastri AR, Frey WH 2nd, Sung JH, Rustan T (1990) Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology 40:251–256
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665. doi:10.1007/BF00307636
Lee S, Park YD, Yen SH, Ksiezak-Reding H, Goldman JE, Dickson DW (1989) A study of infantile motor neuron disease with neurofilament and ubiquitin immunocytochemistry. Neuropediatrics 20:107–111
Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW (2003) Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol 162:213–218
Lipton AM, White CL 3rd, Bigio EH (2004) Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol 108:379–385. doi:10.1007/s00401-004-0900-9
Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH et al (2006) Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 112:539–549. doi:10.1007/s00401-006-0138-9
Mackenzie IR, Foti D, Woulfe J, Hurwitz TA (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293. doi:10.1093/brain/awn061
Matsumoto S, Kusaka H, Murakami N, Hashizume Y, Okazaki H, Hirano A (1992) Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol 83:579–583. doi:10.1007/BF00299405
Munoz-Garcia D, Ludwin SK (1984) Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 16:467–480. doi:10.1002/ana.410160408
Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE et al (2007) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114:221–229. doi:10.1007/s00401-007-0261-2
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. doi:10.1126/science.1134108
Sasaki S, Toi S, Shirata A, Yamane K, Sakuma H, Iwata M (2001) Immunohistochemical and ultrastructural study of basophilic inclusions in adult-onset motor neuron disease. Acta Neuropathol 102:200–206
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H et al (2005) Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 37:806–808. doi:10.1038/ng1609
Yokota O, Tsuchiya K, Terada S, Ishizu H, Uchikado H, Ikeda M et al (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115:561–575. doi:10.1007/s00401-007-0329-z
Disclosure statement
The authors do not have any disclosures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by NIH grants P50-AG16574, P50-NS40256 and Pacific Alzheimer Research Foundation (PARF) grant C06-01.
Rights and permissions
About this article
Cite this article
Josephs, K.A., Lin, WL., Ahmed, Z. et al. Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116, 159–167 (2008). https://doi.org/10.1007/s00401-008-0397-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-008-0397-8