
Abstract
Mutations in the fused in sarcoma gene (FUS) were recently found in patients with familial amyotrophic lateral sclerosis (ALS). The present study aimed to clarify unique features of familial ALS caused by FUS mutation in the Japanese population. We carried out clinical, neuropathological, and genetic studies on a large Japanese pedigree with familial ALS. In six successive generations of this family, 16 individuals of both sexes were affected by progressive muscle atrophy and weakness, indicating an autosomal dominant trait. Neurological examination of six patients revealed an age at onset of 48.2 ± 8.1 years in fourth generation patients, while it was 31 and 20 years in fifth and sixth generation patients, respectively. Motor paralysis progressed rapidly in these patients, culminating in respiratory failure within 1 year. The missense mutation c.1561 C>T (p.R521C) was found in exon 15 of FUS in the four patients examined. Neuropathological study of one autopsied case with the FUS mutation revealed multiple system degeneration in addition to upper and lower motor neuron involvement: the globus pallidus, thalamus, substantia nigra, cerebellum, inferior olivary nucleus, solitary nucleus, intermediolateral horn, Clarke’s column, Onuf’s nucleus, central tegmental tract, medial lemniscus, medial longitudinal fasciculus, superior cerebellar peduncle, posterior column, and spinocerebellar tract were all degenerated. Argyrophilic and basophilic neuronal or glial cytoplasmic inclusions immunoreactive for FUS, GRP78/BiP, p62, and ubiquitin were detected in affected lesions. The FUS R521C mutation in this Japanese family caused familial ALS with pathological features of multiple system degeneration and neuronal basophilic inclusions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by a progressive loss of motor neurons. Approximately, 5–10% of cases are familial forms, which are usually transmitted by an autosomal dominant trait [30]. Genetically, 20–25% of familial ALS pedigrees have missense mutations in the superoxide dismutase 1 gene (SOD1) [29], while TARDBP [10, 31], encoding TAR-DNA binding protein (TDP)-43, and ANG [7], encoding angiogenin, have also been linked to familial ALS. Recently, Kwiatkowski et al. [14] and Vance et al. [33] reported mutations in a gene encoding another DNA/RNA-binding protein called fused in sarcoma (FUS) specific for familial ALS. FUS mutations were detected in ~4% of familial ALS cases (~0.4% of all ALS cases) [15]. FUS is a nucleoprotein that functions in DNA and RNA metabolism [1, 2, 34, 35], and has been implicated in tumorigenesis [3, 28]. However, the precise roles of FUS have not been fully elucidated. Previous studies of FUS-mutation carriers revealed that the site of onset is the cervical region or proximal upper extremities, with subsequent spreading into the lower extremities; but neither bulbar signs nor cognitive deficits are apparent [14, 33]. Histologically, severe lower motor neuron loss in the spinal cord and, to a lesser degree, the brainstem, major pallor of the corticospinal tracts, mild to moderate upper motor neuron loss in the cerebral cortex, and mild myelin loss in the dorsal columns are observed, whereas dorsal horn neurons are unaffected [14, 33].
Herein, we report the clinical and pathological findings in a large Japanese familial ALS pedigree with an autosomal dominant inheritance pattern, and carrying a FUS R521C mutation. Patients demonstrated rapid progression of motor paralysis and pathological features of multiple system degeneration with argyrophilic and basophilic neuronal cytoplasmic inclusions and glial cytoplasmic inclusions containing FUS, revealing a unique phenotype of familial ALS caused by the FUS mutation.
Methods
Patients and clinical study
This family came from the same identical district in Japan. In five successive generations, 16 individuals were affected by progressive muscle atrophy and weakness. Blood samples from 37 individuals, including four affected persons from the present family (Fig. 1a), were obtained with written informed consent. The patients were subjected to a thorough neurological examination. The clinical features of representative cases are described below.

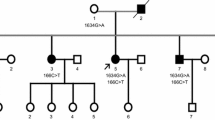
Pedigree tree of the present ALS family with FUS mutation and the mutation in FUS of patient 1. a Filled symbols indicate affected individuals. For confidentiality, gender and birth order are not shown. Five people in the pedigree were suspected to be asymptomatic carriers of a FUS mutation because their children with FUS mutations developed ALS, though their DNA samples were not available. b Direct sequencing shows a single base substitution (C1561T) in FUS of patient 1 (V-4). Antisense strands of FUS of patient 1 and a normal individual are shown
Patient 1 (V-4)
The proband, whose parent died of ALS at 49 years of age, suffered from weakness of the right upper extremity from age 31 years. The patient was diagnosed with ALS by electromyography (EMG) and muscle biopsy at another hospital. The patient consulted our hospital 2 months after onset of weakness. Neurological examination disclosed muscle weakness, atrophy, and fasciculation in the right proximal upper limb, and hyperreflexia in the lower extremities. Subsequently, muscle weakness of the four extremities, gait disturbance, and dysphagia rapidly progressed, culminating in the patient being bedridden within 7 months. Sensory disturbance and cognitive impairment were not evident. The patient’s respiratory function deteriorated and artificial ventilation was initiated 8 months after onset and continued for 14 years. Urinary disturbance and oculomotor paresis developed 4 years after onset and the patient died from hemorrhagic shock at the age of 45 years. Total disease duration was 14 years.
Patient 2 (VI-4)
The patient, a child of patient 1, suffered from weakness of bilateral upper limbs from age 20 years. For the 2 months following onset, bilateral arm weakness progressively worsened and the patient experienced difficulty with running. The patient was admitted to our hospital 6 months after onset. Neurological examination revealed moderate muscle weakness and atrophy of the bilateral trapezius and sternocleidomastoid muscles, with sparing of the cranial nerves innervating other muscles. The patient had a proximal dominant severe weakness, atrophy, and fasciculation in the bilateral upper limb muscles. Mild hyperreflexia and mild muscle weakness without atrophy were also noted in both lower limbs. The plantar response was flexor. Neither cognitive impairment nor sensory disturbance was observed. No abnormal findings were found in serum laboratory tests, except for mild elevation of creatine kinase to 598 U/l (normal range: 62–287). The results of CSF examination were normal. EMG showed chronic neurogenic patterns, including giant motor unit potentials in the right biceps, deltoid, first dorsal interossei, and anterior tibial muscles. The results of nerve conduction tests were essentially normal, except for a slightly prolonged F-wave latency in the left peroneal nerve. Motor-evoked potentials elicited by stimulation of the median nerve showed prolonged peripheral latency and normal central conduction time bilaterally, while those elicited by stimulation of the posterior tibial nerve were normal. Somatosensory-evoked potential studies were normal. Brain and cervical spinal cord MRI findings were normal, while a 99mTc-ethylcysteinate dimer SPECT (99mTc-ECD SPECT) study showed a decrease in cerebral blood flow in the right striatum, thalamus, and fronto-temporal lobe (Fig. 2). There was no mutation in SOD1. Nine months after onset, mechanical ventilation was introduced because of respiratory failure. Disease duration at the last examination was 14 months.

Brain MRI and 99mTc-ECD SPECT of patient 2 on admission obtained 6 months after onset. a T1- (TE 14.0/TR 585.0) and b T2-weighted axial images (TE 96.0/TR 3203.0) demonstrate no abnormality. c SPECT findings show a decrease in cerebral blood flow in the right striatum, thalamus, and fronto-temporal lobe (arrows)
Patient 3 (IV-7)
This patient noticed muscle weakness of the right foot and left arm at age 51 years and was diagnosed with ALS at another hospital using electrophysiological tests including EMG. Weakness in both arms progressively worsened. Dysphagia and dysarthria appeared 6 months after onset. Neurological examination at 8 months after onset showed dysarthria, dysphagia, tongue atrophy, neck weakness and proximal dominant muscle weakness, and atrophy of the bilateral upper limbs. Spasticity, hyperreflexia, and extensor plantar response were noted in the left lower limb. After one year, the respiratory disturbance became worse, resulting in death at age 52. Total disease duration was 21 months.
Patient 4 (IV-29)
At 58 years of age, this patient suffered from left leg weakness that progressively deteriorated during the following 8 months. Neurological examination demonstrated muscle weakness, fasciculation, and atrophy of the left lower limb and hyperreflexia in both the lower limbs. No sensory disturbance was noted. EMG findings showed chronic neurogenic patterns. Muscle weakness spread to both the upper limbs and the patient could not raise the arms. The gait disturbance progressed, and the patient was unable to walk unaided at 19 months after onset. Dysphagia and dysarthria appeared thereafter. Muscle weakness of all four extremities progressed rapidly and the patient was bedridden at 22 months after onset. Respiratory dysfunction became overt at 25 months after onset and the patient started mechanical ventilation at 26 months after onset. The disease duration at the last examination was 7 years.
Neuropathological study
A postmortem examination of patient 1 was performed 17 h after death. Brain and spinal cord specimens were fixed for 2 weeks in buffered 10% formalin, embedded in paraffin, and then sliced into 6-μm-thick sections. The sections were stained with hematoxylin and eosin (HE), Bodian, and Gallyas stains. We tested three anti-FUS antibodies, each of which recognizes a different epitope. Anti-FUS rabbit polyclonal antibody (Ab–TLS–M) was provided by the Laboratory for Structural Neuropathology and the Research Resource Center, RIKEN Brain Science Institute [4]. The anti-FUS antibodies were generated against a peptide encompassing amino acids 260–274 of mouse FUS. Anti-FUS rabbit polyclonal antibodies made by Bethyl Laboratories (Montgomery, TX, USA) and Novus Biologicals (Littleton, CO, USA) recognize an N-terminal epitope, encompassing amino acids 1–50 of human FUS. Immunohistochemistry experiments using these three antibodies revealed equally labeled cytoplasmic inclusions. Therefore, the polyclonal antibody Ab-TLS-M was used for all subsequent immunostaining experiments. The following primary antibodies and dilutions were used for immunohistochemical investigations: anti-FUS rabbit polyclonal antibody (Ab-TLS-M) (1:500) [4], anti-FUS rabbit polyclonal antibody (1:250, Bethyl Laboratories), anti-FUS rabbit polyclonal antibody (1:250, Novus Biologicals), anti-ubiquitin rabbit polyclonal antibody (1:500, DAKO, Glostrup, Denmark), anti-p62 rabbit polyclonal antibody (1:1,000, Biomol, Plymouth Meeting, PA, USA), anti-78-kDa glucose-regulated protein/immunoglobulin heavy-chain binding protein (GRP78/BiP) rabbit polyclonal antibody (1:500, Santa Cruz, Santa Cruz, CA, USA), anti-microtubule-associated protein (MAP)-2 mouse monoclonal antibody (1:500, clone HM-2, Sigma, Saint Luis, MO, USA), anti-phosphorylated neurofilament mouse monoclonal antibody (1:500, clone 2F11, DAKO, Glostrup, Denmark), anti-SOD1 sheep polyclonal antibody (1:500, Binding Site, Birmingham, UK), anti-tau rabbit polyclonal antibody (1:500, DAKO, Glostrup, Denmark), anti-β-actin mouse monoclonal antibody (1:500, clone AC-15, Sigma, Saint Luis, MO, USA), anti-α-synuclein mouse monoclonal antibody (1:500, clone Sf38, Santa Cruz, Santa Cruz, CA, USA), anti-αB-crystallin rabbit polyclonal antibody (1:500) [9], anti-cystatin C rabbit polyclonal antibody (1:1,000, DAKO, Glostrup, Denmark), anti-polyglutamine mouse monoclonal antibody (1:500, clone 2c11, Millipore, Billerica, MA, USA), and anti-TDP43 rabbit polyclonal antibody (1:10,000, Cosmo Bio, Tokyo, Japan). The secondary antibodies were horseradish peroxidase-conjugated anti-rabbit antibody (PI-1000) and horseradish peroxidase-conjugated anti-mouse antibody (PI-2000) (1:500, both from Vector Laboratories, Burlingame, CA, USA). Immunohistochemistry was also performed using an indirect immunoperoxidase method as previously described [11]. The sections were deparaffinized in xylene, dehydrated in ethanol, and then incubated with 0.3% hydrogen peroxide in absolute methanol for 30 min at room temperature to inhibit endogenous peroxidase activity. After rinsing in tap water, the sections were completely immersed in distilled water and then heated in 0.01 M citrate buffer (pH 6.0) in a microwave for 10 min for antigen retrieval. After this pretreatment, the sections were incubated with a primary antibody diluted in 5% normal goat serum in 20 mM Tris–HCl (pH 7.6) containing 0.5 M NaCl, 0.05% NaN3, and 0.05% Tween 20 (TBST) at 4°C overnight, and then with a 1:200 dilution of the appropriate secondary antibody for 1 h at room temperature. A colored reaction product was developed using 3,3′-diaminobenzidine tetrahydrochloride (DAB) solution (0.02% DAB, 0.003% H2O2, 50 mM Tris–HCl, pH 7.6). The sections were lightly counter-stained with hematoxylin. Neuronal and glial cytoplasmic inclusions immunoreactive for FUS were evaluated using a semiquantitative grading system, similar to the one used in a previous study [22] in which pathological lesions were scored as none (–), rare (+), occasional (++), common (+++), or numerous (++++).
Genetic analysis
We performed genetic analysis for FUS mutations using peripheral blood samples obtained from the four patients. Exons 5, 6, 14, and 15 of FUS were amplified by polymerase chain reaction (PCR) using intronic primers [14, 33]. The PCR product was sequenced using a BigDye Terminator Sequencing Kit (Applied Biosystems). Sequencing reactions were performed using an automated Applied Biosystems Model 3100 DNA sequencer.
Results
Clinical findings
The clinical features are summarized in the Table 1. The family history suggests an autosomal dominant inheritance pattern. Both sexes (11 men and 5 women) were affected. Mean age at onset was 40.6 ± 13.8 years (n = 6, mean ± SD), and mean duration from onset to respiratory failure was 11.7 ± 7.3 months (n = 6, mean ± SD). Age at onset in the fourth generation was 48.2 ± 8.1 years (n = 4, mean ± SD) while it was 31 and 20 years in the fifth (n = 1) and sixth (n = 1) generations, respectively. Three out of six patients examined, showed preferential involvement of the proximal upper extremities with flailing arms and subsequent spread of motor weakness to the lower extremities.
General autopsy findings in patient 1
General autopsy examinations demonstrated bronchopneumonia with bilateral pulmonary edema in the upper lobes and pulmonary atelectasis in the right lower lobe. There was also unclotted blood in the small intestine and colon lumen. However, the origin of the bleeding in the gastrointestinal tract was not clear.
Neuropathological findings in patient 1
The patient’s brain weighed 1,070 g at autopsy. A macroscopic examination demonstrated severe atrophy of the bilateral frontal lobes, brainstem, cerebellum, and spinal cord (Fig. 3a–c). Bilateral precentral gyri were mildly atrophic. Anterior roots of spinal nerves were markedly atrophied, while posterior roots were moderately atrophic (Fig. 3d, e). By Klüver–Barrera staining, severe degeneration was found in the central tegmental tracts, medial lemniscus, medial longitudinal fasciculus, superior cerebellar peduncles, lateral corticospinal tracts, posterior columns, and spinocerebellar tracts. Neurons were markedly lost from the anterior horns, intermediolateral horns, Clarke’s columns, and Onuf’s nuclei in the spinal cord (Fig. 4a–c). Ganglion cells also disappeared from the dorsal root ganglia. Moreover, severe neuronal loss was seen in the Betz cells of the primary motor cortex, the inner and outer segments of the global pallidus, the thalami, compact and reticular parts of the substantia nigra, the Purkinje and granular cells and dentate nucleus of the cerebellum, the trochlear and abducens nuclei of the midbrain, and the inferior olivary, hypoglossal, ambiguous, and solitary nuclei of the medulla oblongata (Fig. 4d). All of these nerve cell losses were associated with heavy astrogliosis as determined by GFAP immunostaining and Holzer staining. The hippocampi and parahippocampal gyri were not atrophic, and pyramidal cells in the hippocampi were not lost. Argyrophilic and basophilic neuronal cytoplasmic inclusions were often seen in oculomotor, pontine, and Meynert nuclei, and these were immunoreactive for FUS, ubiquitin, p62, and GRP78/BiP, but not for MAP-2, phosphorylated neurofilament, SOD1, Tau, β-actin, α-synuclein, αB-crystallin, cystatin C, polyglutamine, or TDP43 (Fig. 4e–k). Following HE staining, the basophilic neuronal cytoplasmic inclusions showed an eosinophilic core with HE staining, which was surrounded by a pale basophilic halo similar to those described in a previous report [6]. Additionally, glial cytoplasmic inclusions were revealed by FUS immunostaining (Fig. 4l–n). FUS-immunoreactive glial cytoplasmic inclusions were also immunopositive for ubiquitin and p62, and appeared to extend into the radiating processes. Lewy bodies were not detected, and neither Tau nor α-synuclein immunoreactivity was found in glial cytoplasmic inclusions. No ubiquitinated neuronal inclusions were observed either in the hippocampus or in the cerebral cortex. In all brain and spinal cord regions, FUS-positive neuronal and glial inclusions were more frequently and widely observed than basophilic, ubiquitin-, and silver-positive inclusions (Table 2). Neurons were markedly missing from the anterior horns. Glial cytoplasmic inclusions were detected in the anterior horns, but not neuronal cytoplasmic inclusions. FUS-positive glial inclusion bodies were diffuse in the white matter, but demyelinating lesions were not detected. In the controls, FUS immunoreactivity was more strongly detected in the neuronal nuclei of the anterior horn cells relative to that in the neuronal cytoplasm (Fig. 4r). In patient 1, in neurons without inclusion bodies, faint diffuse nuclear immunostaining to FUS with prominent nucleolar staining was seen, while such FUS nuclear staining was not apparent in neurons without inclusions (Fig. 4k).

Macroscopic findings in patient 1. Macroscopically, bilateral frontal lobes (a), brainstem (b), cerebellum (c), and spinal cord (d, e) show severe atrophy. Anterior roots of the spinal nerves in front view (d, arrows) are markedly atrophic while posterior roots in back view (e, arrows) also show moderate atrophy. f Schematic illustrations indicating the distribution of degeneration based on HE staining and gross findings in the CNS of patient 1. Colors indicate grades of involvement as mild (green), moderate (yellow), and severe (red) in each region examined based on HE staining and gross findings. Scale bars 3 cm (a–c), and 1 cm (d–e)

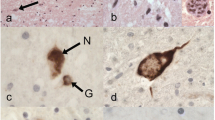
Microscopic findings in patient 1. a–c Klüver–Barrera (KB) staining of axial sections of the spinal cord shows severe degeneration of bilateral lateral corticospinal tracts and posterior columns in the cervical (a), thoracic (b), and lumbar cord (c). d HE staining shows severe neuronal loss with neuropil rarefaction in the anterior horns of the lumbar cord. e HE staining shows basophilic neuronal cytoplasmic inclusions in the oculomotor nucleus. The basophilic neuronal cytoplasmic inclusions are stained with Bodian (f) and Gallyas stains (g). GRP78/BiP (h), ubiquitin (i), p62 (j), and FUS (k) immunoreactivities can be seen in cytoplasmic inclusions in the oculomotor nucleus. Faint diffuse nuclear immunostaining to FUS with prominent nucleolar staining is present in the adjacent neuron without the inclusion (arrow in k), while FUS nuclear staining is not apparent in the red nucleus with a FUS inclusion on the same section (inset in k). j and k are consecutive sections. FUS (l), ubiquitin (m), and GRP78/BiP (n) immunoreactivities are seen in cytoplasmic inclusions in the precentral gyrus. These images (l–n) are of consecutive sections. Glial cytoplasmic inclusions immunoreactive for ubiquitin (o), p62 (p), and FUS (q) can be seen in the cerebral peduncle. Staining of the anterior horn cells of a control individual demonstrates diffuse nuclear staining without inclusions (r). Scale bars 2 mm (a–c), 100 μm (d), 50 μm (e), 10 μm (f–q), and 50 μm (r)
Genotype findings
Mutations in the FUS gene have previously been identified in familial ALS cases [14, 33]. Therefore, we conducted mutation scanning of exons 5, 6, 14, and 15 of FUS by direct sequencing, and found a missense mutation, 1561 C>T (R521C), within exon 15 of FUS in four affected family members (patients 1–4) (Fig. 1b). This missense mutation is identical to one of the FUS mutations previously reported [14, 33]. DNA specimens from the other two patients who were clinically examined (patients 5 and 6) were not available. We also found that 2 of 34 non-affected individuals tested were heterozygous for the relevant mutation.
Discussion
In the present study, we describe the first Japanese family with ALS with the FUS R521C mutation that is one of the most common mutations of the gene located in the highly conserved C-terminus region. Thus, FUS mutation should be considered as one of the causes of familial ALS in Japanese populations.
Average age at onset is reported as around 45 years and average survival is about 33 months after onset [14, 33]. In our patients, average age at onset in the fourth generation was comparable to those reported previously; however, patients in the fifth and sixth generations had average ages at onset that were 20 and 30 years, respectively, earlier than that in the fourth generation patients and showed very rapid progression culminating in respiratory failure within 9 months of onset. Genetic anticipation is usually seen in trinucleotide repeat disorders, but not in the case of missense mutations. Thus, factors other than the FUS mutation may have contributed to the accelerated onset and very rapid progression in the descendant generations of this family. Alternatively, this may be a chance effect. Preferential onset in upper limbs, as found in the family reported here, has also been described in families with FUS mutations [14, 33]. Thus, onset in the proximal upper limbs with subsequent spreading to the lower limbs appears to be a characteristic feature of familial ALS caused by FUS mutation, irrespective of race.
Interestingly, the autopsied case, whose disease duration was 13 years with mechanical ventilation, showed multiple system degeneration in addition to upper and lower motor neuron involvements: neurons in the inner and outer segments of the globus pallidus, thalami, compact and reticular parts of substantia nigra, Purkinje and granular cells and dentate nucleus of the cerebellum, inferior olivary nucleus, solitary nucleus, intermediolateral horns, Clarke’s columns, and Onuf’s nucleus were lost, and the central tegmental tracts, medial lemniscus, medial longitudinal fasciculus, superior cerebellar peduncles, posterior columns, and spinocerebellar tracts showed degeneration. The pathology of the autopsied materials demonstrated extensive cerebellar atrophy, probably because 14 years had passed since onset. Cerebellar ataxia was not apparent clinically before initiating artificial ventilation within a year of disease onset. We could not evaluate whether cerebellar symptoms developed clinically in the patient after initiating artificial ventilation, because of severe weakness of the four limbs. In previous reports, the only extra-motor involvement found has been mild myelin loss in the dorsal columns [33]. The occurrence of widespread neurodegeneration involving multiple systems, may be related to the long disease duration in our patient. For example, sporadic ALS cases in a totally locked-in state show widespread degeneration beyond the motor neuron system [8, 17]. However, widespread degeneration is not always a feature in sporadic ALS patients surviving for long periods with respiratory assistance [24, 27, 36]. In addition, Nishihira et al. [24] reported that the distribution patterns of TDP-43-immunoreactive neuronal cytoplasmic inclusions were not influenced by long-term survival with artificial respiratory support. In patient 2, a decrease in cerebral blood flow in the right striatum, thalamus, and fronto-temporal lobe was evident in the very early course of the disease. We assumed that subclinical neural dysfunction in multiple systems started in the early course of the disease, resulting in widespread neurodegeneration of multiple systems in the late stage of FUS mutation-related ALS.
The present autopsied case showed basophilic neuronal cytoplasmic inclusions in oculomotor nuclei, pontine nuclei, and Meynert nuclei. Perhaps, as anterior horn cells in the spinal cord showed a remarkable disappearance, no basophilic inclusions were seen in the anterior horns. Basophilic inclusions were distributed in regions, where neurons were relatively preserved. The presence of FUS-positive cytoplasmic inclusions that were immunopositive for ubiquitin and p62 in the anterior horn neurons has been described [33]. The inclusions in our case were positive for FUS, ubiquitin, p62, and GRP78/BiP, but not for MAP-2, phosphorylated neurofilament, SOD1, Tau, β-actin, α-synuclein, αB-crystallin, cystatin C, or polyglutamine, suggesting a distinct mechanism from those underlying the formation of Lewy bodies, Bunina bodies, and neurofibrillary tangles. Although neuronal basophilic inclusions are rarely observed in the spectrum of motor neuron diseases, such as juvenile ALS [16, 20, 26], adult-onset ALS [12, 13], frontal lobe syndrome [19], and familial ALS with posterior column involvement [32], these inclusions are negative for ubiquitin and are therefore different from those found in patients with FUS mutation-related ALS. The presence of ubiquitin and GRP78/BiP in FUS-positive inclusions indicates an involvement of protein misfolding and endoplasmic reticulum stress. In patients with the FUS mutation, aberrant cytoplasmic localization and an increase in the level of total insoluble FUS is reported [14, 33]. Doi et al. [4] reported that FUS is a major nuclear aggregate-interacting protein in a model of Huntington’s disease [4]. However, in Huntington’s disease, FUS protein shifts into nuclear aggregates with polyglutamine in neurons [4]. In contrast, in this familial ALS case, FUS protein aggregates were present in the neuronal cytoplasm. FUS-positive neuronal and glial inclusions were found more frequently and widely than basophilic, ubiquitin-, and silver-positive inclusions in all brain and spinal regions. Thus, it was assumed that ubiquitination of FUS is a late event. Therefore, mislocalization of nuclear protein and the presence of cytoplasmic aggregates are similar to the findings in familial ALS patients with mutations in TARDBP, suggesting that both conditions have similar pathogenic mechanisms.
In our cases, FUS-positive inclusions were seen in the glial cytoplasm as well as in neuronal cytoplasm. Multiple system degeneration involving the fronto-temporal cortex, hippocampal formation, neostriatum, and substantia nigra is observed in sporadic ALS cases with long disease duration and on artificial ventilation. In such cases, TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions are widespread in the central nervous system [23, 24]. Neumann et al. [21] found that FUS-labeled ubiquitin-positive neuronal and glial cytoplasmic inclusions are seen in fronto-temporal lobar degeneration (FTLD), with the neuronal inclusions composed of an unidentified ubiquitinated protein (FTLD-U). In addition, abundant FUS immunoreactivity can be found in intermediate filament inclusion disease (NIFID), presenting as FTLD [22]. Recently, Munoz et al. [18] reported FUS pathology in basophilic inclusion body disease, a tau-negative form of FTLD. Inclusion bodies found under these conditions do not contain TDP-43 [18, 21, 22]. Accordingly, Frank and Tolnay [5] proposed that FTLD-U, NIFID, and BIDB can be referred to as FUS proteinopathies (FTLD-FUS). FUS may thus contribute to the development of FTLD and ALS with multiple system degeneration as TDP-43 does. However, distinct molecular pathways may be involved because the protein structures of FUS and TDP-43 are different. In TDP-43 proteinopathy, truncated TDP-43 with a deletion of the C-terminal part has a primary role in the formation of inclusions, leading to neuronal degeneration [25]. In this study we did not evaluate whether truncations in FUS might occur, similar to those described for TDP-43. Therefore, we think determining the existence of such a truncation in FUS would be an interesting future study.
References
Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS, Akhmedov AT (1999) Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem 274:34337–34342
Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS (1999) Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 18:4515–4521
Crozat A, Aman P, Mandahl N, Ron D (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363:640–644
Doi H, Okamura K, Bauer PO et al (2008) RNA-binding protein TLS is a major nuclear aggregate-interacting protein in Huntingtin exon 1 with expanded polyglutamine-expressing cells. J Biol Chem 283:6489–6500
Frank S, Tolnay M (2009) Frontotemporal lobar degeneration: toward the end of conFUSion. Acta Neuropathol 118:629–631
Fujita K, Ito H, Nakano S, Kinoshita Y, Wate R, Kusaka H (2008) Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol 116:439–445
Greenway MJ, Andersen PM, Russ C et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38:411–413
Hayashi H, Kato S (1989) Total manifestations of amyotrophic lateral sclerosis. ALS in the totally locked-in state. J Neurol Sci 93:19–35
Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE (1989) Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell 57:71–78
Kabashi E, Valdmanis PN, Dion P et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kikuchi H, Doh-ura K, Kawashima T, Kira J, Iwaki T (1999) Immunohistochemical analysis of spinal cord lesions in amyotrophic lateral sclerosis using microtubule-associated protein 2 (MAP2) antibodies. Acta Neuropathol 97:13–21
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665
Kusaka H, Matsumoto S, Imai T (1993) Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuropathol 12:215–218
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136:1001–1004
Matsumoto S, Kusaka H, Murakami N, Hashizume Y, Okazaki H, Hirano A (1992) Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol 83:579–583
Mizutani T, Sakamaki S, Tsuchiya N et al (1992) Amyotrophic lateral sclerosis with ophthalmoplegia and multisystem degeneration in patients on long-term use of respirators. Acta Neuropathol 84:372–377
Munoz DG, Neumann M, Kusaka H et al (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627
Munoz-Garcia D, Ludwin SK (1984) Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 16:467–480
Nelson JS, Prensky AL (1972) Sporadic juvenile amyotrophic lateral sclerosis. A clinicopathological study of a case with neuronal cytoplasmic inclusions containing RNA. Arch Neurol 27:300–306
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) Frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Nishihira Y, Tan CF, Hoshi Y et al (2009) Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol 117:45–53
Nishihira Y, Tan CF, Onodera O et al (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182
Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M (2009) Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet 18:3353–3364
Oda M, Akagawa N, Tabuchi Y, Tanabe H (1978) A sporadic juvenile case of the amyotrophic lateral sclerosis with neuronal intracytoplasmic inclusions. Acta Neuropathol 44:211–216
Piao YS, Wakabayashi K, Kakita A et al (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 13:10–22
Rabbitts TH, Forster A, Larson R, Nathan P (1993) Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 4:175–180
Rosen DR, Siddique T, Patterson D et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Shaw CE, Enayat ZE, Powell JF et al (1997) Familial amyotrophic lateral sclerosis. Molecular pathology of a patient with a SOD1 mutation. Neurology 49:1612–1616
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Tsuchiya K, Matsunaga T, Aoki M et al (2001) Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol 20:53–59
Vance C, Rogelj B, Hortobagyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Wang X, Arai S, Song X et al (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454:126–130
Yang L, Embree LJ, Tsai S, Hickstein DD (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273:27761–27764
Yoshida M, Murakami N, Hashizume Y, Itoh E, Takahashi A (1992) A clinicopathological study of two respirator-aided long-survival cases of amyotrophic lateral sclerosis. Rinsho Shinkeigaku 32:259–265
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tateishi, T., Hokonohara, T., Yamasaki, R. et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol 119, 355–364 (2010). https://doi.org/10.1007/s00401-009-0621-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0621-1