
Abstract
Basophilic Inclusion Body Disease (BIBD) is a tau-negative form of frontotemporal lobar degeneration (FTLD), characterized by neuronal cytoplasmic inclusions (NCI) that are visible on hematoxylin and eosin stain (HE), contain RNA, and are inconsistently ubiquitin-immunoreactive (ir). The normal nuclear expression of TDP-43 is not altered. Here we investigate whether the distribution of the structurally and functionally related protein fused in sarcoma (FUS) is altered in BIBD. Mutations in the FUS gene have recently been identified as a cause of familial amyotrophic lateral sclerosis (ALS). In addition to these familial ALS cases, FUS protein has recently been demonstrated in NCI in a subset of FTLD with ubiquitinated inclusions (atypical FTLD-U) and in neuronal intermediate filament inclusion disease (NIFID). We examined seven BIBD brains of patients with average age at onset 46 (range 29–57) and average duration of disease 8 years (range 5–12). Three cases presented with the behavioural variant of fronto-temporal dementia (FTD-bv) and one with FTD-bv combined with severe dysarthria. All four developed motor neuron disease/ALS syndrome (MND/ALS) several years later. In the other three cases, presentation was predominantly with motor symptoms, construed as MND/ALS in two, and progressive supranuclear palsy (PSP) in one. Severity of cortical degeneration varied, but all cases shared severe nigrostriatal atrophy and lower motor neuron pathology. In spared areas of cortex, FUS antibodies showed intense labelling of neuronal nuclei and weak positivity of cytoplasm, whereas, in affected areas, intense labelling of NCI was accompanied by reduction or disappearance of the normal IR pattern. The number of FUS-ir NCI was much greater than the number detected by HE or with ubiquitin or P62 immunohistochemistry. FUS-ir glial cytoplasmic inclusions (GCI) were abundant in the grey and white matter in all cases, whereas neuronal intranuclear inclusions were rare and only seen in 2/7 cases. Thus, BIBD shares with atypical FTLD-U and NIFID the presence of FUS-ir NCI and GCI, and together comprise a new biochemical category of neurodegenerative disease (FUS proteinopathies). The consistent involvement of motorneurons in BIBD indicates that the association of FTLD and MND/ALS can occur on a FUS or TDP-43 pathological substrate.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Frontotemporal lobar degeneration (FTLD) refers to the underlying pathology of a group of diseases characterized by slowly progressive atrophy and neuronal loss involving the frontal and/or temporal lobes and sometimes the parietal lobes in a bilateral, albeit often asymmetrical pattern [10]. The clinical presentations manifest the distribution of the atrophy. In addition to the canonical syndromes of frontotemporal dementia (FTD) with prominent behavioral disturbances (FTD-bv), progressive non-fluent aphasia, and semantic dementia [37, 42], motor neuron disease/amyotrophic lateral sclerosis (MND/ALS) type weakness [31] and extrapyramidal movement disorder [49] can be present. The deposition of protein aggregates as neuronal cytoplasmic inclusions (NCI) in most, if not all cases, constitutes the basis for taxonomy, as well as the best available window into the pathogenesis of these conditions. The classification is based on the dichotomy between NCI that contain tau and those that are ubiquitin-ir only. The latter type, denominated FTLD-U, represents approximately 60% of the cases [34]. There is only a weak correlation between the type of protein deposits, the anatomical pattern of atrophy, and the associated clinical presentation [20, 25, 26]. Exceptions include semantic dementia and FTD with MND/ALS, both of which are almost exclusively associated with FTLD-U pathology [12, 26]. Patients with classical MND/ALS (without dementia) consistently show ubiquitin-ir NCI in lower motor neurons (LMN) and often have FTLD-U type pathology in the cerebral cortex and dentate gyrus [30, 48, 63]. Although most cases are sporadic, familial FTLD-U can result from mutations in genes encoding progranulin, valosin containing protein, and charged multivesicular body protein 2B (CHMP2B), and some families with FTLD-U and MND/ALS show genetic linkage to a region on chromosome 9p [52].
Ubiquitin is only a tag for proteins to be processed through the ubiquitin-proteosome system (UPS). The search for the underlying protein in FTLD-U and MND/ALS led to the identification of the transactive response (TAR) DNA binding protein with Mr 43 kD (TDP-43), a DNA–RNA binding protein as a major constituent of NCI in the majority of sporadic FTLD-U and MND/ALS cases [4, 46], as well as in carriers of mutations in the genes encoding progranulin and valosin containing protein and chromosome 9p linked cases [10]. However, approximately 15% of FTLD-U patients showed no immunohistochemical or biochemical evidence of abnormality in TDP-43. This included familial FTD caused by CHMP2B mutations [21] and a subset of cases with sporadic FTLD-U [11, 24, 50]. The latter represent a well defined clinico-pathological entity, characterized by the additional presence of unusual neuronal intranuclear inclusions (NII) of vermiform shape and have been named atypical FTLD-U (aFTLD-U) [33, 53]. Recently, the term FTLD-UPS has been applied to TDP-43-negative FTLD-U cases in which the NCI label only with antibodies against markers of the UPS, such as ubiquitin and p62 [34]. FTLD-UPS thus includes aFTLD-U and CHMP2B mutation carriers, but leaves out two distinct FTLD-U subtypes, neuronal intermediate filament inclusion disease (NIFID, also known as FTLD-IF) and basophilic inclusion body disease (BIBD) [34].
BIBD is the consensus name [10] for a rare entity that can present with FTD or MND/ALS, or a combination of both [39]. It was first described in juvenile MND/ALS [1, 36, 43, 47], and has been reported in adult-onset MND/ALS [15, 18, 19, 27, 28, 38, 54, 55], including at least one family [59]. The presence of gaze palsy [28] or the development of incontinence [28] hinted to the atypical character of MND/ALS in some of these patients. Cases of BIBD presenting with behavioural symptoms were initially named generalized variant of Pick’s disease [40], and subsequently reported under this [58] and other names [22, 65]. Most of these patients subsequently developed motor weakness.
The pathology of BIBD is characterized by frontotemporal cortical atrophy, which may extend to the parietal lobes and is accompanied by marked degeneration of the neostriatum and substantia nigra. The basophilic NCI (BI), for which the disease is named are similar in size and shape to Pick bodies and are visible with HE and Nissl stains but do not take up silver. BI have been variably described as ubiquitin-positive or ubiquitin-negative, a discrepancy that may be explained by the ultrastructural finding that the randomly oriented 12–25 nm in diameter fibrils forming the BI are coated by dense granular material presumed to be derived from ribosomes [18, 28, 40, 55]. Early research utilizing crude histochemical procedures suggested that BI contained RNA [40, 43] and recently messenger RNA-related proteins have been identified as a constituent [15]. BI are present in the cerebral cortex, basal ganglia, brainstem, and spinal cord in BIBD but are not found in cases of classic MND/ALS [41].
Familial MND/ALS (FALS), which often follows an autosomal dominant inheritance pattern, may be caused by mutations in several genes, including copper/zinc superoxide dismutase 1 (SOD1), angiogenin, dynactin, and TDP-43 (TARDBP) [56], and genetically linked to several chromosomal loci [16, 17, 35]. In parallel with findings in FTLD-U, TDP-43-ir NCI were absent in cases of FALS with SOD1 mutations [32]. A focused search for other genes coding DNA–RNA binding proteins in a section of FALS linked to chromosome 16 (FALS type 6) resulted in the discovery of mutations in the fused in sarcoma (FUS, also known as translated in liposarcoma) gene [29, 61]. FUS is a 526 amino acid, heterogeneous ribonuclear protein that binds to RNA through its C-terminus, whereas its DNA-binding N-terminus participates in transcriptional activation [2, 51]. FUS expression is ubiquitous, and in most cell types it is located in both nucleus and cytoplasm, but nuclear expression predominates in neurons and is exclusive in glia [3]. Cellular processes in which FUS participates include cell proliferation [7], DNA repair [5], transcription regulation, mRNA splicing [64], and transport of RNA between nucleus and cytoplasm [66]. A total of 14 different mutations, mostly missense in the final exon, were reported in the two original studies [29, 61]. All but one caused autosomal dominant, clinically classic MND/ALS. Furthermore, the FUS protein product accumulated in NCI in motor neurons and dystrophic neurites (DN) in FUS mutation carriers [61]. Given the previously noted similarities between MND/ALS and FTLD-U, the presence of FUS pathology was recently investigated in subsets of TDP-43-negative FTLD-U, resulting in the identification of abundant FUS pathology as a highly characteristic and consistent finding in both aFTLD-U and NIFID [44, 45].
Since BIBD represents another subtype of TDP-43-negative FTLD-U [10, 15], we decided to investigate whether the characteristic BI contained FUS.
Methods
Cases
Cases were collected from the authors’ archives. Six cases have been previously reported (see Table 1 for summary and references), and one is presented here. A family history was not identified in any case.
Case 7
This woman presented at age 42 with a hypokinetic rigid syndrome. Neurologic examination 7 years later revealed vertical gaze palsy, saccades, severe dysarthia, dysphagia, blepharospasm, and bradykinesia of upper limbs, in the absence of tremor. EMG showed signs of denervation in the upper limbs. There was no family history. The clinical diagnosis was progressive supranuclear palsy (PSP). She died at age 50. At autopsy, the brain weighed 1,320 g and did not show obvious atrophy of the cerebral hemispheres. The substantia nigra was depigmented and there was mild atrophy of the cerebellum. Microscopic examination revealed subcortical neuronal loss and gliosis involving the neostriatum and globus pallidus, substantia nigra, locus ceruleus, inferior olive and cerebellar dentate, accompanied by BI. In addition, mild Purkinje cell loss and a few torpedos were present in the cerebellar cortex, and some LMN in the anterior horns appeared swollen. Less than 10% of the BI were ubiquitin-ir but numerous ubiquitin-ir NCI of irregular shape with fuzzy margins were present in LMN. The cerebral cortex and hippocampus showed no abnormalities on standard microscopic examination.
Immunohistochemistry
Sections of the frontal cortex, hippocampus, basal ganglia, medulla, and in cases 1–3 and 6 spinal cord were used for immunohistochemistry (IHC). All IHC was performed on 5-μm thick sections of formalin fixed, paraffin embedded tissue using the Ventana BenchMark® XT automated staining system (Ventana, Tuscon, AZ) and developed with aminoethylcarbizole (AEC) or diaminobenzime (DAB). Three primary antibodies recognizing different epitopes of FUS were used: Sigma–Aldrich (aa 86–213) at dilution 1:25 to 1:200, Bethyl Laboratories A300-302A (aa 1–50), at 1:500, and ProteinTech Group 11570-1-AP (aa 52–400) at 1:50, all with overnight incubation at room temperature, following microwave antigen retrieval). Other antibodies used to include ubiquitin (DAKO anti-ubiquitin; 1:300, following microwave antigen retrieval), p62 (BD Transduction Laboratories p62 Lck ligand; 1:500 following microwave antigen retrieval) and TDP-43 (ProteinTech Group anti-TARDBP; 1:400 following microwave antigen retrieval), neurofilament (2F11, DAKO, 1:50), tau (DAKo, 1:1,600), and α-synuclein (Zymed, 1:300, following formic acid pretreatment). Based on the amount of normal physiological staining, it was apparent that the anti-FUS sensitivity was greatly influenced by the degree of tissue fixation and that this was only partially reversed by antigen retrieval. Therefore, the dilution of the Sigma anti-FUS primary antibody was adjusted in each case (from 1:25 to 1:200) to allow for faint physiological staining that ensured sensitivity (internal positive control) but did not compromise visualization of the pathology. The intensity of physiological and pathological staining varied in parallel, and was always greatest in samples with brief fixation. The variation could be observed within a subject (case 1) in which blocks were obtained at different time points.
Immunofluorescence
Double-label immunofluorescence was performed on selected regions from BIBD case #7 using mouse monoclonal anti-p62 (BD Transduction Laboratories) and rabbit polclonal anti-FUS (Sigma–Aldrich). The secondary antibodies were Alexa Fluor 488 conjugated anti-mouse and Alexa Fluor 594 conjugated anti-rabbit (Molecular Probes, Eugene, OR, USA, 1:500). 4-6-diamidino-2-phenylindol (DAPI) (Vector Laboratories, Burlingame, CA, USA) 6 was used for nuclear counterstaining.
Results
Four cases were male and three female. The average age at onset was 46 (range 29–57) and the average duration of disease 8 years (range 5–12). Three cases (cases 1–3) presented with FTD-bv, and one (case 4) with FTD-bv combined with severe dysarthria, followed several years later by MND/ALS in all four cases. The presentation in the other three cases was predominantly motor, construed as MND/ALS in two and PSP in one (case 7) (Table 1).
The common element in the pathology was the severe atrophy of the striatum and substantia nigra. The distribution of cortical atrophy was congruent with the clinical presentation (Table 1). Extensive fronto-temporal atrophy with lesser involvement of the parietal lobes, was restricted to cases 1–4 with FTD-bv presentation. In cases with a predominantly motor presentation, cortical atrophy was restricted to the precentral gyrus in case 5, and absent in case 7. It could not be assessed in relation to the neurodegenerative process in case 6, who had suffered an episode of anoxia 1 month prior to death. Hippocampal sclerosis was severe in all cases presenting with FTD-bv, but was not present in cases presenting with motor symptoms, except case 6, where it may be attributed to the anoxic episode. Loss of brainstem or spinal LMN was prominent in cases 5 and 6 only. In the other two in which cord was available (cases 1, 7), a better correlate of weakness was degeneration of corticospinal tracts (cst) and cytological alterations of LMN. Case 7, with a clinical diagnosis of PSP, showed degeneration restricted to the basal ganglia, without significant cortical or cst involvement.
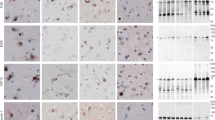
Rounded or crescent shaped BI were identified in HE stained sections of atrophic neocortex, but only rarely in the dentate gyrus or hippocampus. They were most common in the striatum, substantia nigra, periaqueductal grey and cerebellar dentate nucleus in all cases (Fig. 1a, b). In some cases (cases 5, 7), anterior horn LMN were swollen but did not contain visible inclusions. When inclusions were present in LMN (case 1) their irregular shape was suggestive of aggregates of Nissl bodies. A small proportion of BI (~5–10%) showed variable, generally weak immunoreactivity (IR) for ubiquitin (Fig. 1c–f) and intense P62 IR (Fig. 1g). Ubiquitin and P62 IHC also labelled some LMN NCI not visible with HE. None of the NCI was positive for neurofilament, tau or α-synuclein and TDP-43 showed only the normal nuclear IR (Fig. 1h).

Basophilic inclusions (BI). a, b H&E. BI take a variety of shapes, with the side facing the nucleus often adopting a concave outline (a case 1, putamen; b case 2, hippocampal CA1 sector). c–e Ubiquitin. Ubiquitin-IR varied from moderate (c case 1, putamen) to intense (d case 2, dentate gyrus) to negative (e case 1, putamen). f Ubiquitin. Ubiquitin-positive (black arrow) and negative (red arrow) BI could be identified in adjacent neurons, case 1, putamen. g P62. P62-IR (arrow) was seen in a subset of BI, case 7, putamen. h TDP-43. TDP-43-IR was confined to nuclei in all areas, case 1, CA1 sector. Scale bar 10 μm (a, b, c, e), 25 μm (d, f, g, h)
FUS IR was affected by the degree of fixation, but the results in different cases are qualitatively similar. Neurons in unaffected areas showed diffuse FUS IR that was intense in the nucleus and weaker in the cytoplasm (Fig. 2a, b). FUS IHC labelled all BI (Fig. 2c) and also showed many NCI that were not visible with HE, including arciform profiles (Fig. 2d), thin perinuclear rings and flame-shaped NCI, reminiscent of neurofibrillary tangles (Fig. 2e). In some neurons, the entire perikaryon was intensely FUS-immunoreactive (ir) (Fig. 2f). In some cells, the presence of FUS-ir NCI was accompanied by total or partial loss of nuclear FUS IR (Fig. 2f, g), whereas others maintained a normal level of nuclear staining (Fig. 2i). FUS IHC also revealed glial cytoplasmic inclusions (GCI), either flame-shaped reminiscent of coiled bodies, or ovoid. These inclusions were common in the striatum, globus pallidus, cerebellar dentate nucleus, and anterior horn, and relatively rare in the cortex and white matter oligodendroglia (Fig. 2j–l). There was no obvious discrepancy in the distribution of NCI and GCI. Short thick contorted FUS-ir DN was sparse in the cerebral cortex (Fig. 2f). No NII were identified in five cases. Case 2 showed a few bar-shaped NII in the cerebral cortex and a single vermiform example in the CA3 sector of the hippocampus, all in association with NCI (Fig. 3a). A single vermiform NII was found in a LMN in case 1 (Fig. 3b). Of note, case 2 was the only one to have BI in the dentate gyrus.

FUS-IR. a, b FUS-IR is restricted to the nuclei of neurons in unaffected areas, including the dentate (a) and the insular cortex (b) of case 7. c In affected areas, FUS IR was reduced (inset) or lost from nuclei, and rounded neuronal cytoplasmic inclusions (NCI) were labelled (arrows). Case 2, temporal cortex; inset entorhinal cortex. d Arciform NCI in dentate gyrus, case 2. e Flame-shaped NCI (black arrow) in CA1 sector, case 7. Note the markedly reduced nuclear FUS IR despite the presence of intensely labelled nuclei in the neighbourhood (green arrows). FUS nuclear IR is absent in a subset of neurons without NCI (red arrows). f FUS-IR fills the entire perikaryon (black arrow). A contorted neurite (red arrow) is also seen, case 2, insular cortex. g The morphology of these FUS-IR apical dendrites (black arrow) is reminiscent of the contorted neurites. Numerous labelled nuclei (green arrow) are seen in lightly affected areas. h Subcortical NCI (arrow) in the putamen (case 1). i Nucleus ambiguous (case 7), with a rare example of a neuron with NCI (black arrow) and preserved nuclear FUS IR (red arrow). j Glial cytoplasmic inclusions (GCI, red arrows) are seen near NCI (black arrows) in spinal cord anterior horn, case 7. k GCI (red arrows) and NCI (black arrows) in globus pallidus, case 7. l GCI of different shapes (red arrows) in the putamen, case 7. Scale bar 50 μm (j), 25 μm (a, b, c inset, e, f, g, i, k), 10 μm (c, d, h, l)

Intranuclear inclusions. FUS-IR. In two cases only, rare neurons show FUS ir intranuclear inclusions (black arrows), always in association with NCI. a Case 2, temporal cortex. b Case 7, spinal cord anterior horn. GCI (red arrow) Scale bar 10 μm
In order to demonstrate that FUS antibodies were labelling BI, photographs were obtained of neurons containing FUS-labelled NCI before and after removal of AEC chromagen and staining with HE (Fig. 4).

FUS antibodies label BI. Following FUS IHC utilizing AEC, the same neurons were photographed before (a, c) and after (b, d) destaining in water and regular H&E staining. a, b cerebral cortex, case 4. c, d putamen, case 7
Co-localization of P62 and FUS-IR was examined by double-label immunofluorescence. All P62+ NCI and GCI showed FUS-IR in the same distribution; however only a subset of FUS-ir NCI and GCI were double-labelled for p62 (Fig. 5).

Double label immunofluorescence for p62 (green) and FUS (red). Merged images show cell nuclei stained with DAPI (blue). Only a subset of FUS-positive neuronal and glial cytoplasmic inclusions as shown in the putamen (a, c) and spinal cord (b) are double-labeled for p62. Scale bar 40 μm (a, b); 10 μm (c)
A generalized reduction in neuronal nuclear FUS staining as compared to neighbouring regions in the same section was also found in some regions without obvious degeneration and with relatively few NCI, such as hippocampal CA1 sector in case 7 (Figs. 6, 7). The correspondence of decreased FUS-IR with anatomical regions and their close proximity with normal FUS-IR regions argue against attributing this finding to fixation-related variations in IR.

Patient with mild cortical pathology (Case 7). FUS-IR. Regions with numerous NCI showed widespread reduced nuclear FUS IR. a Low power demonstrating the contrast between the near normal insular cortex where most neurons retain nuclear expression with only a rare NCI (bottom inset) and the affected putamen, where FUS IR is restricted to NCI (top inset). b Hippocampus, where the near normal CA3 sector (top inset) with intense nuclear FUS IR contrasts with the CA1 sector (bottom inset) with labelled NCI and reduced nuclear FUS IR. Scale bar 500 μm (a, b), 25 µm (inserts). Arrows mark NCI

Patient with severe cortical pathology (Case 2). FUS-IR. a Lateral temporal cortex, low power, showing the contrast between the near normal superior temporal gyrus with nuclear FUS IR (high power in b) and the middle temporal gyrus (high power in c) where FUS IR is predominantly located in NCI of varied shapes. d Hippocampus, low power (same section as a), showing loss of nuclear FUS IR. FUS IR is almost entirely restricted to NCI in the dentate (high power in e). Scale bar 500 μm (a, d), 25 μm (b, c), 10 μm (e)
The hippocampus showed a spectrum of severity of atrophy (see Table 1), ranging from undetectable (case 7) to virtually complete neuronal loss leaving only a few neurons in the dentate gyrus (cases 3, 4). The cases could be arranged in a hierarchical order, in which atrophy appeared in the subiculum only, followed by the stepwise addition of CA1, CA4, the dentate granular layer and finally CA2–CA3. Even in the absence of detectable atrophy, a profound loss of nuclear FUS IR, affecting over 90% of neurons, was evident in CA1 and the subiculum, but not in CA4-CA2, the dentate, or the presubiculum (case 7, Fig. 3b). This is different from the normal pattern observed in normal control and disease control cases [44]. Loss of nuclear FUS-IR was accompanied by the appearance of flame-shaped FUS-ir NCI in approximately 1% of neurons in CA1 and the subiculum, along with contorted DN from CA2 to the subiculum. At an intermediate stage (case 2), where almost complete neuronal loss in the subiculum and CA1 was accompanied by moderately severe loss in CA4–CA2 and the dentate, loss of nuclear FUS IR was universal in the hippocampus and over 90% of dentate granular neurons bore NCI (Fig. 4d, e). Finally, severe atrophy resulting in virtually complete neuronal loss in the hippocampus was accompanied by loss of FUS-ir elements (cases 3, 4).
Discussion
The main finding in the present report is that BIBD demonstrates an abnormality in the cellular distribution of FUS in affected areas, consisting of partial displacement from nuclei and accumulation in neuronal and glial cytoplasmic inclusions. This represents a major advance in our understanding of the pathobiology of this enigmatic condition, in which the biochemical defect was previously completely unknown.
FUS shares a number of structural and functional similarities with another FTLD/ALS associated protein, TDP-43. Both participate in transport of specific types of mRNA to dendritic spines, where it is locally translated in response to synaptic activation or depolarization. Such transport requires sequestering mRNA from the translation apparatus in the cell body, and involves three types of RNA-containing granules: ribonucleoprotein particles (RNP), processing bodies (PB) and stress granules (SG), each associated with a distinct protein signature [9]. Actin-stabilizing proteins are coded by the transported mRNA associated with FUS and the failure of this mechanism is presumed to account for the observed abnormal dendritic spine morphology of FUS deficient neurons [13, 14]. TDP-43 also functions as neuronal activity-responsive factor in dendrites where, in association with the post-synaptic protein PSD-95, it participates in the formation of RNA granules containing beta-actin mRNA, among others [62]. The presence of TDP-43 in dendritic PB suggests a functional role: in response to neuronal activation, TDP-43 would deliver specific mRNAs to P bodies for switching-off their translation [62]. Thus, both FUS and TDP-43 seem to be involved in the regulation of synaptic plasticity in a role similar to that previously established for other RNA-binding proteins, including fragile X mental retardation protein and Staufen 1 [23].
The basophilia of BI can be attributed to their RNA content, as determined by histochemical procedures [40, 43]. Ultrastructural observations suggesting that the RNA is derived from ribosomes [40, 47] have been recently confirmed by the demonstration of ribosomal protein S6, indicating the presence of the smaller, 40S subunit of ribosomes [15]. However, the absence of ribosomal protein L28 indicates that the larger 60S subunit does not participate in the formation of BI. The mRNA-binding proteins poly(A)-binding protein 1 (PABP1) and T cell intracellular antigen 1 (TIA-1) are also present in BI, whereas decapping enzyme 1 (Dcp1) is not [15]. This pattern indicates that BI are related to SG, rather than RNP or PB, the other two types of neuronal RNA granules. In SG, the presence of TIA-1 and other proteins blocks transcription and the binding of the 60S subunit [9]. Thus, FUS and TDP-43 are associated with two different types of RNA granules, both of which appear in response to stress [9]: FUS with SG and TDP-43 with PB [62]. The latter structures, associated with mRNA decay, do not participate in the formation of BI, as the Dcp1 protein, a P body marker, is absent [15]. The relationship of FUS to stress granules in normal conditions and in BIBD remains a matter for future research. It is possible that dendritic translation is involved in the pathogenesis of both FUS and TDP-43 proteinopathies.
The consistent involvement of both anterior horn motor neurons and supratentorial structures in BIBD, regardless of the initial clinical presentation, is similar to the pattern observed in FTLD-TDP and lends further support to the intimate relationship between ALS and FTLD-U. The pattern of hippocampal sclerosis, starting in CA1-subiculum junction and spreading to involve the dentate gyrus, is also similar in BIBD, aFTLD-U and some ALS patients with and without memory impairment [57], but different from that of Alzheimer’s disease. This finding further supports the concept of shared mechanism between TDP-43 and FUS proteinopathies.
The presence of FUS-ir pathology in BIBD is shared with two other major forms of sporadic TDP-43-negative FTLD-U, aFTLD-U [44], and NIFID [45]. These conditions can now be grouped in the broad class of FTLD-FUS. It is possible that aFTLD-U, NIFID and BIBD might represent a continuous spectrum of disease or even a single pathological entity. However, comparison with previous reports of aFTLD-U and NIFID suggests that although there is some overlap, BIBD retains some distinct features. The most obvious is the basophilia of NCI in BIBD. Although some NCI in NIFID are visible with HE, they tend to be slightly eosinophilic and those in aFTLD-U are only detected with IHC [33, 53]. Vermiform NII are a consistent and prominent feature of both aFTLD-U and NIFID, but are absent or rare in BIBD. Although there is overlap, these conditions also show some differences in the anatomical pattern of degeneration and distribution of NCI. Loss of LMN is a more prominent and consistent feature of BIBD than in aFTLD-U.
The displacement of FUS from nuclei in both NCI bearing and normal appearing neurons, is a feature of BIBD that appears to be more prominent than in aFTLD-U and NIFID. It suggests that alterations in normal nuclear transport of FUS in BIBD, leading to increased cytoplasmic localization, might be involved in pathogenesis. This is in accordance with findings in cells transfected with mutant FUS which show a relative increase of cytoplasmic compared with nuclear FUS [29, 61]. However, the loss of nuclear FUS in inclusion bearing cells in FUSopathies does not appear to be as prominent or as consistent as in TDP-43 proteinopathies.
Finally, since TARDBP mutations have been found in a small subset of subjects with presumed TDP-43 proteinopathy presenting clinically as sporadic or familial ALS- [60], FTD with MND/ALS [6] and even FTD-bv [8] it is possible that FUS mutations will appear among patients with BIBD with or without a family history.
The demonstration that BIBD is a FUSopathy closes two circles: it completes the search for the protein accumulating in the NCI of all major forms of FTLD, and provides a tentative explanation for the unique presence of RNA in the NCI of BIBD.
References
Aizawa H, Kimura T, Hashimoto K et al (2000) Basophilic cytoplasmic inclusions in a case of sporadic juvenile amyotrophic lateral sclerosis. J Neurol Sci 176:109–113
Aman P, Panagopoulos I, Lassen C et al (1996) Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 37:1–8
Andersson MK, Stahlberg A, Arvidsson Y et al (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 9:37
Arai T, Hasegawa M, Akiyama H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611
Baechtold H, Kuroda M, Sok J et al (1999) Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem 274:34337–34342
Benajiba L, Le BI, Camuzat A et al (2009) TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65:470–473
Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS (1999) Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 18:4515–4521
Borroni B, Bonvicini C, Alberici A et al (2009) Mutation within TARDBP leads to Frontotemporal Dementia without motor neuron disease. Hum Mutat
Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8:776–789
Cairns NJ, Bigio EH, Mackenzie IR et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 114:5–22
Cairns NJ, Neumann M, Bigio EH et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240
Davies RR, Hodges JR, Kril JJ et al (2005) The pathological basis of semantic dementia. Brain 128:1984–1995
Fujii R, Okabe S, Urushido T et al (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15:587–593
Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118:5755–5765
Fujita K, Ito H, Nakano S et al (2008) Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol 116:439–445
Greenway MJ, Andersen PM, Russ C et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38:411–413
Gros-Louis F, Gaspar C, Rouleau GA (2006) Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 1762:956–972
Hamada K, Fukazawa T, Yanagihara T et al (1995) Dementia with ALS features and diffuse Pick body-like inclusions (atypical Pick’s disease?). Clin Neuropathol 14:1–6
Hilton DA, McLean B (2002) December 2001: rapidly progressive motor weakness, starting in pregnancy. Brain Pathol 12:267–268
Hodges JR, Davies RR, Xuereb JH et al (2004) Clinicopathological correlates in frontotemporal dementia. Ann Neurol 56:399–406
Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol 66:884–891
Ishihara K, Araki S, Ihori N et al (2006) An autopsy case of frontotemporal dementia with severe dysarthria and motor neuron disease showing numerous basophilic inclusions. Neuropathology 26:447–454
Jin P, Alisch RS, Warren ST (2004) RNA and microRNAs in fragile X mental retardation. Nat Cell Biol 6:1048–1053
Josephs KA, Lin WL, Ahmed Z et al (2008) Frontotemporal lobar degeneration with ubiquitin-positive, but TDP-43-negative inclusions. Acta Neuropathol 116:159–167
Kertesz A, Blair M, McMonagle P, Munoz DG (2007) The diagnosis and course of frontotemporal dementia. Alzheimer Dis Assoc Disord 21:155–163
Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG (2005) The evolution and pathology of frontotemporal dementia. Brain 128:1996–2005
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665
Kusaka H, Matsumoto S, Imai T (1993) Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuropathol 12:215–218
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Leigh PN, Whitwell H, Garofalo O et al (1991) Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain 114:775–788
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079
Mackenzie IR, Bigio EH, Ince PG et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434
Mackenzie IR, Foti D, Woulfe J, Hurwitz TA (2008) Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131:1282–1293
Mackenzie IR, Neumann M, Bigio EH et al (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18
Mackenzie IR, Rademakers R (2008) The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 21:693–700
Matsumoto S, Kusaka H, Murakami N et al (1992) Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol (Berl) 83:579–583
McKhann GM, Albert MS, Grossman M et al (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol 58:1803–1809
Mizutani T, Sakamaki S, Tsuchiya N et al (1992) Amyotrophic lateral sclerosis with ophthalmoplegia and multisystem degeneration in patients on long-term use of respirators. Acta Neuropathol 84:372–377
Munoz DG (1998) The pathology of Pick complex. In: Kertesz A, Munoz DG (eds) Pick’s disease and Pick complex. Wiley-Liss, New York, pp 211–241
Munoz-Garcia D, Ludwin SK (1984) Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 16:467–480
Murayama S, Mori H, Ihara Y et al (1990) Immunocytochemical and ultrastructural studies of lower motor neurons in amyotrophic lateral sclerosis. Ann Neurol 27:137–148
Neary D, Snowden JS, Gustafson L et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Nelson JS, Prensky AL (1972) Sporadic juvenile amyotrophic lateral sclerosis. A clinicopathological study of a case with neuronal cytoplasmic inclusions containing RNA. Arch Neurol 27:300–306
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain (in press)
Neumann M, Roeber S, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol (Berl). doi:10.1007/s00401-009-0581-5
Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Oda M, Akagawa N, Tabuchi Y, Tanabe H (1978) A sporadic juvenile case of the amyotrophic lateral sclerosis with neuronal intracytoplasmic inclusions. Acta Neuropathol (Berl) 44:211–216
Okamoto K, Murakami N, Kusaka H et al (1992) Ubiquitin-positive intraneuronal inclusions in the extramotor cortices of presenile dementia patients with motor neuron disease. J Neurol 239:426–430
Padovani A, Agosti C, Premi E, Bellelli G, Borroni B (2007) Extrapyramidal symptoms in Frontotemporal Dementia: prevalence and clinical correlations. Neurosci Lett 422:39–42
Pikkarainen M, Hartikainen P, Alafuzoff I (2008) Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol 67:280–298
Prasad DD, Ouchida M, Lee L, Rao VN, Reddy ES (1994) TLS/FUS fusion domain of TLS/FUS-erg chimeric protein resulting from the t(16;21) chromosomal translocation in human myeloid leukemia functions as a transcriptional activation domain. Oncogene 9:3717–3729
Rademakers R, Hutton M (2007) The genetics of frontotemporal lobar degeneration. Curr Neurol Neurosci Rep 7:434–442
Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M (2008) TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 116:147–157
Sam M, Gutmann L, Schochet SS Jr, Doshi H (1991) Pick’s disease: a case clinically resembling amyotrophic lateral sclerosis. Neurology 41:1831–1833
Sasaki S, Toi S, Shirata A et al (2001) Immunohistochemical and ultrastructural study of basophilic inclusions in adult-onset motor neuron disease. Acta Neuropathol 102:200–206
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Takeda T, Uchihara T, Arai N, Mizutani T, Iwata M (2009) Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: distinction from Alzheimer disease. Acta Neuropathol 117:35–44
Tsuchiya K, Ishizu H, Nakano I et al (2001) Distribution of basal ganglia lesions in generalized variant of Pick’s disease: a clinicopathological study of four autopsy cases. Acta Neuropathol (Berl) 102:441–448
Tsuchiya K, Matsunaga T, Aoki M et al (2001) Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol 20:53–59
Valdmanis PN, Daoud H, Dion PA, Rouleau GA (2009) Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep 9:198–205
Vance C, Rogelj B, Hortobagyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Wang IF, Wu LS, Chang HY, Shen CK (2008) TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem 105:797–806
Wilson CM, Grace GM, Munoz DG, He BP, Strong MJ (2001) Cognitive impairment in sporadic ALS: a pathologic continuum underlying a multisystem disorder. Neurology 57:651–657
Yang L, Embree LJ, Tsai S, Hickstein DD (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273:27761–27764
Yokota O, Tsuchiya K, Terada S et al (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115:561–575
Zinszner H, Sok J, Immanuel D, Yin Y, Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110(Pt 15):1741–1750
Acknowledgments
This work was supported by grants from Canadian Institutes of Health Research (DM, IM); the Pacific Alzheimer Research Foundation (IM); the Deutsche Forschungsgemeinschaft (MN); the Stavros-Niarchos Foundation (MN); and the Synapsis Foundation (MN). We thank Margaret Luk, Mareike Schroff, Nahid Nelson and Vidya Beharry for their excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Munoz, D.G., Neumann, M., Kusaka, H. et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118, 617–627 (2009). https://doi.org/10.1007/s00401-009-0598-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0598-9