
Abstract
An improved method of Agrobacterium-mediated transformation of cowpea was developed employing both sonication and vacuum infiltration treatments. 4 day-old cotyledonary nodes were used as explants for co-cultivation with Agrobacterium tumefaciens strain EHA105 harbouring the binary vector pSouv-cry1Ac. Among the different injury treatments, vacuum infiltration and their combination treatments tested, sonication for 20 s followed by vacuum infiltration for 5 min with A. tumefaciens resulted in highest transient GUS expression efficiency (93% explants expressing GUS at regenerating sites). After 3 days of co-cultivation, the explants were cultured in 150 mg/l kanamycin-containing selection medium and putative transformed plants were recovered. The presence, integration and expression of nptII and cry1Ac genes in T0 transgenic plants were confirmed by polymerase chain reaction (PCR), genomic Southern and qualitative reverse transcription (RT)-PCR analysis. Western blot hybridization and enzyme-linked immunosorbent assay (ELISA) detected and demonstrated the accumulation of Cry1Ac protein in transgenic plants. The cry1Ac gene transmitted in a Mendelian fashion. The stable transformation efficiency increased by 88.4% using both sonication-assisted Agrobacterium-mediated transformation (SAAT) and vacuum infiltration than simple Agrobacterium-mediated transformation in cowpea.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cowpea (Vigna unguiculata L. Walp) is widely cultivated in Africa, India, Middle East and, South America mostly for dry grain and fodder (Ehlers and Hall 1997; Timko et al. 2007), and is a major source of high-quality dietary protein for millions of local poor people (Singh 2002; Diouf and Hilu 2005; Xu et al. 2010). Cowpea production is seriously affected by a number of biotic and abiotic constraints, of which notably insect pests and viral diseases cause substantial yield loss worldwide (Solleti et al. 2008a). Despite its economic importance, progress in genetic improvement of cowpea for insect pest and disease resistance through conventional breeding is slow primarily due to narrow genetic base and barriers in crossing with distant wild species (Gomathinayagam et al. 1998; Fang et al. 2007). Consequently, the transfer of insect pest and virus resistance genes by genetic transformation could potentially aid plant breeders in overcoming these constraints and accelerate the development of resistant cultivars for breeding programs. Furthermore, efficient genetic transformation system would provide a valuable tool for functional genomics studies of cowpea. Agrobacterium-mediated transformation has been extensively applied to many crop plants including grain legumes, because this method offers several advantages such as the defined integration of transgenes, potentially low copy number, and preferential integration into transcriptional active regions of the chromosome (Koncz et al. 1989; Hiei et al. 1994). Agrobacterium-mediated transformation of cotyledonary explants has led to the generation of stable transgenic plants in cowpea (Muthukumar et al. 1996; Popelka et al. 2006; Chaudhury et al. 2007; Solleti et al. 2008a, b). Cotyledonary explants are preferred for Agrobacterium-mediated transformation of cowpea as T-DNA delivery to axillary meristem followed by regeneration via adventitious bud formation minimizes the risks of chimeras and somaclonal variation (Tzfira et al. 1997). However, cowpea transformation still remains inefficient and consequently, production of transgenic cowpea is far from being a routine procedure due to poor transformation efficiency and low numbers of regenerated transgenic plants. Sonication-assisted Agrobacterium-mediated transformation (SAAT) (Joersbo and Brunstedt 1992; Trick and Finer 1998; Santare′m et al. 1998) and vacuum infiltration (Charity et al. 2002; Park et al. 2005; Paz et al. 2006) methods have been reported to enhance the efficiency of Agrobacterium-mediated transformation of recalcitrant plant species. Exposure of the explants to short periods of sonication in the presence of Agrobacterium carrying desired T-DNA vector is thought to produce large numbers of micro wounds across the tissue which permits the Agrobacterium to penetrate deeper and more completely throughout the tissue as compared to the natural infection obtained during co-cultivation (Trick and Finer 1997; Santare′m et al. 1998; Tang et al. 2001; Liu et al. 2005), thus enhancing the bacterial colonization and infection of the tissue. Performed scanning electron and light microscopy observations revealed that ultrasound treatment produces small and uniform fissures and channels throughout the plant tissue, which allows Agrobacterium access to internal plant tissue (Trick and Finer 1997). SAAT method has been successfully employed in improving transformation of a number of recalcitrant plants (Oliveira et al. 2009).
Agroinfiltration is an effective method in enabling the regenerating cells, often located a few cell layers beneath the surface of explants, rapid access to Agrobacterium and consequently increasing transient transgene expression in many recalcitrant plant species (Bechtold and Pelletier 1998; Tague and Mantis 2006). This method has been adapted for the successful transformation of number of recalcitrant plants (Subramanyam et al. 2011).
Although the benefits of sonication and vacuum infiltration during A. tumefaciens-mediated transformation methods are evident, no effort has been made to apply these methods to cowpea. In order to improve the Agrobacterium-mediated transformation in cowpea for routine generation of transgenic plants with candidate genes, we investigated the effect of sonication and vacuum infiltration on Agrobacterium-mediated transformation of cowpea cotyledonary node explants. Stable transgenic cowpea plants expressing cry1Ac were recovered using both SAAT and vacuum infiltration, which showed presence, integration, expression and inheritance of transgenes.
Materials and methods
Plant material and explant preparation
The mature seeds of cowpea cultivar Pusa Komal (IARI, New Delhi) were surface sterilized with 70% ethanol (v/v) for 30 s followed by 0.2% of mercuric chloride (w/v) for 5 min. The sterilized seeds were rinsed 5 times with sterile water and cultured on MSB5 medium [MS salts (Murashige and Skoog 1962) + B5 vitamins (Gamborg et al. 1968)] supplemented with 3% sucrose (w/v), 0.8% agar agar (w/v) and 10 μM TDZ. The cultures were incubated at 26 ± 2°C under 16 h-photoperiod regime provided by cool white fluorescent lamps (36 μmol m−2 s−1). Cotyledonary node explants (5–6 mm) were excised from 4-day-old seedlings by removing both the cotyledons, and decapitating epicotyls as close as possible and hypocotyls 3 mm below the nodal region, and used for transformation experiments.
Binary plasmid, bacterial strain and culture conditions
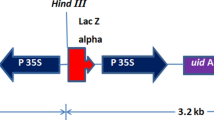
The binary plasmid pSouv:cry1Ac (Btcry1Ac expression cassette cloned in binary vector pCAMBIA2301) (Fig. 1) was mobilized into the disarmed hypervirulent Agrobacterium tumefaciens strain EHA105 and used for transformation experiments. The T-DNA of pCAMBIA2301 includes neomycin phosphotransferase gene (nptII) and β-glucuronidase gene (gus) interrupted by catalase intron, both driven by the cauliflower mosaic virus (CaMV) 35S promoter. The A. tumefaciens strain harboring pSouv:cry1Ac was maintained on solid YEP medium (An et al. 1988) supplemented with 10 mg/l of rifampicin, and 50 mg/l of kanamycin. Single bacterial colony was inoculated into 25 ml of liquid AB minimal medium (Chilton et al. 1974) with appropriate antibiotics and grown overnight at 28°C on a rotary shaker at 180 rpm, until optical density at 600 nm reached to 0.8. The cells were collected by centrifuging at 5,000 rpm for 5 min, and then the pellet was resuspended in liquid co-cultivation medium, LCM (MSB5 medium containing 1 μM BAP, pH adjusted to 5.5) supplemented with 100 μM acetosyringone and used for inoculation.

Schematic construction of pSouv:cry1Ac (14.5 kb). The 2.9 kb (EcoRI–HindIII) fragment containing Btcry1Ac under the control of CaMV 2X35S promoter and NOS terminator was cloned at the EcoRI–HindIII sites of T-DNA of pCAMBIA2301. LB and RB left border and right border of T-DNA region, NOS T nos terminator, 2X35SP double 35S promoter, nptII neomycin phosphotransferase II
Inoculation of explants with A. tumefaciens
For each experiment, 30–40 explants were subjected to wounding treatment either by mechanical injury with needle or by sonication, and inoculated in bacterial suspension by occasional shaking for 30 min or by vacuum infiltration. The explants inoculated in bacterial suspension without prior wounding treatment were considered as control. After inoculation in all cases, explants were blotted on a sterile filter paper to remove excess liquid and co-cultivated for 3 days under dark condition at 22°C, in petri dishes lined with filter paper moistened with LCM supplemented with 100 μM acetosyringone. Following co-cultivation, the explants were rinsed three to four times with LCM and blotted dry on sterile filter paper and placed onto initial multiple shoot induction and selection medium, SISM (MSB5 medium containing 5.0 μM BAP and 0.5 μM kinetin supplemented with 150 mg/l kanamycin and 500 mg/l cefotaxime) for 20 days with three rounds of subculture at an interval of 5, 7 and 8 days, respectively.
Wounding and SAAT treatments
The cotyledonary node explants were wounded at axils by puncturing approximately 1.5 mm in depth with a sterile hypodermic needle (0.56 mm in diameter.) prior to inoculation with Agrobacterium cell suspension.
For SAAT, the explants were immersed in 15 ml flat bottom glass culture tubes (Borosil, India) containing 6 ml of Agrobacterium cell suspension. The tubes were capped, placed in a float at the center of a bath-type sonicator (Telsonic ultrasonic TPC-40, Switzerland) and then subjected to ultrasound at a frequency of 30 kHz. The treatments differed as to sonication duration (5, 10, 15, 20, 25, and 30 s). Following sonication, explants were removed from the tubes, placed on sterile filter paper surface to blot off excess bacteria and then transferred to co-cultivation medium.
For vacuum infiltration experiment, the explants with or without wounding and 20 s sonication treatments were placed in vacuum system consisted of a vacuum pump at 600 mm Hg (Rocker 400, Tarson, India) to which a desiccator was attached. Glass petri dishes containing explants immersed in Agrobacterium cell suspension were placed in the desiccator and vacuum was applied for different durations (2.5, 5, 10, 15 and 20 min).
The best treatments achieved in SAAT and vacuum infiltration experiments were combined to evaluate the effect of sonication followed by vacuum infiltration in contrast to the use of these methods alone.
In all experiments, the frequency of transient GUS expression was analyzed after 3 days of co-cultivation. The optimal wounding, sonication and vacuum infiltration treatments were determined as the levels that led to a perceived increase in GUS positive foci in explants at the site of regeneration without any perceived decrease in explant viability. Control treatments consisted of explants either uninoculated or inoculated with Agrobacterium without wounding, sonication and vacuum infiltration treatments.
Histochemical GUS assays
GUS activity was visualized using the histochemical assay (Jefferson 1987). Transient expression was examined after 3 days of co-cultivation (Solleti et al. 2008a). The explants were bleached with 100% ethanol for 24 h prior to examination under a stereomicroscope. Transient expression of GUS was scored on a per explant basis by estimating the number of blue foci visible on the axillary region of each cotyledonary node explant. The blue foci were the discrete areas of cells with GUS activity.
Shoot recovery
Following three rounds of kanamycin selection on SISM, the survived explants were transferred to SIEM [shoot induction and elongation medium (MSB medium containing 5.0 μM BAP, 0.5 μM kinetin and 500 mg/l cefotaxime)] and cultured for 10 days for optimal elongation and selective regeneration of transformants. Elongated putative transformed shoots (>1.5 cm) were transferred to rooting medium (MS + 2.5 μM IBA) devoid of any antibiotics for root induction. Rooted putative transformed plants were transferred to pots containing sterile soil:compost (1:1) and were acclimatized in greenhouse containment for 3 weeks.
Evaluation of transgenic plants
Molecular characterization of the transformants was carried out by PCR, Southern hybridization, GUS histochemical analysis of different plant tissues, RT-PCR, ELISA and Western blot hybridization analysis for confirmation of the presence, integration, expression and inheritance of the introduced genes.
Stable GUS assay
Stable gus expression was detected in various plant parts including flower, anthers, pollens and pistils following the histochemical procedure as described previously.
Screening of putative transformed plants using polymerase chain reaction (PCR)
Genomic DNA was isolated from the young leaves of T0 putative transformants and T1 transgenic plants using the modified CTAB method (Solleti et al. 2008a). PCR amplification was carried out with gene specific primers for nptII and Btcry1Ac using genomic DNA from putative transformed plants, non-transformed control plants (negative control) and pSouv:cry1Ac (positive control) as templates. The 540 bp region of nptII and 1 kb coding region of Btcry1Ac were amplified using respective 20 mers (nptII Fw: CCACCATGATATTCGGCAAC; Rv: GTGGAGAGGCTATTCGGCTA) and 24 mers (Btcry1Ac Fw: CCCAGAAGTTGAAGTACTTGGTGG; Rv: CCGATATTGAAGGGTCTTCTGTAC) oligonucleotide primers. The amplification reaction was carried out under the following conditions: 94°C for 5 min (1 cycle), 94°C for 1 min (denaturation), 58°C for 1 min (annealing), 72°C for 1 min (extension) for 35 cycles followed by the final extension at 72°C for 7 min (1 cycle). PCR was performed using ~100 ng of purified genomic DNA and Taq DNA polymerase (Genei, Bangalore, India) according to manufacturer’s instruction. The amplified products were resolved by electrophoresis on 1% agarose gel and visualized by ethidium bromide staining (Sambrook et al. 1989).
Southern hybridization
Randomly selected PCR-positive T0 transgenic cowpea plants were further analyzed by Southern hybridization for the integration of the cry1Ac. 10 μg samples of genomic DNA from non-transformed control and transgenic plants were digested with HindIII. The digested samples were fractioned on a 0.8% agarose gel and transferred to Zeta-Probe membrane (Bio-Rad, USA). The blot was hybridized with DIG-labeled 1 kb PCR product, corresponding to the coding region of cry1Ac gene. The probe labeling and Southern hybridization were performed using the non-radioactive DIG Labeling and Detection system (Roche, Germany) following supplier’s instructions. Pre-hybridization and hybridization were carried out using high hybridization buffer containing 5XSSC, 1% blocking solution, 0.1% (w/v) N-lauroyl sarcosine and 0.02% (w/v) sodium dodecyl sulfate. Washing and detection were performed according to the instruction of the DIG labeling and detection system (Roche Diagnostics, Mannheim, Germany).
Qualitative reverse transcription (RT)-PCR analysis
Total RNA was isolated from the PCR-positive transgenic T0 plants using Trizol Reagent (Invitrogen, USA) from 100 ng of leaf tissue according to the manufacturer’s instructions. The integrity of RNA was verified by visualizing the RNA bands on 1.5% denaturing agarose gel (Sambrook et al. 1989). RT-PCR was carried out using First Strand cDNA Synthesis Kit (Fermentas, USA) according to the manufacturer’s instructions. PCR of the coding sequences of Btcry1Ac gene in the cDNA was carried out using respective primers as described earlier.
Western blot hybridization
Proteins were extracted from 1 g of young leaves of T0 transgenic plants using an extraction buffer containing 100 mM potassium phosphate buffer (pH 7.8), 1 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, 10% glycerol, 1 mM dithiothreitol (DTT). The protein concentration was determined by the method of Bradford (1976). 30 μg of protein was fractionated on 12% acrylamide gels with sodium dodecyl sulfate (SDS–PAGE) and blotted on to a PVDF membrane by electro transfer blotting unit. Blots were blocked for 2 h at room temperature in 5% blocking buffer (non-fat powdered milk in Tris-buffered saline with 0.1% Tween-20). Goat polyclonal antibodies (Amar Diagnostics, India) were used at 1/500 dilution in blocking buffer and incubated for overnight at 4°C. The samples were washed three times in TBST (tris-buffered saline tween-20) for 5 min each. A secondary rabbit anti-goat antibody alkaline phosphatase conjugate (Amar Diagnostics, India) was then used for final detection, at a dilution of 1/1,000. Blots were incubated for 40 min at 4°C, washed 5 times for 5 min each with TBST followed by development in nitro blue tetrazolium/bromo chloro indolyl phosphate (NBT/BCIP) substrate solution (Sigma, USA) for 15–20 min. The reaction was stopped by washing the membrane with distilled water.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed to quantify the accumulated levels of Cry1Ac protein in T0 transgenic plants using Desigen Quan T-ELISA-96 well plate kit (Desigen, Maharashtra, India) following manufacturer’s protocol. Total protein was extracted from 5 mg of dry leaf powder using 500 μl of sample extraction buffer. The sample was chilled and spun at 8,000 rpm for 15 min and 100 μl of supernatant was used for loading to anti-Cry1Ac pre-coated plate. For the estimation of Cry1Ac, the 96-well titre plate was coated with 150 μl per well (1:1,000) of goat anti-Cry1Ac antibodies. The plate was then loaded with 100 μl samples and buffer was used in control wells. The plate was incubated at 37°C for 1.5 h, followed by washing with wash buffer twice. After washing, the plate was incubated with alkaline phosphatase conjugated secondary antibodies at a dilution of 1:1,000 with 250 μl per well for 45 min at 37°C. The plate was then washed with wash buffer twice and 250 μl of freshly prepared substrate (p-nitro phenyl phosphate, 1 mg/ml) was added per well. The plate was incubated at room temperature in the dark for 30 min and reaction was stopped and readings recorded at 405 nm in a micro plate reader (Tecan, Switzerland).
Segregation analysis
The leaves of T1 transgenic plants generated from eight independent transformation events were analyzed for the presence of nptII and Btcry1Ac genes using PCR, as described earlier. Segregation patterns were analyzed with the Chi-square test (χ2) as described by Solleti et al. (2008b).
Data analysis
Data were subjected to analysis of variance (ANOVA) and mean separation by Duncan’s multiple-range test (DMRT) using single-factor completely randomized block design in order to study the effect of different treatments on frequencies of transient expression. All experiments were performed at least three times with a minimum of 30–40 explants per treatment.
Results and discussion
Effect of wounding
Efficient Agrobacterium-mediated transformation requires optimal delivery of the T-DNA to regenerable cells of the explants. Wounding of explants allows Agrobacterium to better access plant cells as it stimulates the production of potent vir gene inducers, like phenolic substances and enhances the plant cell competence for transformation (Stachel et al. 1985; Shimoda et al. 1990; Bidney et al. 1992). Only plants with an appropriate wound response develop larger populations of wound adjacent competent cells for regeneration and transformation (Potrykus 1991). Although excessive wounding is probably detrimental to stable transformation, the frequency of gene transfer via Agrobacterium-mediated transformation in recalcitrant species can be significantly enhanced by inducing wounds in the target tissue (Bidney et al. 1992). In cowpea, infection of cotyledonary node explants with most effective supervirulent A. tumefaciens strain EHA105, in absence of injury treatment resulted in 85% transient GUS expression frequency (Solleti et al. 2008a). However, the accounted GUS foci were located mostly at the cotyledons detachment site of cotyledonary node explants, and not at the regenerating site. The low stable transformation efficiency, 1.64% in cowpea (Solleti et al. 2008a) could be attributed to poor conversion of transient transformation to stable transformation. Wounding of regenerating sites of the cotyledonary node explants of cowpea by a hypodermic needle and co-cultivation with A. tumefaciens resulted in more efficient transient expression especially on needle-wounded explants, mainly in terms of the percentage of explants showing GUS foci at the regenerating sites as compared to unwounded explants infected with A. tumefaciens (Fig. 2). This clearly indicated that higher transient transformation of regenerating cells of meristematic tissue-based explants such as cotyledonary nodes, required an efficient wounding treatment. Wounding at the regenerating sites before co-cultivation allowed better bacterial penetration into the regenerating cells of cotyledonary node explants, facilitating the accessibility of plant cells for Agrobacterium infection. Such mechanical wounding treatments greatly enhanced transformation efficiency in a number of plant species including recalcitrant grain legumes (Roome 1992; Rohini et al. 2005; Supartana et al. 2006; Saini and Jaiwal 2007).

Effect of mechanical injury by hypodermic needle on transient transformation of cowpea cotyledonary nodes as evaluated with GUS assay. Control–injury is omitted in explants. The bars indicate ± standard errors. Means followed by the same letter are not statistically significant at P < 0.05
Effect of sonication and vacuum infiltration
To identify more efficient methods to improve access of Agrobacterium and also to create an area of wounding to induce cotyledonary node cells and to produce phenolic compounds for vir gene induction in cowpea, we evaluated the effect of sonication and vacuum infiltration on A. tumefaciens-mediated transformation of cotyledonary node explants. These treatments have the potential to increase transformation efficiency by improving penetration of Agrobacterium cells into the cell layers beneath the epidermis of cotyledonary node region. This is an important criterion as regenerating cells of cotyledonary node explants are positioned a few cells layers beneath the surface at the axils in Vigna species including cowpea, mungbean and blackgram (Sahoo and Jaiwal 2009). A control experiment with explants without inoculation with Agrobacterium was designed to determine whether these treatments could be used without a negative effect on shoot regeneration from cotyledonary node explant. Sonication was very effective in increasing transient GUS expression frequency (Figs. 3, 4a). With the increase in sonication treatment time, the number of transiently transformed explants increased significantly with a maximum of 79% of the explants showing GUS foci at the regenerating sites when sonication treatment was prolonged to 20 s (Figs. 3, 4a). The number of GUS foci appeared to be quite variable among cotyledonary node explants (data not shown). At lower sonication treatment time (10–20 s), the GUS foci were well defined, corresponding to probably one or a collection of small individual spots (Fig. 3a–f). With the increase in sonication treatment time beyond 20 s, a diffuse GUS expression was presented all over the surface of the cotyledonary node explants, making the quantification of the number of foci difficult (Fig. 3g–h). Moreover, with increase in sonication treatment time to 30 s, the untransformed explants showed a decrease in their bud-forming capacity indicating that longer sonication treatment compromised viability of regenerating cells (data not shown). SAAT has been used to enhance stable transformation of many recalcitrant plant species including soybean (Trick and Finer 1998), loblolly pine (Tang 2003), black locust (Zaragoza’ et al. 2004), sweetpotato (Wang et al. 2006), rice (Yookongkaew et al. 2007), Chenopodium rubrum (Flores Solı′s et al. 2007), chickpea (Pathak and Hamzah 2008), flax (Beranova′ et al. 2008) and Theobroma cacao (Silva et al. 2009).

Transient expression of GUS at the regenerating sites of sonication-treated cotyledonary node explants after 3 days of co-culture. a Control (untransformed). b Agrobacterium-treated explants (without sonication treatment). c–h sonication-treated cotyledonary nodes (c 5 s, d 10 s, e 15 s, f 20 s, g 25 s and h 30 s). Bar (in all figures) 1 mm

a Effect of SAAT treatment duration and b vacuum infiltration treatment duration on transient transformation of cowpea cotyledonary nodes as evaluated with GUS assay. c Effect of different wounding methods on transient transformation of cowpea cotyledonary nodes as evaluated with GUS assay. C Without wounding. I Injury treatment by hypodermic needle. S 20 s sonication treatment. V Vacuum infiltration treatment for 5 min. SV 20 s sonication followed by vacuum infiltration treatment for 5 min. The bars indicate ± standard errors. Means followed by the same letter are not statistically significant at P < 0.05
We attempted various time intervals of vacuum infiltration of explants at 600 mmHg in an Agrobacterium suspension, and of the different time intervals tested, a 5 min vacuum infiltration resulted in a maximum of 93% transient transformation efficiency as accounted on the basis of number of explants showing GUS foci at the regenerating sites (Fig. 4b). Vacuum infiltration of cotyledon explants of Pinus radiata in an Agrobacterium suspension has allowed Agrobacterium to penetrate several layers deep through the sub-epidermal layer to mesophyll cells and vascular tissues (Charity et al. 2002), although the cells buried several layers deep, were not necessarily those that would induce shoots (Yeung et al. 1981). The vacuum infiltration of Agrobacterium has been successfully used to produce transgenic plants of model plant Arabidopsis (Clough and Bent 1998), and recalcitrant crop species including wheat (Cheng et al. 1997), mungbean (Jaiwal et al. 2001), pinus (Charity et al. 2002), cotton (Leelavathi et al. 2004), kidney bean (Liu et al. 2005), coffee (Canche-Moo et al. 2006), chickpea (Indurker et al. 2010) and banana (Subramanyam et al. 2011).
Combined treatment of sonication and vacuum infiltration
In order to evaluate the combined action of sonication and vacuum infiltration on transient transformation, the effect of 20 s sonication and 5 min vacuum infiltration was tested as compared to the two treatments separately. The combination of 20 s sonication followed by 5 min vacuum infiltration resulted in maximum frequency of cotyledonary node explants expressing GUS at the regenerating sites (Fig. 4c). Sonication coupled with vacuum infiltration has increased transient and stable transformation of radish (Park et al. 2005), kidney bean (Liu et al. 2005), citrus (Oliveira et al. 2009), Fraxinus pennsylvanica (Du and Pijut 2009), chickpea (Indurker et al. 2010) and banana (Subramanyam et al. 2011).
Production of transgenic cowpea plants carrying cry1Ac gene
Putative transformed plants were regenerated from cotyledonary node explants, which were subjected to a combination of 20 s sonication followed by 5 min vacuum infiltration prior to Agrobacterium co-cultivation, on kanamycin selection medium and established in greenhouse containment (Fig. 5a–g). A strong, uniform and stable gus expression was detected in flower, anthers, pollens and pistils of PCR-positive T0 plants and no endogenous gus expression was detected in the tissues of control plants (Fig. 5h–o).

Transient and stable gus expression and regeneration of transgenic plants. a cotyledonary node explants. Bar 2 mm. (b and c) Transient GUS expression, non-transformed (control) explants not showing GUS activity (b), cotyledonary node explants showing transient GUS activity after 3 days of co-cultivation (c). Bar 4 mm. d Shoot induction from axils of explant after 5-day culture on SISM. Bar 2 mm. e Proliferation of multiple shoots within 4 weeks of culture. Bar 10 mm. f In vitro rooting of elongated transformed shoot. Bar 12 mm. g Acclimatized plant maintained in transgenic green house. Bar 10 cm. h Non-transformed control flower. Bar 7 mm. i Transformed flower. Bar 7 mm. j Control anthers. Bar 8 mm. k Transformed anthers. Bar 8 mm. l Control pistil. Bar 8 mm. m Transformed pistil. Bar 8 mm. n Control pollens. Bar 3 mm. o Transformed pollens. Bar 3 mm
Analysis of transgenic cowpea plants
The detection of the expected 540 bp and 1 kb amplified products corresponding to nptII and cry1Ac in PCR analysis confirmed the presence of the transgenes in T0 transformed plants (Fig. 6a, b). No amplification was detected in the control untransformed plants.

Molecular analysis of T0 transgenic plants. a PCR amplification of the 1 kb fragment of the cry1Ac gene, b PCR amplification of the 540 bp fragment of the nptII gene. Lane M λ DNA/EcoRI + HindIII marker, lane P pSouv:cry1Ac plasmid DNA (positive control), lane C DNA from untransformed plant (negative control), lane B blank, lanes 1–7 DNA from independently transformed plants. c Southern blot hybridization analysis of junction fragments of four randomly selected PCR-positive T0 lines. The plasmid and genomic DNA were digested with HindIII, and hybridized with cry1Ac probe. Lanes 1–4 genomic DNA from four T0 lines, lane C genomic DNA from untransformed plant, lane P cry1Ac PCR amplicon. d RT-PCR analysis of cry1Ac gene, e RT-PCR analysis of nptII gene. Lane M λ DNA/EcoRI + HindIII marker; lane C untransformed plant (negative control); lane B blank; lanes 1–8 T0 transgenic plants
Four randomly selected PCR-positive T0 transgenic cowpea plants were further screened by Southern analysis to confirm the integration of cry1Ac gene. Southern blot analyses of four T0 transgenic plants are shown in Fig. 6c. Hybridizations of DIG-labeled cry1Ac probe to total genomic DNA digested with HindIII were expected to identify DNA fragments unique to individual integration events greater than 5.0 kb (Fig. 1). All the four randomly selected T0 transgenic plants were found positive for cry1Ac gene and furthermore, they showed differential integration events, confirming that these plants were derived from independent transformation events (Fig. 6c, lanes 1, 2, 3 and 4). The T0 transgenic plants exhibited simple hybridization patterns that ranged from single integration event to three loci and, in general, most fragments were greater than 5.0 kb (Fig. 6c). A signal of size less than 5.0 kb was detected in lane 3 (Fig. 6c), suggesting the possibility of rearrangement of the T-DNA near the left border upon integration into the plant genome. No hybridization signal was detected in the untransformed plant (Fig. 6c, lane C).
The expression of the cry1Ac genes in leaves of T0 transgenic plants was determined by RT-PCR analysis. RT-PCR showed the presence of expected transcripts of transgenes in different T0 transgenic plants. The amplification of a 1 kb fragment of cry1Ac confirmed the accumulation of transcripts of cry1Ac in T0 transgenic plants (Fig. 6d, e) indicating the absence of gene silencing events. Furthermore, the amplification of the cry1Ac sequence from plant cDNA templates in RT-PCR ruled out the possibility of Agrobacterium contamination.
The stable transformation efficiency was determined based on the number of T0 plants PCR-positive for Btcry1Ac and nptII divided by the total number of explants co-cultivated. An average stable transformation efficiency of 3.09 was recorded (Table 1), which was significantly higher than the previously published report on Agrobacterium-mediated transformation of cowpea using extra copies of vir genes (Solleti et al. 2008a).
Cry1Ac expression analysis
The randomly chosen PCR-positive T0 transgenic lines were subjected to Cry1Ac protein expression analysis by Western hybridization and ELISA. The expression of the Cry1Ac protein was analyzed in T0 transgenic lines generated from four independent transformation events by Western blot hybridization. A single band of 68 kDa corresponding to Cry1Ac toxin protein was detected immunologically in T0 transgenic plants confirming stability of cry1Ac expression. Protein extracts of control non-transformed plants did not show the 68 kDa protein band (Fig. 7a).

a Detection of Cry1Ac protein by Western blotting analysis in transgenic cowpea leaves. M Protein molecular weight marker, lanes 1–4 cry1Ac transgenic lines (CT1A, CT1B, CT1C and CT1D), respectively, lane 5 non-transformed plant. b Expression level of BtCry1Ac protein in transgenic cowpea lines (CT1A, CT1B, CT1C and CT1D) from enzyme-linked immunesorbent assay (ELISA). Error bars represent ± standard error of the means. Means followed by the same letter are not statistically significant at P < 0.05
The level of expression of Cry1Ac protein in transgenic lines ranged from 0.001 to 0.089% of the total leaf soluble protein (Fig. 7b). The results described above demonstrated that expression of the cry1Ac regulated by the double 35S-promoter led to the accumulation of Cry1Ac protein in transgenic plants.
Segregation analysis
The seeds from T0 generation were advanced to T1 generation and the T1 transgenic lines generated from eight independent transformation events were analyzed for the segregation pattern of cry1Ac by PCR analysis. Presence of the expected 1 kb amplified product corresponding to cry1Ac in T1 transgenic lines confirmed the inheritance of cry1Ac gene (Fig. 8). The segregation pattern of these selected transgenic events showed typical 3:1 Mendelian ratio as expected for single dominant gene inheritance (Table 2).

PCR amplification of the 1 kb fragment of the cry1Ac gene of T1 plants. Lane M λ DNA/EcoRI + HindIII marker, lane P pSouv:cry1Ac plasmid DNA (positive control), lane C DNA from untransformed plant (negative control), lane B blank, lanes 1–5 DNA from T1 transgenic plants
In conclusion, an improved Agrobacterium-mediated transformation system was developed for cowpea by employing sonication and vacuum infiltration was enhanced by 88.4% using SAAT in combination with vacuum infiltration as compared to simple Agrobacterium-mediated transformation. This is the first report on cowpea transformation using SAAT and vacuum infiltration. Furthermore, cowpea transgenics expressing cry1Ac is reported for the first time.
Abbreviations
- BAP:
-
6-Benzylaminopurine
- TDZ:
-
Thidiazuron
- GUS:
-
β-Glucuronidase
- nptII :
-
Neomycin phosphotransferase II
- SAAT:
-
Sonication-assisted Agrobacterium-mediated transformation
References
An G, Ebert PR, Mitra A, Ha SB (1988) Binary vectors. In: Gelvin SB, Schilperoot RA, Verma DPS (eds) Plant molecular biology manual, vol A3. Kluwer, The Netherlands, pp 1–19
Bechtold N, Pelletier G (1998) In planta Agrobacterium-mediated transformation of adult Arabidopsis thaliana plants by vacuum infiltration. Methods Mol Biol 82:259–266. doi:10.1385/0-89603-391-0:259
Beranova′ M, Rakousky′ S, Va′vrova′ Z, Skalicky′ T (2008) Sonication assisted Agrobacterium-mediated transformation enhances the transformation efficiency in flax (Linum usitatissimum L.). Plant Cell Tiss Organ Cult 94:253–259. doi:10.1007/s11240-007-9335-z
Bidney D, Scelonge C, Martich J, Burrus M, Sims L, Huffman G (1992) Micro projectile bombardment of plant tissues increases transformation frequency by Agrobacterium tumefaciens. Plant Mol Biol 18:301–313. doi:10.1007/BF00034957
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein using the principle of protein-dye binding. Anal Biochem 72:248–254
Canche-Moo RLR, Ku-Gonzalez A, Burgeff C, Loyola-Vargas VM, Rodrı′guez-Zapata LC, Castan˜o E (2006) Genetic transformation of Coffea canephora by vacuum infiltration. Plant Cell Tiss Org Cult. doi:10.1007/s11240-005-9036-4
Charity JA, Holland L, Donaldson SS, Grace L, Walter C (2002) Agrobacterium mediated transformation of Pinus radiata organogenic tissue using vacuum infiltration. Plant Cell Tissue Organ Cult 70:51–60. doi:10.1023/A:1016009309176
Chaudhury D, Madanpotra S, Jaiwal R, Saini R, Kumar PA, Jaiwal PK (2007) Agrobacterium tumefaciens-mediated high frequency genetic transformation of an Indian cowpea (Vigna unguiculata L. Walp.) cultivar and transmission of transgenes into progeny. Plant Sci 172:692–700. doi:10.1016/j.plantsci.2006.11.009
Cheng M, Fry JE, Zhou H, Hironaka CM, Duncan DR, Coner TW, Wan Y (1997) Genetic transformation of wheat mediated by Agrobacterium tumefaciens. Plant Physiol 115:971–980
Chilton MD, Currier TC, Farrand SK, Bendich AJ, Gordon MP, Nester EW (1974) Agrobacterium tumefaciens DNA and PS8 bacteriophage DNA not detected in crown gall tumors. Proc Natl Acad Sci USA 71:3672–3676
Clough J, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743. doi:10.1046/j.1365-313x.1998.00343.x
Diouf D, Hilu KW (2005) Microsatellite and RAPD markers to study genetic relationships among cowpea breeding lines and local varieties in Senegal. Genet Resour Crop Evol 52:1957–1967
Du N, Pijut PM (2009) Agrobacterium-mediated transformation of Fraxinus pennsylvanica hypocotyls and plant regeneration. Plant Cell Rep 28:915–923. doi:10.1007/s00299-009-0697-z
Ehlers JD, Hall AE (1997) Cowpea (Vigna unguiculata L. Walp). Field Crops Res 53:187–204
Fang J, Chao CT, Roberts PA, Ehlers JD (2007) Genetic diversity of cowpea [Vigna unguiculata (L.) Walp.] in four West African and USA breeding programs as determined by AFLP analysis. Genet Resour Crop Evol 54:1197–1209. doi:10.1007/s10722-006-9101-9
Flores-Solı′s JI, Mlejnek P, Studena′ K, Procha′zka S (2007) Application of sonication-assisted Agrobacterium-mediated transformation in Chenopodium rubrum L. Plant Soil Environ 49:255–260
Gamborg OL, Miller RA, Ojima K (1968) Nutrient requirements of suspension cultures of soybean root cell. Exp Cell Res 50:151–158. doi:10.1016/0014-4827(68)90403-5
Gomathinayagam P, Ganeshram S, Rathnaswamy R, Ramaswamy NM (1998) Interspecific hybridization between Vigna unguiculata (L.) Walp. and V. vexillata (L.) A. Rich. through in vitro embryo culture. Euphytica 102:203–209. doi:10.1023/A:1018381614098
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6(2):271–282. doi:10.1046/j.1365-313X.1994.6020271.x
Indurker S, Misra HS, Eapen S (2010) Agrobcterium-mediated transformation in chickpea (Cicer arietinum L.) with an insecticidal protein gene: optimization of different factors. Physiol Mol Biol Plants 16(3):273–284. doi:10.1007/s12298-010-0030-x
Jaiwal PK, Kumari R, Ignacimuthu S, Potrykus I, Sautter C (2001) Agrobacterium tumefaciens-mediated transformation of mungbean (Vigna radiata)- a recalcitrant grain legume. Plant Sci 161:239–247. doi:10.1016/S0168-9452(01)00352-1
Jefferson RA (1987) Assaying chimearic genes in plants: the GUS gene fusion system. Plant Mol Biol 204:387–405
Joersbo M, Brunstedt J (1992) Sonication: a new method for gene transfer to plants Physiol. Plant. 85(2):230–234. doi:10.1111/j.1399-3054.1992.tb04727.x
Koncz C, Martini N, Mayerhofer R, Koncz-Kalman Z, Korber H, Redei GP, Schell J (1989) High frequency T-DNA- mediated gene tagging in plants. Proc Natl Acad Sci USA 86(21):8467–8471
Leelavathi S, Sunnichan SG, Kumria R, Vijaykanth GP, Bhatnagar RK, Reddy VS (2004) A simple and rapid Agrobacterium-mediated transformation protocol for cotton (Gossypium hirsutum L.): Embryogenic calli as a source to generate large numbers of transgenic plants. Plant Cell Rep 22:465–470. doi:10.1007/s00299-003-0710-x
Liu Z, Park BJ, Kanno A, Kameya T (2005) The novel use of a combination of sonication and vacuum infiltration in Agrobacterium-mediated transformation of kidney bean (Phaseolus vulgaris L.) with lea gene. Mol Breed 16:189–197. doi:10.1007/s11032-005-6616-2
Murashige T, Skoog S (1962) A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol Plant 15:473–497
Muthukumar B, Mariamma M, Veluthambi K, Gnanam A (1996) Genetic transformation of cotyledon explants of cowpea (Vigna unguiculata L. Walp) using Agrobacterium tumefaciens. Plant Cell Rep 15:980–985. doi:10.1007/BF00231600
Oliveira MLP, Febres VJ, Costa MGC, Moore GA, Otoni WC (2009) High-efficiency Agrobacterium-mediated transformation of citrus via sonication and vacuum infiltration. Plant Cell Rep 28:387–395. doi:10.1007/s00299-008-0646-2
Park BJ, Liu Z, Kanno A, Kameya T (2005) Transformation of radish (Raphanus sativus L.) via sonication and vacuum infiltration of germinated seeds with Agrobacterium harboring a group 3LEA gene from B. napus. Plant Cell Rep 24:494–500. doi:10.1007/s00299-005-0973-5
Pathak MR, Hamzah RY (2008) An effective method of sonication-assisted Agrobacterium-mediated transformation of chickpeas. Plant Cell Tiss Organ Cult 93:65–71. doi:10.1007/s11240-008-9344-6
Paz MM, Martinez JC, Kalvig AB, Fonger TM, Wang K (2006) Improved cotyledonary node method using an alternative explant derived from mature seed for efficient Agrobacterium-mediated soybean transformation. Plant Cell Rep 25(3):206–213. doi:10.1007/s00299-005-0048-7
Popelka JC, Gollasch S, Moore A, Molvig L, Higgins TJV (2006) Genetic transformation of cowpea (Vigna unguiculata L.) and stable transmission of the transgenes to progeny. Plant Cell Rep 25:304–312. doi:10.1007/s00299-005-0053-x
Potrykus I (1991) Gene transfer to plants: Assessment of Published Approaches and Results. Annu. Rev. Plant Physiol. Plant Mol. Biol. 42:205–225
Rohini VK, Manjunath NH, Rao KS (2005) Studies to develop systems to mobilize foreign genes into parasitic flowering plants. Physiol Mol Biol Plants 11(1):111–120
Roome WJ (1992) Agrobacterium-mediated transformation of two forest tree species Prunus serotina and Fraxinus pennsylvanica. MS Thesis, State University of New York, College of Environmental Science and Forestry
Sahoo L, Jaiwal PK (2009) Asiatic Beans. In: Kole C, Hall TC (eds) Compendium of Transgenic Crop Plants. Wiley, New York, pp 115–132 doi:10.1002/9781405181099.k0307
Saini R, Jaiwal PK (2007) Agrobacterium tumefaciens - mediated transformation of blackgram: an assessment of factors influencing the efficiency of uidA gene transfer. Biol Plantarum 51(1):69–74. doi:10.1007/s10535-007-0014-z
Sambrook KJ, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York
Santare′m ER, Trick HN, Essing JS, Finer JJ (1998) Sonication assisted Agrobacterium-mediated transformation of soybean immature cotyledons: optimization of transient expression. Plant Cell Rep 17:752–759
Shimoda N, Toyoda-Yamamoto A, Nagamine J, Usami S, Katayama M, Sakagami Y, Machida Y (1990) Control of expression of Agrobacterium vir genes by synergistic action of phenolic signal molecules and monosaccharides. Proc Natl Acad Sci USA 87:6684–6688. doi:10.1073/pnas.87.17.6684
Silva TER, Cidade LC, Alvim FC, Cascardo JCM, Costa MGC (2009) Studies on genetic transformation of Theobroma cacao L.: evaluation of different polyamines and antibiotics on somatic embryogenesis and the efficiency of uidA gene transfer by Agrobacterium tumefaciens. Plant Cell Tiss Organ Cult 99:287–298. doi:10.1007/s11240-009-9603-1
Singh BB (2002) Recent genetic studies in cowpea. In: Fatokun CA, Tarawali SA, Singh BB, Kormawa PM, Tamo M (eds) Challenges and opportunities for enhancing sustainable cowpea production. International Institute of Tropical Agriculture, Ibadan, pp 3–13
Solleti SK, Bakshi S, Purkayastha J, Panda SK, Sahoo L (2008a) Transgenic cowpea (Vigna unguiculata) seeds expressing a bean α-amylase inhibitor 1 confer resistance to storage pests, bruchid beetles. Plant Cell Rep 27:1841–1850. doi:10.1007/s00299-008-0606-x
Solleti SK, Bakshi S, Sahoo L (2008b) Additional virulence genes in conjunction with efficient selection scheme, and compatible culture regime enhance recovery of stable transgenic plants in cowpea via Agrobacterium tumefaciens-mediated transformation. J Biotech 135:97–104. doi:10.1016/j.jbiotec.2008.02.008
Stachel SE, Messens E, Van Montagu M, Zambryski P (1985) Identification of the signal molecules produced by wounded plant cells which activate the T-DNA transfer process in Agrobacterium tumefaciens. Nature 318:624–629. doi:10.1038/318624a0
Subramanyam K, Subramanyam K, Sailaja KV, Srinivasulu M, Lakshmidevi K (2011) Highly efficient Agrobacterium-mediated transformation of banana cv. Rasthali (AAB) via sonication and vacuum infiltration. Plant Cell Rep 30(3):425–436. doi:10.1007/s00299-010-0996-4
Supartana P, Shimizu T, Nogawa M, Shioiri H, Nakajima T, Haramoto N, Nozue M, Kojima M (2006) Development of simple and efficient in planta transformation method for wheat (Triticum aestivum L.) using Agrobacterium tumefaciens. J Biosci Bioeng 102(3):162–170. doi:10.1263/jbb.102.162
Tague BW, Mantis J (2006) In planta Agrobacterium-mediated transformation by vacuum infiltration. Methods Mol Biol 323:215–223. doi:10.1385/1-59745-003-0:215
Tang W (2003) Additional virulence genes and sonication enhance Agrobacterium tumefaciens-mediated loblolly pine transformation. Plant Cell Rep 21:555–562. doi:10.1007/s00299-002-0550-0
Tang W, Sederoff R, Whetten R (2001) Regeneration of transgenic loblolly pine (Pinus taeda L.) from zygotic embryos transformed with Agrobacterium tumefaciens. Planta 213:981–989. doi:10.1007/s004250100566
Timko MP, Ehlers JD, Roberts PA (2007) Cowpea. In: Kole C (ed) Pulses, sugar and tuber crops. Theoretical and applied genetics, genome mapping and molecular breeding in plants, vol 3. Springer, Berlin, pp 49–67
Trick H, Finer JJ (1997) SAAT:sonication-assisted Agrobacterium-mediated transformation. Transgenic Res 6:329–336. doi:10.1023/A:1018470930944
Trick HN, Finer JJ (1998) Sonication-assisted Agrobacterium-mediated transformation of soybean [Glycine max (L.) Merrill] embryogenic suspension culture tissue. Plant Cell Rep 17:482–488. doi:10.1007/s002990050429
Tzfira T, Jensen CS, Wang W, Zuker A, Vinocur B, Altman A, Vainstein A (1997) Transgenic Populus tremula: a step-by-step protocol for its Agrobacterium-mediated transformation. Plant Mol Biol Rep 15:219–235. doi:10.1023/A:1007484917759
Wang X, Zhou Z, Li Q, Gu XH, Ma DF (2006) Optimization of genetic transformation using SAAT in sweetpotato. Jiangsu J Agric Sci 22(1):14–18
Xu P, Wu X, Wang B, Liu Y, Qin D, Ehlers JD, Close TJ, Hu T, Lu Z, Li G (2010) Development and polymorphism of Vigna unguiculata ssp. Unguiculata microsatellite markers used for phylogenetic analysis in asparagus bean (Vigna unguiculata ssp. sesquipedialis (L.) Verdc.). Mol breeding 25:675–684. doi:10.1007/s11032-009-9364-x
Yeung EC, Aitken J, Biondi S, Thorpe TA (1981) Shoot histogenesis in cotyledon explants of radiata pine. Botan. Gaz. 142:494–501
Yookongkaew N, Srivatanakul M, Narangajavana J (2007) Development of genotype-independent regeneration system for transformation of rice (Oryza sativa ssp. indica). J Plant Res 120(2):237–245. doi:10.1007/s10265-006-0046-z
Zaragoza’ C, Munoz-Bertomeu J, Arrillaga I (2004) Regeneration of herbicide-tolerant black locust transgenic plants by SAAT. Plant Cell Rep 22:832–838. doi:10.1007/s00299-004-0766-2
Acknowledgments
We thank Prof. I. Altossar, University of Ottawa, Canada for providing p1AcPRD, Prof. K. Veluthambi, MKU, Madurai, India for Agrobacterium strain and Center for Application of Molecular Biology to International Agriculture (CAMBIA), Australia for pCAMBIA2301. The research was supported by the grants from Department of Biotechnology, Government of India. Souvika Bakshi is grateful to Council of Scientific and Industrial Research (CSIR) for Senior Research Fellowship, and Ayan Sadhukhan and Sagarika Mishra to Indian Institute of Technology Guwahati for doctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Toriyama.
Rights and permissions
About this article
Cite this article
Bakshi, S., Sadhukhan, A., Mishra, S. et al. Improved Agrobacterium-mediated transformation of cowpea via sonication and vacuum infiltration. Plant Cell Rep 30, 2281–2292 (2011). https://doi.org/10.1007/s00299-011-1133-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-011-1133-8