
Abstract
Rice thaumatin-like protein (Rtlp1) is a high-molecular-weight antimicrobial pathogenesis-related protein that plays a role in plant stress response. This study examines transcriptional regulation of Rtlp1 using wild type and transgenic rice plants carrying a β-glucuronidase (GUS) reporter gene driven by the Rtlp1 promoter (pRtlp1GUS). The Rtlp1 promoter is induced within 6 h after infection with rice blast fungus (Magnaporthe grisea). The Rtlp1 promoter is also induced by salicylic acid (SA), methyl jasmonate (MeJA), wounding or an elicitor from rice blast fungus. The function of the pRtlp1GUS reporter gene was analyzed by deletion mapping and transient expression assays in cell culture. A 120 bp truncated fusion construct with six W-boxes (5′-TGAC-3′) demonstrated a strong dose-dependent elicitor-response. These results suggest that W-box elements are required for the response of the Rtlp1 promoter to fungal elicitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Thaumatin-like proteins (TLP) are high-molecular-weight antimicrobial proteins closely related to osmotin that are classified as class 5 pathogenesis-related proteins (PR-5). TLP may exert its antifungal activity by permeabilizing the fungal cell membrane (Vigers et al. 1991, 1992; Malehorn et al. 1994; Abad et al. 1996). Yun et al. (1998) suggested that expression of tobacco osmotin in yeast kills microbes rapidly by subverting signal transduction during infection. Datta et al. (1999) reported that transgenic rice overexpressing Tlp exhibited enhanced resistance to Rhizoctonia solani, which causes rice sheath blight disease. Rice plants overexpressing Tlp also have enhanced resistance to rice blast (Magnaporthe grisea) (data not shown).
The signal transduction pathways involved in plant pathogen defense are complex. Pathogenesis-related (PR) proteins are key elements in pathogen defense, and it is important to understand mechanisms that regulate PR gene expression. PR gene expression has been studied in monocots including maize, rice and wheat; for example, Northern blot analysis has been carried out on maize proteinase inhibitor (Cordero et al. 1994), rice chitinase (Xu et al. 1996; Nishizawa et al. 1999), wheat lipoxygenase (Mauch et al. 1997), and rice PR1 (Agrawal et al. 2000). However, the function of promoters of rice PR-genes have not been characterized in detail. This study presents characterization of rice Tlp1 expression in wild type and transgenic rice expressing an Rtlp1 promoter—β-glucuronidase (GUS) fusion reporter gene. Deletion analysis was used to identify functionally important regions in the Rtlp1 promoter and promoter activity was studied in transgenic plants challenged with rice blast fungus.
Materials and methods
Plant materials
Genomic DNA and total RNA were isolated from roots and leaves of 14 day-old rice seedlings (Oryza sativa L. cv. Sasanishiki) grown in a greenhouse. Cells from scutellum of mature rice seed were cultured in liquid AA medium (Toriyama and Hinata 1985) containing 3% sucrose and 1 mg/l 2,4-dichlorophenoxy acid (2,4-d) at 25°C. These cells were used to generate transgenic plants.
cDNA library construction and isolation of Rtlp1 cDNA
Total RNA was prepared 4 days after subculture from cultured cells grown in suspension according to the method of Sambrook et al. (1989) with slight modifications. Poly(A) + RNA was purified using an oligo(dT)-binding latex particle, and cDNA was synthesized with either cDNA Synthesis System Plus (Amersham) or ZAP-cDNA Synthesis (Stratagene). cDNA was cloned by ligating double-stranded cDNA to linearized pBluescript SK-(Stratagene) or by in vivo excision from a cDNA library in ZAP II phage vector. Randomly selected cDNA clones were isolated using the alkali lysis method, and partial nucleotide sequences of cDNA inserts were determined using a Model 370A DNA Autosequencer (Applied Biosystems). The GenBank DNA database was searched for homologous DNA sequences using Blastn. A total of 2,000 clones were sequenced. One of them contained the full-length cDNA of Rtlp1 (GenBank no. X68197)
Isolation of total RNA and northern blot analysis
Total RNA was isolated from root, leaf and callus tissue using guanidine-thiocianate/phenol extraction (Sambrook et al. 1989). For northern blot analysis, 20 μg total RNA was separated by electrophoresis in a formaldehyde/1.5% agarose gel, and transferred to a nylon membrane (Hybond N+, Amersham). Filters were hybridized with Rtlp1 cDNA 32P-labeled using the Random Primer DNA Labeling System (Amersham). Filters were visualized and hybridization signal quantified using a BAS-2000 phosphor image analyzer (Fuji).
Isolation of Rtlp1 promoter using modified inverse PCR
Genomic DNA was isolated from rice leaves by urea-phenol extraction (Shure et al. 1983). Inverse PCR (Ochman et al. 1988) was used with modifications to isolate the 5′-flanking region of the Rtlp1 gene. One μg genomic DNA was double-digested with restriction enzymes, protruding ends were converted to blunt-ends and the DNA fragments were circularized by self-ligation for 16 h at 16°C in 400 μL containing 66 mM Tris–HCl (pH7.6), 6.6 mM MgCl2, 10 mM dithiothreitol, 0.1 mM ATP and 10 U T4 DNA ligase. Samples were extracted with phenol/chloroform and precipitated with ethanol, resuspended in buffer and used as a PCR template. The 5′-flanking region of Rtlp1 was amplified using primer pairs oriented in opposite directions in the cDNA sequence. PCR was carried out in a 100 μl reaction volume containing 20 μM primers (5′-AGTAAATTGTTAATGGCGTCTCCGGCCACC-3′, 5′-TGAACGAGCACCGGTTGGTGATGG TGAAGG-3′), 250 μM dNTPs, 10 mM Tris–HCl (pH 9.0), 50 mM KCl, 0.1% Triton X-100, 1.5 mM MgCl2 and 2.5 U Ex Taq polymerase (Takara). Thermal cycling conditions were 94°C for 2 min, followed by 30 cycles of 94°C/60 s, 62°C/90 s, 72°C/120 s, and final extension at 72°C for 7 min. A 1.3 kb PCR fragment was amplified from genomic DNA, double-digested with PvuII and DraI and self-ligated. This fragment contained the Rtlp1 5′-flanking region, and was cloned into the pT7Blue vector (Invitrogen). Nucleotide sequence of the DNA insert was determined using a Model 377A DNA Autosequencer (Applied Biosystems).
Construction of a binary vector and transformation of rice plants
The 5′-flanking region of Rtlp1 was cloned immediately upstream of the GUS reporter gene in vector pBI221 (Clontech) replacing the CaMV 35 S promoter. This fusion gene reporter plasmid is called pRtlp1GUS. pRtlp1GUS was digested with HindIII, and inserted into the HindIII-site of a binary vector EBisKH2 (Kanzaki et al. 2002) (pEKH/Rtlp1GUS). The binary plasmid pEKH/Rtlp1GUS contains the GUS gene driven by the rice Rtlp1 promoter and the hygromycin phosphotransferase (Hpt) gene driven by the CaMV 35S promoter. pEKH/Rtlp1GUS was transferred into Agrobacterium tumefaciens strain EHA105 by electroporation. Rice plants were transformed and regenerated (O. sativa L. cv. Sasanishiki) according to Hiei et al. (1994) with slight modifications. Fifty microgram per milliliter hygromycin (Hyg) was used to select transformants . After acclimatization, transformants were grown in soil in a greenhouse.
Characterization of Rtlp1 promoter using GUS assay
Seedlings of transgenic rice harboring pEKH/Rtlp1GUS (T0 generation) were transferred into an inoculation chamber at the four- to five-leaf stages (30 days after sowing), and each pot was inoculated by spraying 250 μl of a conidial suspension of the rice blast fungus M. grisea (race 007.0) adjusted to 5 × 105 spores/ml (containing 0.05% Tween-20). Inoculated plants were kept in a closed chamber (25°C, 100% relative humidity) for 20 h and transferred to a moist incubator at 25°C until the lesion developed. Leaves were frozen in liquid nitrogen and used for GUS assay. Three replicates were taken for each treatment.
Seedlings of transgenic rice (T0 generation) were transferred into an inoculation chamber at the four- to five-leaf stage (30 days after sowing), and plants were treated with either 5 mM salicylic acid (SA), sprayed with 100 μM methyl jasmonate (MeJA) (containing 0.05% Tween-20) or wounded mechanically with forceps. After each treatment, plants were kept in a closed chamber (25°C, 100% relative humidity). Leaves were collected 1, 2, 6 or 24 h after treatment, frozen immediately in liquid nitrogen and stored at −80°C. Rice cultured cells harboring pEKH/Rtlp1GUS were maintained as hygromycin-resistant calli and assayed for GUS assay. Each treatment was repeated 3 times. Elicitor was prepared from rice blast hyphae according to the method of Koga et al. (1998). Water soluble cell wall extract was isolated from 70 g fungal hyphae, and dissolved in 210 ml 20 mM potassium phosphate buffer (pH 6.5). Cell wall extract (330 μl) was applied to 20 ml cultured cells. Potassium phosphate buffer (20 mM, pH 6.5) was used as the negative control.
Construction of promoter fragment/TATAintGUS vectors and GUS transient assay
A DNA fragment containing the TATA-box and the GUS reporter gene was PCR-amplified using pIG121Hm (Tanaka et al. 1990) as template and ligated into pBI221 (Clontech) digested with HindIII and SacI (pTATAintGUS). Different regions of the Rtlp1 promoter were amplified from genomic DNA. PCR primers were used to create fragment termini to match HindIII or SphI cleaved DNA ends, and promoter fragments were cloned into pT7Blue vector (Invitrogen). Fragments from the Rtlp1 promoter were digested with HindIII and SphI, and inserted immediately upstream of the TATA-box of pTATAintGUS. The constructs were called pRtlp1M5intGUS, pRtlp1M4intGUS, pRtlp1M3intGUS, pRtlp1M2intGUS and pRtlp1M1intGUS. A cDNA fragment containing the parsley (Petroselinum crispum) gene encoding WRKY1 transcription factor was PCR amplified from pBT-35S-WRKY1 (a kind gift from Dr. I. Somssich, Rushton et al. 1996) and placed downstream of the maize Ubi-1 promoter in pAHC17 (Christensen et al. 1992). The resulting plasmid pUbiPcWRKY1 was used to study transactivation of the Rtlp1 promoter.
Transient GUS activity assay was carried out in embryogenic calli of japonica rice from scutella of mature seed embryo (O. sativa L. cv. Sasanishiki). The calli were transformed by particle bombardment using the Bio-Rad PDS 1000/He device as described in Zhang et al. (1996).
In young transgenic plants, GUS activity was detected in hygromycin-resistant calli using histochemical and fluorometric assays essentially as described by Jefferson et al. (1987). Protein concentration was determined by the method of Bradford (1976) with a Bio-Rad kit (Protein Assay kit; Bio-Rad).
Results
Pathogen-induced Rtlp1 expression
A rice EST library was constructed from rice calli exposed to salt stress, and a cDNA clone, PSAL005, was isolated that has high sequence homology with Tlp genes in the PR-5 family. This gene was called Rtlp1. The coding region of Rtlp1 is 723 bp in length and encodes a protein of 177 amino acids with a molecular mass of 18 kDa.
Expression of Rtlp1 was examined in susceptible (Sasanishiki) and resistant (Toyonishiki) cultivars infected with rice blast fungus race 007.0 (Fig. 1). Northern blot analysis indicated that Rtlp1 transcripts increased in susceptible cultivar Sasanishiki within 6 h after inoculation, and was expressed constitutively in the field-resistant cultivar Toyonishiki. A weak signal was detected in the buffer-treated sample (basal transcription from Rtlp1).

Induction of Rtlp1 by infection with rice blast fungus. At the four- to five-leaf stage, rice seedlings were transferred to an inoculation chamber and pots were inoculated by spraying with a suspension of conidia of the rice blast fungus (race 007.0). Control samples were sprayed with buffer. Total RNA was isolated from infected and uninfected leaves and used for northern blot analysis. Blots were probed with 32P-labeled Rtlp1 cDNA. minus symbol uninfected; plus symbol infected. Hours from treatment to harvest of RNA are shown. Sasanishiki is susceptible and Toyonishiki is field-resistant to the blast fungus race 007.0
Isolation of the Rtlp1 5′-flanking region
The upstream flanking region of Rtlp1 was isolated using modified inverse PCR (Ochman et al. 1988). Genomic DNA was digested with restriction enzymes that recognize AT-rich sequences and the termini were converted to blunt-ends for cloning. A 1.2 kb fragment of the 5′-flanking region of Rtlp1 was isolated, sequenced and analyzed for motifs that are associated with transcriptional regulation (Fig. 2). A putative TATA-box motif is located 150 bp upstream of the start codon. Several other putative regulatory motifs were identified including eight W-box motifs (elicitor responsive element, consensus sequence 5′-(T)TGAC(C)-3′; Rushton et al. 1996), a GCC-box motif (ethylene responsive element, consensus sequence 5′-(A)GCCGCC-3′; Ohme-Takagi and Shinshi 1995) and a MYB recognition element (SA-responsive element, MRE consensus sequence 5′-A(A/C)C(A/T)A(A/C)C-3′; Lois et al. 1989).

Nucleotide sequence of the Rtlp1 5′-flanking region. Nucleotide sequence is numbered with coordinate +1 at the translation start site. Double underline indicates putative TATA-box. Boxes indicate putative cis-acting consensus sequence elements: W-box (5′-TGAC-3′); GCC-box (5′-GCCGCC-3′); MRE (MYB recognition-element, 5′-A(A/C)C(A/T)A(A/C)C-3′). The coding region is underlined
Rtlp1 promoter activity enhanced by rice blast infection
The Rtlp1 promoter was fused with a GUS reporter gene (pRtlp1GUS) and a binary vector, pEKH/Rtlp1GUS, was constructed and transformed into rice embryogenic calli. Transformed rice plants were regenerated from hygromycin-resistant calli and GUS activity was detected by histochemical stain. Transgenic plants were challenged with rice blast infection and stained for GUS activity (Fig. 3). Kinetics of the response of the Rtlp1 promoter and GUS expression are shown in Fig. 4. In uninfected plants, GUS activity was absent in leaves except for near sites of leaf wounding. In infected plants, GUS activity was strong near foci on rice blast-infected leaves 7 days after inoculation (Fig. 3). In addition, GUS expression was tenfold higher in cultured cells exposed to rice blast than in control uninfected cells. GUS activity was weak in the anther and the lemma in young panicle and in the lower part of the sheath (data not shown). A fluorometric GUS assay was used to quantify expression of pRtlp1GUS; the results indicate that expression from the Rtlp1 promoter increases 20-fold in leaves treated with rice blast conidia within 6 h after infection (Fig. 4).

Histochemical analysis of GUS expression from a Rtlp1GUS reporter gene. a The leaves of transgenic rice harboring pEKH/Rtlp1GUS were inoculated with rice blast and incubated for 7 days. b Histochemical stain for GUS activity in infected leaves. c Histochemical stain for GUS activity in intact leaves of uninfected transgenic plants harboring pEKH/Rtlp1GUS

Activation of Rtlp1 promoter by blast fungus infection. Transgenic rice were treated as described in legend to Fig. 3. GUS activity was quantified fluorometrically in infected or uninfected leaves. The transgenic plants were inoculated with a suspension of conidia (containing 0.05% Tween20) of the rice blast fungus race 007.0. Plants were sprayed and incubated under conditions of high humidity at 25°C. Incubation times are indicated. Mean specific activity and standard error is shown
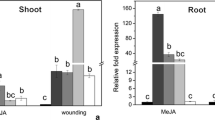
Rtlp1 promoter activity in transgenic plants enhanced by jasmonic acid, wounding and elicitor
Transgenic plants harboring pEKH/Rtlp1GUS were sprayed with SA (5 mM) or MeJA (100 μM), which act as second messengers in plant defense signal transduction. GUS activity was three- to fourfold higher in SA- or MeJA-treated plants than in untreated control plants; GUS activity increased within 2 h after treatment (Fig. 5). As mentioned above, GUS activity also increased in plants wounded mechanically with forceps; wounding increased GUS activity fourfold within 1 h after treatment.

Activation of Rtlp1 promoter by elicitor or wounding. Transgenic rice plants harboring pEKH/Rtlp1GUS were treated with 5 mM SA or 100 μM MeJA by spraying, or mechanical wounding using forceps. Plants were incubated under conditions of high humidity at 25°C. GUS activity was quantified fluorometrically in treated or untreated leaves. Mean specific activity and standard error is shown
Transgenic rice cells grown in suspension culture harboring pEKH/Rtlp1GUS were also tested for responsiveness to elicitor. Even without elicitor-treatment the basal GUS activity was high in cultured cells. Nevertheless, GUS activity was higher in elicitor-treated cells than in control cells; GUS expression increased twofold within 2 h after treatment (Fig. 6).

Inducibility of Rtlp1 promoter-GUS fusion gene in rice cultured cells. Rice cultured cells harboring pEKH/Rtlp1GUS were maintained as hyg-resistant calli, and then used for GUS assay after elicitor treatment. Elicitor from rice blast race 007.0 was prepared according to the method of Koga et al. (1998). Mean specific activity and standard error is shown
A 120-bp Rtlp1 promoter fragment confers strong elicitor responsiveness
Elicitor-responsive regions in the Rtlp1 promoter were identified by transiently expressing promoter deletion constructs in cultured rice cells. Four truncated forms of the Rtlp1 promoter were prepared and inserted upstream of the TATA-box in pTATAintGUS. Five constructs (one full-length and four truncated) were transfected into rice cells and transfected cells were tested for transient GUS-expression in response to elicitor (Fig. 7a). A truncated construct including region M3 (262 bp) (pRtlp1M3) was insensitive to elicitor-treatment (no increase in GUS activity); this truncated promoter lacks W-boxes. Truncated promoters including regions M2 + M3 (647 bp; pRtlp1M4) or M2 (385 bp; pRtlp1M2) had two- to threefold stronger GUS activity in elicitor-treated cells than the TATA-only promoter; these truncated promoters have two W-boxes and one GCC-box. A promoter construct including region M1 (120 bp) (pRtlp1M1) had the highest induction level and had sixfold higher GUS activity in elicitor-treated cells than the TATA-only promoter; the 120 bp region M1 includes six W-boxes. This truncated promoter construct responded to elicitor in a dose-dependent manner (Fig. 7b). In this expression system, the construct including the full-length 767 bp Rtlp1 promoter (eight W-boxes and one GCC-box; pRtlp1M0) supported a threefold increase in GUS expression after elicitor-treatment, which was a smaller induction than pRtlp1M1 (120 bp promoter construct).

Determination of elicitor-responsive regions in the Rtlp1 promoter. a Schematic representation of the Rtlp1 promoter region with W-boxes (left) and transient GUS activity of each region of Rtlp1 promoter after particle bombardment delivery of promoter-GUS fusion into elicitor-treated cells. Nucleotide sequence numbers are coordinated from +1 at the translation start site. b Elicitor inducibility of M1 fragments (pRtlp1M1intGUS). Bar indicates elicitor inducibility of TATAintGUS. Mean specific activity and standard error is shown
WRKY transcription factor confers elicitor-responsiveness to the 120 bp Rtlp1 promoter fragment
WRKY is a transactivation factor that bind to W-box sequences and activates transcription of the gene downstream of the W-box. Thus, it is possible that WRKY stimulates transcription from the Rtlp1 promoter. This idea was tested using WRKY1 cDNA from parsley and transient transactivation of pRtlp1GUS in rice cells. WRKY1 cDNA (Rushton et al. 1996) was fused with maize Ubi-1 promoter (Fig. 8a), and the construct was co-transformed with pRtlp1M1intGUS into rice calli using particle bombardment. The transient assay indicated that GUS expression was fourfold higher in cells co-expressing parsley WRKY1 than in cells without WRKY1 (Fig. 8b).

Transactivation capacity of M1 fragments (pRtlp1M1intGUS) during co-transformation with parsley WRKY1 cDNA. WRKY1 expression was driven by the maize ubiquitin1 promoter. GUS expression was measured in transformed cultured cells. a Constructs used for co-transformation. H HindIII; E EcoRI; S SacI., b GUS activity was measured in at least three independent experiments. Mean specific activity and standard error is shown
Discussion
This study presents a functional analysis of the promoter of Rtlp1 in wild type rice, transgenic rice and in cultured rice cells. The results indicate that the Rtlp1 promoter is rapidly induced by several agents including rice blast fungus and that W-box motifs in the Rtlp1 promoter are required for regulation of Rtlp1 expression.
The Rtlp1 promoter induces expression of Rtlp1 or a Rtlp1GUS fusion gene within 6 h after infection with rice blast fungus (Figs. 1, 4). Previous studies report that rice blast infection causes hyphal invasion of rice epidermal cells 19 h after infection (Koga et al. 1998) indicating that Rtlp1 is induced before hyphae invade and overgrow leaf cells. It is possible that plant wounding or an elicitor generates a signal that flows from the penetration hole of the rice blast appressorium to the host cells; these signals could be transmitted downstream to activate transcription of PR genes including Rtlp1.
Promoter regions have been isolated by several methods including inverse-PCR (Ochman et al. 1988), TAIL-PCR (Liu et al. 1995; Terauchi and Kahl 2000) or conventional screening of genomic DNA libraries. The 5′-flanking regions of plant genes are usually AT-rich. Therefore, the 5′-flanking region of Rtlp1 was amplified by inverse-PCR using template genomic DNA digested with restriction enzymes that recognize AT-rich regions. This modified inverse-PCR method was highly efficient for recovery of the target promoter sequence, and this method may be generally useful for isolation of promoter regions of plant genes.
SA and JA may play key roles in signal transduction cascades that contribute to plant defense mechanisms. SA and JA may be antagonistic to each other (Niki et al. 1998; Pieterse and van Loon 1999). Schweitzer et al. (1997) indicated that PR-proteins accumulate in plants treated with JA, but pathogen attack does not enhance the level of endogenous JA. The role of SA in host defense is not clear, in part because the endogenous level of SA is very high in rice, especially in leaves (Silverman et al. 1995; Yang and Qi 2000). Results presented here indicate that the activity of the Rtlp1 promoter increases 1 h after SA-treatment and 2 h after MeJA-treatment (Fig. 5). Transcription of Pbz1, a rice PR-protein gene, is also enhanced 2 h after SA-treatment and 4 h after MeJA-treatment (Lee et al. 2001). These results suggest that rice PR-proteins can be induced by SA or JA. Silverman et al. (1995) found a correlation between the level of leaf SA and generalized blast resistance in 28 varieties and suggested that SA plays a role as a constitutive defense compound. Rice Pbz1 was also induced by blast fungus even in SA-deficient transgenic plants harboring the NahG gene (Lee et al. 2001). However, SA may not play a role in signaling during the defense response in rice. Alternatively, exogenous SA may enhance production of H2O2 despite the high level of endogenous SA in rice (Rao et al. 1997; Ganesan and Thomas 2001); thus, Rtlp1 and Pbz1 could be induced by H2O2 that accumulates in plants treated with exogenous SA. A recent microarray gene expression study examined 2,375 Arabidopsis thaliana genes and showed that 25% of MeJA-inducible genes are also induced by SA (Schenk et al. 2000). This observation is consistent with the hypothesis that defense signaling in plants may have a different transcriptional network from wounding and JA or pathogen attack and SA. Brown et al. indicated that in addition to the roles in regulating ethylene-responsiveness, ethylene response factors (ERFs) also play important roles in Jasmonate-responsiveness, possibly by interaction with the GCC-box in in A. thaliana (2003). One GCC-box sequence founnd in M2 region of Rtlp1 promoter may interact with transcriptional factor ERFs and lead to Jasmonate-responsiveness of Rtlp1 promoter.
The Rtlp1 promoter was also induced 1 h after leaf wounding and 2 h after elicitor-treatment of cultured cells (Figs. 5, 6). Basal Rtlp1 promoter activity was high in cultured cells even in the absence of elicitor; this may indicate mechanical wounding of cells during suspension culture. However, Rtlp1 promoter activity increased in cultured cells exposed to elicitor, suggesting that the fungal elicitor might induce Rtlp1 in infected cells. Rushton et al. (1996) and Euglem et al. (1999) indicated that the W-box (TGAC core motif) is a key element in the mechanism of fungal-elicitor stimulated transcription in parsley. There are eight W-boxes in the 1.2 kb 5′-flanking region of Rtlp1. In addition, the W-boxes are required for inducible transcription from the Rtlp1 promoter. Transient expression using a truncated promoter–reporter fusion construct showed that a 262 bp fragment (pRtlp1M3) without W-boxes does not respond to elicitor. Furthermore, the highest elicitor-response was achieved with a 120 bp fragment (pRtlp1M1) containing six W-boxes and the response was dose-dependent. These results strongly suggest that the W-box element is essential to the fungal elicitor responsiveness of the rice Rtlp1 promoter.
Rushton et al. (1996) and Eulgem et al. (1999) showed that the WRKY zinc-finger transcription factor binds to the W-box element. WRKY mRNA rapidly accumulates near sites of fungal infection and WRKY activates expression of downstream genes by binding to promoters containing W-box elements. Results presented here indicate that the 120 bp fragment (pRtlp1M1) containing six W-boxes was activated by co-expression of parsley WRKY1 (Fig. 8). These data suggests that a WRKY may be one of trans-acting factors which activate the W-box elements in the promoter region of rice PR-protein genes including Rtlp1. In A. thaliana, genes encoding WRKY proteins form a superfamily with approximately 100 members (Eulgem et al. 2000). Recently, rice WRKY has begun to be studied in detail. In rice, 98 WRKY genes in japonica and 102 in indica rice were identified (Ross et al. 2007). An elicitor-inducible WRKY gene, OsWRKY1, was isolated using cDNA differential display in cells exposed to blast fungal elicitor (Kim et al. 2000). Gene expression of OsWRKY10 and OsWRKY82 increased by SA- and JA-treatments, similaly to Fig. 5 in this paper (Ryu et al. 2006). Overexpressing the elicitor-induced OsWRKY71 upregulated defense-regulated genes (Chujo et al. 2008). In addition, rice WRKY45 is suggested to play a role in BTH-induced and SA-mediated defense signaling based on RNAi and overexpression experiments (Shimono et al. 2007). Among these OsWRKYs, OsWRKY1 and OsWRKY82 belong to WRKY group I-a as well as parsley WRKY1. In addition, OsWRKY1 has the highest sequence homology to parsley WRKY1. Therefore, OsWRKY1 may be one of transcriptinal factor candidates, which activate transcription of Rtlp1 in response to elicitor.
References
Abad LR, D’Urzo MP, Liu D, Narasimhan ML, Reuveni M, Zhu JK, Niu X, Singh NK, Hasegawa PM, Bressan RA (1996) Antifungal activity of tobacco osmotin has specificity and involves plasma membran permeabilization. Plant Sci 118:11–23
Agrawal GK, Rakwal R, Jwa NS (2000) Rice (Oryza sativa L.) OsPR1b gene is phytohormonally regulated in close interaction with light signals. Bioch Biophys Res Commun 19:290–298
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. J Biochem 72:248–254
Brrown RL, Kazan K, McGrath KC, Maclean DJ, Manners JM (2003) A role for the GCC-Box in Jasmonate-mediated activation of the PDF1.2 gene of arabidopsis. Plant Physiol 132:1020–1032
Christensen AH, Sharrock RA, Quail PH (1992) Maize polyubiquitin genes: structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol Biol 18:675–689
Cordero MJ, Raventos D, San Segundo B (1994) Expression of a maize proteinase inhibitor gene is induced in response to wounding and fungal infection: systemic wound-response of a monocot gene. Plant J 6:141–150
Chujo T, Kato T, Yamada K, Takai R, Akimoto-Tomiyama C, Minami E, Nagamura Y, Shibuya N, Yasuda M, Nakashita H, Umemura K, Okada A, Okada K, Nojiri H, Yamane H (2008) Characterization of an elicitor-induced rice WRKY gene, OsWRKY71. Biosci Biotech Biochem 72:240–245
Datta K, Velazhahan R, Oliva N, Ona I, Mew T, Khush GS, Muthukrishnan S, Datta SK (1999) Over-expression of the cloned rice thaumatin-like protein (PR-5) gene in transgenic rice plants enhances environmental friendly resistance to Rhizoctonia sonali causing sheath blight disease. Theor Appl Genet 98:1138–1145
Eulgem T, Rushton PJ, Schmelzer E, Hahlbrock K, Somssich IE (1999) Early nuclear events in plant defense signaling: rapid gene activation by WRKY transcription factors. EMBO J 18:4689–4699
Eulgem T, Rushton PJ, Robatzek S, Schmelzer IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Ganesan V, Thomas G (2001) Salicylic acid response in rice: influence of salicylic acid on H2O2 accumulation and oxidative stress. Plant Sci 160:1095–1106
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6:271–282
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: β-glucronidase as a sensitive and versatile gene fusion marker in higher plant. EMBO J 6:3901–3907
Kanzaki H, Nirasawa S, Saitoh H, Ito M, Nishihara M, Terauchi R, Nakamura I (2002) Overexpressing of wasabi defensin gene confers enhanced resistance to blast fungus (Magnaporthe grisea) in transgenic rice. Theor Appl Genet 105:809–814
Kim CY, Lee SH, Park HC, Bae CG, Cheong YH, Choi YJ, Han C, Lee SY, Lim CO, Cho MJ (2000) Identification of rice blast fungal elicitor-responsive genes by differential display analysis. Mol Plant Microbe Interact 13:470–474
Koga J, Oshima K, Ogawa N, Ogasawara N, Shimura M (1998) A new bioassay for measuring elicitor activity in rice leaves. Annu Phytopathol Soc Jpn 64:97–101
Lee MW, Qi M, Yang Y (2001) A novel jasmonic acid inducible rice myb gene associates with fungal infection and host cell death. Mol Plant Microbe Interact 14:527–535
Liu YG, Mitsukawa N, Oosumi T, Whittier RF (1995) Efficient isolation and mapping of Arabidopsis thaliana T-DNA insert junctions by thermal asymmetric interlaced PCR. Plant J 8:457–463
Lois R, Dietrich A, Hahlbrock K, Schulz W (1989) A phenylalanine ammonia-lyase gene from parsley: structure, regulation and identification of elicitor and light responsive cis-acting elements. EMBO J 88:1641–1648
Malehorn DE, Borgmeyer JR, Smith CE, Shah DM (1994) Characterization and expression of an antifungal zeamatin-like protein (Zlp) gene from Zea mays. Plant Physiol 106:1471–1481
Mauch F, Kmecl A, Schaffrath U, Volrath S, Gorlach J, Ward E, Ryals J, Dudler R (1997) Mechanosensitive expression of a lipoxygenase gene in wheat. Plant Physiol 114:1561–1566
Niki T, Mitsuhara I, Seo S, Ohtsubo N, Ohashi Y (1998) Antagonistic effect of salicylic acid and jasmonic acid on the expression of pathogenesis-related (PR) protein genes in wounded mature tobacco leaves. Plant Cell Physiol 39:500–507
Nishizawa Y, Kawakami A, Hibi T, He DY, Shibuya N, Minami E (1999) Regulation of the chitinase gene expression in suspension-cultured rice cells by N-acetylchitooligosaccharides: differences in the signal transduction pathways leading to the activation of elicitor-responsive genes. Plant Mol Biol 39:907–914
Ochman H, Gerber AS, Hartl DL (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120:621–625
Ohme-Takagi M, Shinshi H (1995) Ethylene-inducible DNA binding proteins that interact with an ethylene-responsive element. Plant Cell 7:173–182
Pieterse SM, van Loon LC (1999) Salicylic acid-independent plant defense pathways. Trends Plant Sci 4:52–58
Rao MV, Paliyath G, Ormrod DP, Murr DP, Watkins CB (1997) Influence of salicylic acid on H2O2-metabolizing enzymes. Plant Physiol 115:137–149
Ros CA, Liu Y, Shen QJ (2007) The WRKY gene family in rice (Oryza sativa). J Integ Plant Biol 49:827–842
Rushton PJ, Torres JT, Parkinske M, Wernert P, Hahlbrock K, Somssich IE (1996) Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J 15:5690–5700
Ryu HS, Han M, Lee AK, Cho JI, Ryoo N, Heu S, Lee YH, Bhoo SH, Wang GL, Hahn SH, Jeon JS (2006) A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep 25:836–847
Sambrook J, Fritsch EF, Manniatis T (1989) Molucular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schenk PM, Kazan K, Wilson I, Anderson P, Richmond T, Somerville SC, Manners JM (2000) Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc Natl Acad Sci USA 97:11655–11660
Shimono M, Sugano S, Nakayama A, Jiang CJ, Ono K, Toki S, Takatsuji H (2007) Rice WRKY45 plays a crucial role in benzothiadiazole-inducible blast resistance. Plant Cell 19:2064–2076
Shure M, Wessler S, Fedoroff N (1983) Molecular identification and isolation of the Waxy locus in maize. Cell 35:225–233
Silverman P, Seskar M, Kanter D, Schweitzer P, Metraux J-P, Raskin I (1995) Plant Physiol 108:633–639
Schweitzer P, Buchala A, Silverman P, Seskar M, Raskin I., Metraux J-P (1997) Jasmonate-inducible genes are activated in rice by pathogen attack without a concomitant increase in endogenous jasmonic acid levels. Plant Physiol 114:79–88
Tanaka A, Mita S, Ohta S, Kyozuka J, Shimamoto K, Nakamura K (1990) Enhancement of foreign gene expression by a dicot intron in rice but not in tobacco is correlated with an increased level of mRNA and an efficient splicing of the intron. Nucleic Acids Res 18:6767–6770
Terauchi R, kahl G (2000) Rapid isolation of promoter sequences by TAIL-PCR: the 5′-flanking regions of Pal and Pgi genes from yams (Dioscorea). Mol Gen Genet 263:554–560
Toriyama K, Hinata K (1985) Cell suspension and protoplast culture in rice. Plant Sci 41:179–183
Vigers AJ, Roberts WK, Selitrennikoff CP (1991) A new family of plant antifungal proteins. Mol Plant Microbe Interact 4:315–323
Vigers AJ, Wiedemann S, Roberts WK, Legrand M, Selitrennikoff CP, Fritig B (1992) Thaumatin-like pathogesis-related proteins are antifungal. Plant Sci 83:155–161
Xu Y, Zhu Q, Panbangred W, Shirasu K, Lamb C (1996) Regulation, expression and function of a new basic chitinase gene in rice (Oryza sativa L.). Plant Mol Biol 30:387–401
Yang Y, Qi M (2000) Potential role salicylic acid in maintaining redox balance of rice plants. Phytopathology 90:S86
Yun DJ, Ibeas JI, Lee H, Coca MA, Narashimhan ML, Uesono Y, Hasegawa PM, Bressan R (1998) Osmotin, a plant antifungal protein, subverts signal transduction to enhance fungal cell susceptibility. Mol Cell 1:807–817
Zhang S, Chen Lili, Qu R, Marmey P, Beachy R, Fauquet C (1996) Regeneration of fertile transgenic indica (group 1) rice plants following microprojectile transformation of embryogenic suspension culture cells. Plant Cell Rep 15:465–469
Acknowledgments
We thank Dr. I Somssich (Max-Planck-Institut für Züchtungsforschung, Germany) for kindly supplying pBT-35S-WRKY1 plasmid. Thanks are extended to Ms. M Fujiwara (Iwate Agricultural Research Center) for assistance in producing transgenic rice plants. We are grateful to Dr. S Koizumi (Tohoku Agricultural Research Center MAFF) for the kind supply of rice blast fungal races. We would like to thank to Dr. I. Nakamura of Graduate School of Science and Technology, Chiba University for excellent advise.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Toriyama.
Rights and permissions
About this article
Cite this article
Hiroyuki, K., Terauchi, R. Regulation of expression of rice thaumatin-like protein: inducibility by elicitor requires promoter W-box elements. Plant Cell Rep 27, 1521–1528 (2008). https://doi.org/10.1007/s00299-008-0536-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-008-0536-7