
Abstract
Purpose
A prospective multicenter phase I/II trial was performed to evaluate the clinical safety and efficacy of radiofrequency ablation (RFA) for metastatic bone tumors.
Materials and Methods
Thirty-three patients (27 men, 6 women, mean age 61 years) with metastatic bone tumors were enrolled. In phase I, nine patients were enrolled, and the safety of RFA was evaluated. In phase II, 23 patients were included, and an intent-to-treat analysis was performed. The primary endpoint was to evaluate the treatment’s safety. The secondary endpoint was to evaluate the efficacy of pain relief at 1 week after RFA.
Results
RFA was performed in 32 of 33 enrolled patients. No serious complications were observed during the phase I, so phase II was performed. Four patients exhibited adverse events, including one case each of Grade 3 pain and, Grade 2 hypotension, and one patient developed Grade 1 burns at the grounding pad and puncture site. One patient died of liver failure on day 7 after RFA due to the progression of the primary lesion. The efficacy was excellent (no increase in analgesic dosage, post-RFA VAS score of 0–2 or decreased by not less than 5 compared to before RFA) in 20 patients (60.6%), good (no increase in analgesic dosage, post-RFA VAS score decreased by not less than 2 but by < 5 compared to before RFA) in 3 (9.1%), and poor in 10 patients (30.3%). Thus, the response rate was 69.7%.
Conclusion
RFA is a safe and effective method for treating painful metastatic bone tumors.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Radiofrequency ablation (RFA) is a treatment method in which a needle electrode is percutaneously inserted into a target tissue, and heat generated by dielectric heating at the needle tip causes coagulation necrosis of the target tissue. RFA can be performed at any site into which the needle electrode can be percutaneously inserted, but is characterized by variability in the local temperature distribution and the scope of cauterization as a function of the tissue environment surrounding the target site. The clinical application of RFA to liver tumors has already been explored, and good therapeutic results have been reported [1,2,3,4,5].
The primary treatment for painful bone metastases of cancer is currently radiotherapy; however, although partial improvement in pain is observed in 90% of patients, the rate of complete pain remission is only 54%. In addition, 30% of patients experience recurrence of pain within 12 weeks, and the response rate of patients with recurrent disease to radiotherapy is poor [6, 7]. Some patients receiving radiotherapy alone experienced uncontrollable clinical symptoms. The application of RFA to those patients in combination with radiotherapy might have therapeutic significance [8]. Studies have also been performed on the application of RFA to organs other than the liver, and some reports have described efficacy. However, most of these studies have been case-controlled studies [9,10,11,12], and there have been only few prospective and randomized studies for RFA application outside the liver [13,14,15,16]. Thus, the present study was designed as a multicenter phase I/II trial to evaluate the clinical safety and efficacy of RFA for the treatment of metastatic bone tumors.
Materials and Methods
Patient Selection
Patients were selected and enrolled in this study based on all the inclusion and exclusion criteria described below. The inclusion criteria were as follows: (1) metastatic bone tumors with clinical symptoms, of which pain is the chief complaint; (2) conventional local therapeutic methods, such as external irradiation or, injection of local anesthesia not indicated; (3) either pain that can only be controlled with the dose escalation of analgesics or pain uncontrolled by medication; (4) target lesions confirmed as malignant based on histopathologic findings or diagnostic imaging; (5) target lesions that are evaluable by computed tomography (CT) or magnetic resonance imaging (MRI); (6) adequate function of the principle organs (i.e., bone marrow, heart, liver, lungs, kidneys), specifically, white blood cell count ≥ 3000/mm3; platelet count ≥ 50,000/mm3; prothrombin time − international ratio ≥ 1.5; creatinine levels ≤ 3.0 mg/dl; and a normal electrocardiogram (although arrhythmias and ischemic changes that do not represent a clinical problem were permitted); (7) Eastern Cooperative Oncology Group performance status of 0, 1, 2, or 3; (8) life expectancy ≥ 4 weeks.
The exclusion criteria were as follows: (1) tumors in the vertebral body, skull, or bones of the hands or fingers; (2) use of a cardiac pacemaker; (3) imaging techniques demonstrating the clear presence of an artery or nerve in the needle insertion pathway; (4) pregnancy or possible pregnancy; (5) mild pain that can be self-controlled without any special treatment or a preoperative visual analogue scale (VAS) score ≤ 2; and (6) determination that the patient is unsuitable for inclusion in the study by the treating physician.
Both the ethics committee of the Japanese Society of Interventional Radiology and the Institutional Review Board from each participating hospital approved the study protocol prior to patient entry. All patients provided written informed consent.
Study Design and Statistical Analysis
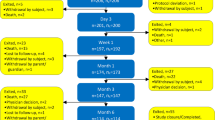
This was a multi-institutional, single-arm, non-comparative phase I/II trial. During the phase I trial and through phase II, the study was designed to detect adverse events (AEs) with an incidence of at least 10%, a power of 80%, a predicted rate of 10%, and an unacceptable rate of 30%. We anticipated a protocol dropout rate of 10%, so the target number of patients for accrual was calculated to be 33. The study design of the phase I trial incorporated the Japan Interventional Radiology in Oncology Study Group (JIVROSG) 3 × 3 method, as previously described [17]. Namely, this study was a step-by-step safety evaluation in nine patients, in which a cohort of three patients was treated with RFA. If no severe AEs occurred during the 4-week observation period, the next cohort of three patients was treated followed by an observation period, and finally the third cohort of three patients was treated. An additional 24 patients were enrolled in the phase II trial. To determine the study outcomes, all the enrolled patients were included in the intention-to-treat analysis.
Interventional Procedure for RFA
The RFA procedure was performed under conscious sedation. The grounding pad was attached to the thigh. Then, the target tumor lesion was confirmed by CT images, and local anesthesia was administered to the skin and subcutaneous tissue around the planned puncture point. Next, the RF needle electrode was inserted into the target tumor lesion. The RF needle used was a 17-gauge Cool-tip electrode (Valleylab, Boulder, CO, USA) or a 14-gauge LeVeen electrode (Boston Scientific, Natick, MA, USA). After confirming the position of the RF needle electrode in the target tumor lesion, the RF application was performed using a generator (Cool-tip RF, Valleylab; or RF3000, Boston Scientific).
With the Cool-tip system, when the tumor diameter was less than 3 cm, it started at 20–30 W output and was increased at a rate of 5–10 W/min. When the tumor diameter was 3 cm or more, the system started at 30–60 W output and was increased at a rate of 10 W/min. The endpoint of RFA was automatic output stop due to the impedance rise. The ablation for 12 min was taken as one course. If the temperature inside the tumor did not reach 60 °C in 12 min, the ablation with the final output power continued until the tumor temperature reached 60 °C or until automatic output stop due to the rise in impedance.
With the LeVeen needle, output was started at 30 W for a development diameter of 2 cm, 40 W for a development diameter of 3 cm, and 50 W for a development diameter of 3.5 cm, and the output was raised at 10 W/min. The procedure stopped at the time of output stop due to the rise in impedance. If automatic output stop due to the impedance rise did not occur in 12 min, the LeVeen needle was set at approximately 1/2–3/4 of the final expansion, and energization was performed again.
If the tumor was large and full cauterization could not be achieved in a single ablation, the puncture needle was placed at another location based on the shape of the lesion, so that as much of the lesion as possible could be coagulated. However, when coagulating the entire tumor was difficult despite multiple punctures, coagulation was focused on the interface between bone and soft tissue. The needle was withdrawn after treatment completion.
Combined and Supportive Therapies
The administration of analgesics including a narcotic was recommended to alleviate the pain during the RFA procedure. When deemed necessary for the patient, the use of general anesthesia, epidural anesthesia, and narcotics was also permitted. Except for cases requiring emergency treatment for AEs and poor responders (i.e., bone pain was unchanged or worsened after 1 week), other treatment procedures for the treated metastatic bone lesions such as another puncture therapy, local therapy, or surgical treatment were not permitted. Antiplatelet medication was stopped 3–14 days before the day of the procedure.
Evaluation Methods and Observation Items
Patients were continuously monitored for the manifestation of AEs from the time of treatment initiation to 4 weeks after the end of treatment. AEs were evaluated using the National Cancer Institute-Common Toxicity Criteria, version 2.0 [18]. The following parameters were recorded as procedural results: rate of completion of the treatment protocol, procedure time from the start of local anesthesia until removal of the inserted electrode needle, number of RFAs, the duration of current delivery, and the maximum power output. Pain was evaluated using a VAS on the day after RFA and at 3 days, within 1 week, and 4 weeks after RFA. Clinical efficacy was rated using the three categories defined below based on the pain 1 week after completion of RFA. That is, using VAS scores, scores before treatment and at 1 week after completion of treatment were compared and evaluated using the following three criteria: excellent: no increase in analgesic dosage, post-RFA VAS score of 0–2 or decreased by ≥ 5 compared to before RFA; good: no increase in analgesic dosage, post-RFA VAS score decreased by ≥ 2 but by < 5 compared to before RFA; and poor: all cases other than those mentioned above. Moreover, pain was evaluated continuously for 1 year after treatment in the cases in which follow-up for more than 4 months was possible.
The change in the amount of analgesia used within 1 month was investigated. For imaging evaluation of target lesions, contrast-enhanced CT or contrast-enhanced MRI of the target lesions was performed within 4 weeks of RFA, and 1 and 4 weeks after RFA, and the images were inspected for changes.
Results
A total of 33 patients were enrolled in this study at 11 participating institutions (Table 1). Patient characteristics are presented in Table 2. No serious complications occurred during the phase I study, so the phase II study was performed. Four patients exhibited adverse events, including one case each of Grade 3 pain and, Grade 2 hypotension, and two Grade 1 burns at the grounding pad and puncture site. The one remaining patient died of liver failure on day 7 after RFA, but the cause was liver failure due to progression of hepatocellular carcinoma, which was the primary lesion.
Procedural results revealed that 29 patients completed the treatment protocol. One patient deviated from the protocol by being treated for two lesions on the same day. Another two patients were able to begin RFA, but could not complete the treatment protocol due to pain experienced during the procedure. RFA could not be performed in the final patient because of the worsening overall physical condition due to hepatocellular carcinoma, which was the primary lesion. RFA treatment was performed in 32 patients with a mean procedure time of 61.3 ± 29.6 min (range, 15–120 min), a mean total number of RFAs of 2.8 ± 1.7 (range, 1–7), a mean duration of current delivery of 30.9 ± 20.4 min (range, 4.8–85.4 min), and a mean maximum power output of 117.6 ± 43.4 W (range, 40–200 W). Figure 1 presents (#19) the changes in VAS scores until 4 weeks. Pain relief from the therapy was (#15) obtained by the first day, with a slow decrease, in the VAS values also subsequently observed. The long-term results are presented in Fig. 2. The treatment effect of RFA remained stable.

Changes in visual analog scale score before and 4 weeks after procedure. The changes in the VAS values at various observation time points are listed. The curve indicates the changes in mean values and the vertical line indicates the standard deviation

Changes in visual analog scale score before and 1 year after procedure. Values are presented as the mean and standard deviation. The changes in the VAS values at various observation time points are listed. The curve indicates the changes in mean values, and the vertical line indicates the standard deviation. The treatment effect of RFA remains stable
The results of the efficacy evaluation were excellent in 20 patients (60.6%), good in 3 (9.1%), and poor in 10 patients (30.3%). Thus, the response rate was 69.7% (95% confidence interval: 51.3–84.4%). Analgesics were administered prior to RFA in 29 patients (87.5%): analgesics including narcotics were administered in 7, non-narcotic analgesics were administered in 7, and a combination of both were given to 15. The dose of analgesics was reduced within 4 weeks in 13 patients, but had to be increased in 2 patients. Diagnostic imaging prior to RFA revealed osteolytic lesions in 28 patients (84.9%), osteoblastic lesions in 3 (9.1%), and mixed lesions in 2 patients (6.1%). Destruction of the bone cortex around the lesion was observed in 32 patients (97%). An intratumoral contrast enhancement was observed in 29 patients (87.9%): an enhancement for the entire tumor was observed in 18 patients (54.6%) and a partial enhancement was observed in 11 (33.3%). Contrast-enhanced CT or MRI performed 1 week after RFA revealed decreased enhanced area in 26 patients (78.8%). This change remained the same even on diagnostic imaging performed 4 weeks later. The mean tumor diameter was 6.1 ± 3.1 cm prior to RFA, 6.26 ± 3.2 cm 1 week after RFA, and 6.0 ± 2.9 cm 4 weeks after RFA. Thus, there were no significant differences in tumor size prior to RFA and the size 1 week after RFA (p = 0.906), or in the size prior to RFA and 4 weeks after RFA (p = 0.618).
Discussion
In this study, one patient exhibited a Grade 5 AE. This patient died of liver failure on day 7 after RFA, but the cause of death was determined to be liver failure caused by the progression of hepatocellular carcinoma, which was the primary lesion. There may have been a problem in the indication of RFA for bone metastasis in this case. Four other patients exhibited AEs that included Grade 3 pain, Grade 2 hypotension, Grade 1 burns at the grounding pad, and Grade 1 burns at the puncture site. Skin burns at the grounding pad site were also observed in other reports [16]. No severe adverse events of RFA for metastatic bone tumor were reported [16, 19]. Therefore, RFA can be deemed a safe procedure for the treatment of metastatic bone lesions.
After enrollment in the study, four patients did not complete the procedure. Specifically, one patient was unable to undergo the procedure due to a worsening overall condition, two others were unable to complete the treatment protocol due to pain experienced during the procedure, and another patient deviated from the treatment protocol by being treated for two lesions on the same day. These situations occurred because this study was a prospective investigation. In a prospective study by Callstrom et al. [19], 5 of the 60 candidates were unable to undergo the procedure or the procedure was terminated midway through.
The clinical efficacy was evaluated by intention-to-treat analysis and was found to be 69.7%. Although this efficacy rate is lower than that reported in previous retrospective studies [20,21,22], it is acceptable for the present study as this was a prospective investigation. In addition, the changes in VAS score demonstrate that the analgesic effects were both immediate and lasting.
Most patients in our study had osteolytic lesions, and destruction of the bone cortex around the lesion as well as intratumoral contrast enhancement was observed in most of the lesions. These results are consistent with data from a previous study that also examined osteolytic lesions [23]. It is worth noting that similar analgesic effects were observed in patients with osteoblastic bone metastasis and in those with osteolytic lesions.
Contrast-enhanced CT or MRI performed after RFA revealed a decrease in the enhancement effect, possibly due to the necrotic changes with radiofrequency ablation. No statistically significant changes in tumor size were observed at 1 or 4 weeks after RFA. Although not statistically significant, tumor size increased slightly 1 week after RFA. This increase may have been due to reactive edema or intratumoral hemorrhage caused by the heat.
Vertebral tumors were excluded from the present clinical study, because percutaneous vertebroplasty already exists as an effective treatment [17]. However, although a combination of RFA and cementoplasty is sometimes used to treat vertebral tumors, it is difficult to determine whether the analgesic effects are due to RFA or cementoplasty in those cases.
There were some limitations in this study. The sample size was small, and the design was a single-arm, non-controlled randomized study. Due to the small sample size and short follow-up period, it is possible that AEs were not adequately identified. In addition, the long time period required to enroll patients may also be considered a limitation of the present study. A randomized controlled study is needed to determine whether this treatment method should be the first choice for painful bone metastases. In conclusion, the results of this study demonstrate that RFA is a safe and effective method for treating painful metastatic bone tumors.
References
McGahan JP, Browning PD, Brock JM, et al. Hepatic ablation using radiofrequency electrocautery. Invest Radiol. 1990;25:267–70.
Rossi S, Di Stasi M, Buscarini E, et al. Percutaneous RF interstitial thermal ablation in the treatment of hepatic cancer. AJR. 1996;167:759–68.
Rossi S, Buscarini E, Garbagnati F, et al. Percutaneous treatment of small hepatic tumors by an expandable RF needle electrode. AJR. 1998;170:1015–22.
Livraghi T, Goldberg SN, Lazzaroni S, et al. Small hepatocellular carcinoma: treatment with radio-frequency ablation versus ethanol injection. Radiology. 1999;210:655–61.
Curley SA, Izzo F, Ellis LM, et al. Radiofrequency ablation of hepatocellular cancer in 110 patients with cirrhosis. Ann Surg. 2000;232:381–91.
Tong D, Gillick L, Hendrickson FR. The palliation of symptomatic osseous metastases: final results of the study the radiation therapy oncology group. Cancer. 1982;50:893–9.
Ratanatharathon V, Powers WE, Moss WT, Peres CA. Bone metastasis: review and critical analysis of random allocation trials of local field treatment. Int J Radiat Oncol Phys. 1999;44:1–18.
Di Staso M, Zugaro L, Gravina GL, et al. A feasibility study of percutaneous radiofrequency ablation followed by radiotherapy in the management of painful osteolytic bone metastases. Eur Radiol. 2011;21:2004–10.
Dupuy DE, Zagoria RJ, Akerley W, et al. Percutaneous radiofrequency ablation of malignancies in the lung. AJR. 2000;174:57–9.
Putnam JB Jr. New and evolving treatment methods for pulmonary metastases. Semin Thorac Cardiovasc Surg. 2002;14:49–56.
Barei DP, Moreau G, Scarborough MT, et al. Percutaneous radiofrequency ablation of osteoid osteoma. Clin Orthop Relat Res. 2000;373:115–24.
Izzo F, Thomas R, Delrio P, et al. Radiofrequency ablation in patients with primary breast carcinoma. Cancer. 2001;92:2036–44.
Dupuy DE, Fernando HC, Hillman S, et al. Radiofrequency ablation of stage IA non-small cell lung cancer in medically inoperable patients: results from the American College of Surgeons Oncology Group Z4033 (Alliance) trial. Cancer. 2015;121:3491–8.
Mimura H, Arai Y, Yamakado K, et al. Phase I/II study of radiofrequency ablation for malignant renal tumors: Japan interventional radiology in Oncology Study Group 0701. Cardiovasc Intervent Radiol. 2016;39:717–23.
Miyazaki M, Arai Y, Myoui A, et al. Phase I/II multi-institutional study of percutaneous radiofrequency ablation for painful osteoid osteoma (JIVROSG-0704). Cardiovasc Intervent Radiol. 2016;39:1464–70.
Goetz MP, Callstrom MR, Chaboneau JW, et al. Percutaneous image-guided radiofrequency ablation of painful metastases involving bone: a multicenter study. J Clin Oncol. 2004;22(2):300–6.
Kobayashi T, Arai Y, Takeuchi Y, et al. Phase I/II clinical study of percutaneous vertebroplasty (PVP) as palliation for painful malignant vertebral compression fractures (PMVCF): JIVROSG-0202. Ann Oncol. 2009;20:1943–7.
Cancer Therapy Evaluation Program. Common toxicity criteria, version 2.0.1998. http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcv20_4-30-992.pdf. Accessed 15 April 2009.
Callstrom MR, Dupuy DE, Solomon SB, et al. Percutaneous image-guided cryoablation of painful metastases involving bone: multicenter trial. Cancer. 2013;119:1033–41.
Nakatsuka A, Yamakado K, Maeda M, et al. Radiofrequency ablation combined with bone cement injection for the treatment of bone malignancies. J Vasc Interv Radiol. 2004;15:707–12.
Toyota N, Naito A, Kakizawa H, et al. Radiofrequency ablation therapy combined with cementoplasty for painful bone metastases: initial experience. Cardiovasc Intervent Radiol. 2005;28:578–83.
Kojima H, Tanigawa N, Kariya S, et al. Clinical assessment of percutaneous radiofrequency ablation for painful metastatic bone tumors. Cardiovasc Interv Radiol. 2006;29:1022–6.
Callstrom MR, Charboneau JW, Goetz MP, et al. Painful metastases involving bone: feasibility of percutaneous CT- and US-guided radio-frequency ablation. Radiology. 2002;224:87–97.
Acknowledgements
This research was supported by the Practical Research for Innovative Cancer Control (16ck0106058h0003) from the Japan Agency for Medical Research and Development (AMED), Health and Labor Sciences Research Grant (H26-055) from the Ministry of Health, Labour and Welfare of Japan, and the National Cancer Center Research and Development Fund (26-A-27).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
The ethics committee of the Japanese Society of Interventional Radiology and the Institutional Review Board from each participating hospital approved the study protocol prior to patient entry. All of the patients provided written informed consent.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Tanigawa, N., Arai, Y., Yamakado, K. et al. Phase I/II Study of Radiofrequency Ablation for Painful Bone Metastases: Japan Interventional Radiology in Oncology Study Group 0208. Cardiovasc Intervent Radiol 41, 1043–1048 (2018). https://doi.org/10.1007/s00270-018-1944-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00270-018-1944-x