
Abstract
The araA gene encoding an L-arabinose isomerase (L-AI) from the acido-thermophilic bacterium Acidothermus cellulolytics ATCC 43068 was cloned and overexpressed in Escherichia coli. The open reading frame of the L-AI consisted of 1,503 nucleotides encoding 501 amino acid residues. The recombinant L-AI was purified to homogeneity by heat treatment, ion-exchange chromatography, and gel filtration. The molecular mass of the enzyme was estimated to be approximately 55 kDa by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The purified enzyme was optimally active at 75°C and pH 7.5. It required divalent metal ions, either Mn2+ or Co2+, for both enzymatic activity and thermostability improvement at higher temperatures. The enzyme showed relatively high activity and stability at acidic pH. It exhibited over 90% of its maximal activity at pH 6.0 and retained 80% of activity after 12 h incubation at pH 6.0. Catalytic property study showed that the enzyme had an interesting catalytic efficiency. Its apparent K m, V max, and catalytic efficiency (k cat/K m) for D-galactose was 28.9 mM, 4.9 U/mg, and 9.3 mM−1 min−1, respectively. The enzyme carried out the isomerization of D-galactose to D-tagatose with a conversion yield over 50% after 12 h under optimal conditions, suggesting its potential in D-tagatose production.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
D-Tagatose is a hexoketose monosaccharide sweetener, which is an isomer of D-galactose and rarely found in nature (Kim 2004). D-Tagatose occurs naturally in small quantities in Sterculia setigera gum, and it is also found in dairy products (Troyono et al. 1992; Mendoza et al. 2005). The sweetness of D-tagatose is 92% that of sucrose when compared in 10% solutions, but with only 38% of the calories. It has been shown to have numerous health and medical benefits, including treatment of obesity (Donner et al. 1999), prevention of dental caries, regulation of intestinal flora (Kim 2004; Buemann et al. 2000), improvement of pregnancy and fetal development, and reduction of symptoms of type 2 diabetes, hyperglycemia, anemia, and hemophilia (Levin 2002; Oh 2007; Lu et al. 2008; Rosenplenter and Mende 2004; Kim 2004). Based on these properties, D-tagatose has attracted a great deal of attention in recent years as a low calorie sugar-substituting sweetener, an intermediate for synthesis of other optically active compounds, and an additive in detergent, cosmetic, as well as in pharmaceutical formulations (Oh 2007; Lu et al. 2008; Kim 2004).
Recently, there has been great interest in the biological manufacture of D-tagatose from D-galactose. Several enzymes involved in the biotransformation of D-tagatose have been investigated (Rollini and Manzoni 2005; Ishida and Kamiya 1997; Kim et al. 2006). L-Arabinose isomerase (L-AI, EC 5.3.1.4) is considered to have the most potential use for D-tagatose production, since it can catalyze the isomerization of D-galactose to D-tagatose and convert L-arabinose to L-ribulose, based on the similarity in configuration of the substrates (Cheetham and Wootton 1993). A number of L-AIs have been identified from various microorganisms (Chouayekh et al. 2007; Jørgensen et al. 2004; Kim et al. 2002, 2003b; Kim and Oh 2005; Lee et al. 2004, 2005a, b; Nakamatu and Yamanaka 1969; Patrick and Lee 1968), while previous studies on the effect of reaction temperature on the conversion of D-galactose to D-tagatose by L-AIs showed that conversion increased as the incubation temperature was raised (Lee et al. 2004). At higher temperatures, the equilibrium of isomerization reaction shifted toward D-tagatose (Kim et al. 2002). Thus, L-AI with high activity and stability at higher temperatures would have the most potential for D-tagatose production.
To attain a thermostable enzyme for better manipulation, new thermotolerant organisms carrying the target enzyme need to be screened. The organism used as source of L-AI in this study is Acidothermus cellulolytics ATCC 43068, an acido-thermotolerant bacterium that thrives at an optimum condition of 55°C, pH 5.2 (Mohagheghi et al. 1986). Although L-AIs have been characterized from various microorganisms and the mechanism regarding isomerization has been suggested, L-AI has never been reported from A. cellulolytics. The elucidation of the A. cellulolytics genome sequence (NCBI accession number: NC_008578) in 2006 opened the door for genome scale research on this important microbial strain (Barabote et al. 2009). In this paper, we identified the L-AI in A. cellulolytics ATCC 43068 (ACAI) and reported the gene cloning, expression, purification, and characterization of this L-AI. The feasibility of the recombinant enzyme used for D-tagatose production was assessed and discussed in comparison with previously described L-AIs.
Materials and methods
Materials
Taq Plus (Taq DNA polymerase + Pfu DNA polymerase) DNA polymerase, deoxynucleoside triphosphate, and chemicals for polymerase chain reaction (PCR) were obtained from Sangong (Shanghai, China), and the pMD19-T Simple vector, restriction endonuclease, and T4 DNA ligase were obtained from Takara (Dalian, China). The pET-22b(+) vector was purchased from Novagen (Darmstadt, Germany); electrophoresis reagents and all chemicals used for enzyme assays and characterization were obtained from Sigma (St. Louis, MO, USA). Oligonucleotides were synthesized by Sangong (Shanghai, China).
Bacterial strains and culture conditions
Acidothermus cellulolyticus (ATCC 43068) was obtained from the American Type Culture Collection (ATCC), Manassas, VA, USA. A. cellulolyticus was grown in an ATCC medium (ATCC medium: 1473 LPBM acido-thermophile medium, pH 5.2) at 55°C. Escherichia coli JM109 and E. coli BL21 were respectively used as hosts for cloning and expression. Strains were grown in Luria-Bertani (LB) medium with ampicillin (50 μg/ml) in a rotary shaker at 37°C. The plasmid pMD19-T Simple vector was used as a cloning and sequencing vector, and pET-22b(+) was used for expression.
Cloning and expression of the araA gene from A. cellulolyticus ATCC 43068
The genomic DNA of A. cellulolyticus ATCC 43068 was prepared with the method described by Saito and Miura (1963). The whole genome of this strain was sequenced and released in GenBank (NCBI accession number: NC_008578), which revealed that there is a putative araA gene encoding an L-AI in the genome (NCBT gene ID: 4485651, protein ID: YP_872632). Based on the putative gene, two primers were designed as follows: Primer 1, 5′-TATATAAGCTTTCACCACGGCCGCGATGC-3′ and Primer 2, 5′-TTATCCATATGACCGACCTGCCCTATCCG-3′. PCR amplification was performed by Taq Plus DNA polymerase for 30 cycles consisting of 94°C for 30 s, 58°C for 1 min, and 72°C for 1 min, followed by a final extension step of 10 min at 72°C. A DNA fragment about 1,500 bp was amplified and then sequenced by Sangong (Shanghai, China). Sequence analysis showed that it contains a 1,503-bp open reading frame encoding an L-AI. An expression plasmid (pET-ACAI) was constructed by ligation of gene araA, digested by NdeI and HindIII, into the corresponding restriction sites of the pET-22b(+) plasmid and transformed into E. coli BL21(DE3) competent cells. The transformed strain was cultured in LB medium supplemented with 0.05 mg/ml ampicillin at 37°C. When the culture reached an optical density of 0.6 at 600 nm, isopropyl-β-D-thiogalactopyranoside was added to a final concentration of 0.4 mM, and the cells were grown for an additional 6 h and harvested by centrifugation at 10,000 × g, 4°C for 15 min.
Purification of ACAI
The centrifuged cells were resuspended in 50 mM Tris-HCl buffer (pH 8.0) and disrupted by sonication. The lysates were centifugated (12,000 × g, for 20 min at 4°C) to remove cell debris, and the supernatants were heated at 70°C for 15 min. The supernatants were collected as crude extract and heated at 70°C for 15 min. After centrifugation (12,000 × g, 20 min, 4°C), the supernatant was loaded onto a Q-Sepharose column (3.5 × 15 cm; GE Healthcare, Sweden), pre-equilibrated with 20 mM Tris–HCl buffer (pH 8.0). Elution was performed with a linear gradient of NaCl (0 to 1 M) at a flow rate of 2 ml/min. Protein fractions containing L-AI activity were pooled, concentrated, and passed through a Superdex 200 column (HiLoad 10/300 prep grade; GE Healthcare, Sweden), pre-equilibrated with 20 mM Tris–HCl buffer (pH 7.5) containing 150 mM NaCl, at a flow rate of 0.5 ml/min. All purification steps were carried out using an Äkta Purifier System (GE Healthcare, Sweden) at 4°C. The purified enzymes were dialyzed against 10 mM Tris–HCl (pH 7.5) and analyzed by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Enzyme assay and protein determination
L-AI activity was measured by the determination of the amount of produced ketose (D-tagatose). The reaction mixture of 1 ml contained D-galactose (50 mM), MnCl2 (1 mM), CoCl2 (0.5 mM), Tris–HCl buffer (50 mM, pH 7.5), and 100 μl of enzyme preparation at a suitable dilution. The reaction mixture was incubated at 75°C for 30 min, followed by cooling samples on ice to stop the reaction. The generated D-tagatose was determined by the cysteine-carbazole sulfuric acid method, and the absorbance was measured at 560 nm (Dishe and Broenfreund 1951). One unit of L-AI activity was defined as the amount of enzyme catalyzing the formation of 1 μmol D-tagatose per minute.
Protein concentration was measured by the method of Lowry using bovine serum albumin as a standard (Lowry et al. 1951). During chromatographic purification steps, the protein concentration was estimated by observing the absorbance at 280 nm.
Effect of temperature and pH
The optimum temperature of enzyme activity was measured by assaying the enzyme samples over the range of 40–90°C for 30 min. Three buffer systems, sodium acetate (50 mM, pH 4.0–5.0), sodium phosphate (50 mM, pH 6.0–7.0), and Tris–HCl (50 mM, pH 7.5–9.0), were used for measuring the optimum pH of enzyme activity. The thermal stability of the enzyme was studied by incubating the enzyme in Tris–HCl buffer (50 mM, pH 7.5) with or without added Mn2+ and Co2+ ions (1 mM) at 75°C. At given time intervals, samples were withdrawn, and the residual activity was measured under standard assay conditions. To determine the pH stability, the enzyme was incubated at pH 6.0, 7.5, and 8.0 at room temperature (approximately 25°C) for up to 48 h, and the remaining enzyme activity was measured at time intervals under standard assay conditions.
Effect of various metallic ions
Before studying the effects of various metallic ions on ACAI activity, the enzyme solution was dialysed against 50 mM Tris–HCl buffer (pH 7.5) containing 10 mM ethylenediamine tetraacetic acid (EDTA) for 48 h at 4°C. Subsequently, the enzyme was dialyzed against 50 mM EDTA-free Tris–HCl buffer (pH 7.5). Enzyme activity was then assessed as described above in the presence of the following ions at 1 mM: Co2+, Mn2+, Mg2+, Fe2+, Ca2+, Ba2+, Ni2+, Zn2+, and Cu2+, respectively. The activity without any metal ion addition was set to 100%. The measured activities were compared with the activity of the enzyme without metal ion addition under the same conditions.
Determination of kinetic parameters
Kinetic parameters of ACAI were determined in 50 mM Tris–HCl buffer (pH 7.5), 1 mM Mn2+, 0.5 mM Co2+, and 1–800 mM substrate (D-galactose). The samples were incubated at 75°C for 20 min. The enzyme reaction was stopped by chilling on ice, and the amount of D-tagatose was determined by the cysteine-carbazole sulfuric acid method. Kinetic parameters, such as K m (mM) and V max (U/mg protein) for substrates were obtained using the Lineweaver–Burk equation. All assays were performed in triplicate at least two separate times. The kinetic data presented represent averages of statistically relevant measurements with their associated standard deviations.
Analysis of the isomerization of D-galactose to D-tagatose by ACAI
The conversion mixture (10 ml) contained 50 mM of D-galactose, 0.5 mM Co2+, 1 mM Mn2+, and 1.0 mg of purified enzyme (19.29 U) in 50 mM Tris–HCl buffer (pH 7.5). The study of the kinetic conversion of D-galactose was investigated until 36 h at 70°C, 75°C, and 80°C. Samples were taken periodically, and the concentration of the generated D-tagatose was determined by the cystein-carbazol-sulfuric acid method.
Results
Cloning and sequence analysis of the araA gene encoding an L-AI
The DNA sequence analysis of araA revealed an open reading frame of 1,503 bp, encoding a polypeptide of 501 amino acid residues with a calculated isoelectric point of pH 5.63 and molecular mass of 54,768 Da. It shows 100% identity with the nucleotide sequence corresponding to the putative araA gene (NCBI Gene ID: 4485651) from A. cellulolytics ATCC 43068 whole genome (GenBank accession number: NC_008578), suggesting a perfect conservation of the gene among A. cellulolytics strains. A homology search revealed that the deduced araA gene product ACAI showed 55% amino acid identity with L-AI from Geobacillus stearothermophilus (AAD45718); 54% identity with L-AIs from Bacillus stearothermophilus (AAD45718), Thermus sp. (AY225311), and Alicyclobacillus acidocaldarius (AAY68209); 53% identity with L-AI from Thermotoga neapolitama (AY028379); 52% identity with L-AI from Thermotoga maritime (NP_228089); and 51% identity with L-AI from E. coli (AAA23463). Four highly conserved residues, E303, E333, H350, and H450, which are considered to be crucial for the catalytic activity of L-AI (Manjasetty and Chance 2006), were found in four conserved regions (Fig. 1).

Multiple sequence alignment of L-arabinose isomerase (L-AI) from Escherichia coli (ECAI), Alicyclobacillus acidocaldarius (AAAI), Bacillus stearothermophilus (BSAI), Thermus sp. (TSAI), Geobacillus stearothermophilus (GSAI), Thermotoga maritime (TMAI), and Thermotoga neapolitama (TNAI). The alignment was performed using ClustalW program. Amino acid residues that are identical in all the displayed sequences are marked by asterisks; strongly conserved or weakly conserved residues are indicated by colons or dots, respectively. The residues involved in the active site of L-AI from E. coli are indicated with red letters
Expression and purification of the araA gene
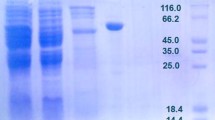
E. coli expressed recombinant L-AI exhibited a strong protein band of close to 55 kDa on SDS-PAGE, compared to that of the control E. coli BL21(DE3), which was in agreement with the predicted molecular mass for the L-AI protein (Kim et al. 2002; Kim and Oh 2005; Lee et al. 2004, 2005b; Patrick and Lee 1968; Poonperm et al. 2007; Prabhu et al. 2008; Rhimi and Bejar 2006). Recombinant ACAI was purified to electrophoretical homogeneity by heat treatment followed by ion-exchange chromatography and gel filtration with a yield of 27% (Table 1). The molecular mass of the purified enzyme was estimated to be approximately 55 kDa by SDS-PAGE (Fig. 2). The purified enzyme exhibited L-AI activity, which strongly supported the assumption that the putative araA gene in A. cellulolytics ATCC 43068 corresponded to that of L-AI protein.

Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of recombinant ACAI from each purification step. Lanes: 1, molecular standard marker (20 μg/ml); 2, cell extract after heat treatment (μg/ml); 3, sample from lane 2 after Q-sepharose ion exchange (89.6 μg/ml); 4, sample from lane 3 after Superdex 200 gel filtration (37.3 μg/ml). The volume loaded on SDS-PAGE for each sample was 15 μl
Effect of temperature, pH, and metal ions on ACAI activity
The temperature dependence of the recombinant enzyme was determined in the presence of Mn2+ (1 mM) and Co2+ (0.5 mM) after 30 min of incubation at various temperatures. The optimum temperature of the L-AI from A. cellulolytics was 75°C. Investigation of pH effect on ACAI activity showed that the enzyme was optimally active at pH 7.5, while more than 90% of its maximum activity was maintained at the acidic pH of 6.0.
To determine the effect of metal ions, ACAI activity was measured in the presence of various metal ions. The enzyme had a requirement for metal ion for activity stimulation. Its activity was significantly activated by the addition of 1.0 mM Mn2+ (4.5-fold) and 0.5 mM Co2+ (5.5-fold). The full activity (6.1-fold) was achieved when both of Mn2+ and Co2+ were added at the concentration of 1 and 0.5 mM, respectively. Compared to Mn2+ and Co2+, other metal ions, such as Mg2+, Ni2+, and Ba2+, were poor activators (data not shown). Fe2+, Ca2+, and Zn2+ had no effect on ACAI activity, while Cu2+ inhibited the enzyme up to approximately 30%.
Effect of temperature, pH, and metal ions on ACAI stability
The ACAI was perfectly stable at 65°C, and no significant activity decrease was observed after 2 h incubation without metal ion addition. At higher temperatures (>65°C), the thermostability of ACAI was proved to be metal-dependent. In the absence of metallic ions, the enzyme was inactivated up to 50% activity under the same conditions (Fig. 3a), while the enzyme maintained 90% and 76% activity after 2 h incubation at 75°C in the presence of 1 mM Co2+ and 1 mM Mn2+, respectively. The pH stability of ACAI was measured at pH ranging from 6.0 to 8.0 at room temperature (approximately 25°C). The enzyme showed relatively high stability at acidic pH, since 80% and 70% of its activity were retained after 12 and 24 h of incubation at pH 6.0, respectively (Fig. 3b).

Effect of temperature (a) and pH (b) on ACAI stability. a The thermal stability profiles of the purified ACAI. Closed squares: thermal stability at 65°C in the absence of metallic ions; open squares: thermal stability at 75°C in the absence of metallic ions; closed triangles: thermal stability at 75°C in the presence of 1 mM Mn2+; open triangles: thermal stability at 75°C in the presence of 1 mM Co2+. b The pH stability profiles of the purified ACAI. Closed squares: pH 7.5; open squares: pH 8.0; closed triangles: pH 6.0. Residual activity was measured under standard conditions (75°C, pH 7.5). Activity measured with non-incubated enzyme was set to 100%
Kinetic studies of ACAI for D-galactose
On the basis of the Lineweaver–Burk plots, the kinetic parameters of ACAI for D-galactose were determined under optimal temperature and pH conditions (75°C, pH 7.5). The apparent K m, V max, and catalytic efficiency k cat/K m of ACAI using D-galactose as a substrate were 28.9 mM−1, 4.9 μmol min−1 mg−1, and 9.3 min−1 mM−1, respectively.
Production of D-tagatose by ACAI at different temperatures
The isomerization of D-galactose (50 mM) to D-tagatose by ACAI at 70°C, 75°C, and 80°C (pH 7.5) demonstrated that the isomerization reaction of D-galactose to D-tagatose reached equilibrium after 12 h incubation, and the conversion ratio was 45%, 53%, and 43% at 70°C, 75°C, and 80°C, respectively (Fig. 4). The production of D-tagatose from D-galactose was further proved by high-performance liquid chromatography analysis, and no by-product was observed.

Conversion of D-galactose to D-tagatose by ACAI at different temperatures. Open squares: conversion curve at 70°C; closed squares: conversion curve at 75°C; closed triangles: conversion curve at 80°C
Discussion
Based on the analysis of the genome sequence of A. cellulolytics ATCC 43068, an isomerase-encoding gene (araA) was proposed as an L-AI. The identified araA gene was cloned from A. cellulolytics ATCC 43068, overexpressed in E. coli, and it was confirmed that the gene product ACAI exhibited L-AI activity for D-tagatose production. Sequence analysis showed that ACAI had higher identity to thermophilic L-AIs than mesophilic L-AIs, and the content of hydrophobic amino acid of ACAI was also higher than mesophilic L-AIs, indicating that ACAI is much more hydrophobic than other mesophilic AIs. In addition, most of the amino acids in the conserved regions were hydrophobic. ACAI was characterized to be thermostable at high temperatures. This result is consistent with the fact that hydrophobic interactions are important for protein thermostability.
Generally, a major consideration in a biotransformation process is the development and/or improvement of suitable biological catalysts (Illanes 1999). It has been reported that thermotolerant enzymes, which can provide higher reaction rates and process yields, higher solubility of substrates and products, and fewer contamination problems, are an important aspect of biocatalytic processes (Poonperm et al. 2007; Kim 2004). The ACAI from A. cellulolytics ATCC 43068 was optimally active at 75°C with Mn2+ and Co2+ additions and perfectly stable below 65°C without metal ion addition, suggesting that this enzyme is remarkably thermostable.
On the other hand, industrial application requires a slightly acidic pH range to reduce browning reaction and the formation of by-products (Kim 2004). Therefore, thermostable L-AIs with high activity and stability at a moderately low pH will be interesting (Lee et al. 2005b). ACAI from Acido-thermophile bacterium A. cellulolytics ATCC 43068 exhibits high activity and stability at a broad pH range of 6.0–8.0. Although the ACAI showed maximum activity at pH 7.5, it maintained more than 90% of its maximum activity at pH 6.0, and no significant activity decrease was observed after 48 h incubation at pH 6.0. Generally, the isomerization of D-galactose to D-tagatose reached equilibrium after 12 h of incubation. Indeed, the ACAI retained more than 80% of its maximum activity after 12 h incubation at pH 6.0, making ACAI a good candidate for industrial applications at acidic pH (approximately 6) values.
The majority of L-AIs are metalloproteins involving metal ions for their optimal activity and thermostability (Rhimi and Bejar 2006). It was reported that mesophilic and thermophilic L-AIs require Mn2+ as a cofactor to enhance the isomerization reaction rate (Patrick and Lee 1968), while the hyperthermophilic L-AIs require Co2+ (Kim et al. 2002; Lee et al. 2004). The ACAI required a divalent metal ion, either Mn2+ or Co2+, for both enzymatic activity and thermostability improvement at high temperature. The addition of 1 mM Mn2+ plus 0.5 mM Co2+ greatly improved its activity (6.1-fold). The thermostability of ACAI at higher temperatures (≥65°C) was proved to be metal-dependent. In the absence of metallic ions, the enzyme was inactivated up to 50% activity after 2 h incubation at 75°C (Fig. 3a). While in the presence of 1 mM Co2+ and 1 mM Mn2+, the activity increased by 40% and 26%, respectively, after 2 h of incubation at 75°C compared to that in the absence of metallic ions (Fig. 3a). These results suggested the both of Mn2+ and Co2+ ions played important roles in the enzyme stabilization at high temperatures and in the isomerization reaction.
Although L-AIs catalyzes the reversible isomerization of both L-arabinose to L-ribulose and D-galactose to D-tagatose, respectively, L-arabinose is the preferred substrate of L-AI (Oh 2007), which limits the industrial application for D-tagatose production. ACAI catalyzes the isomerization of L-arabinose to L-ribulose (data not shown), also showed high substrate affinity towards D-galactose. Compared with other L-AIs, ACAI exhibits a high catalytic efficiency (k cat/K m; 9.3 min−1 mM−1; Table 2), which makes it potential for D-tagatose production. This enzyme efficiency was related to the high yield of D-tagatose production which reached 53% after 12 h at 75°C. Structural and mechanistic studies, to clarify the reason for substrate choosing of ACAI, are presently under way.
The L-AI from A. cellulolytics ATCC 43068 was characterized to be thermostable, active, and stable at acidic pH, and be efficient for D-tagatose biotransformation, which makes it a good candidate to produce D-tagatose in ideally industrial condition.
References
Barabote RD, Xie G, Leu DH et al (2009) Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations. Genome Res. doi:10.1101/gr.084848.108
Buemann B, Toubro S, Raben A, Blundell J, Astrup A (2000) The acute effect of D-tagatose on food intake in human subjects. Br J Nutr 84:227–231
Cheetham P, Wootton A (1993) Bioconversion of D-galactose into D-tagatose. Enzyme Microb Technol 15:105–108
Chouayekh H, Bejar W, Rhimi M, Jelleli K, Mseddi M, Bejar S (2007) Characterization of an L-arabinose isomerase from the Lactobacillus plantarum NC8 strain showing pronounced stability at acidic pH. FEMS Microbiol Lett 277:260–267
Dishe Z, Broenfreund E (1951) A new spectrophotometric method for the detection of keto sugars and trioses. J Biol Chem 192:583–587
Donner TW, Wilber JF, Ostrowski D (1999) D-Tagatose, a novel hexose: acute effects on carbohydrate tolerance in subjects with and without type 2 diabetes. Diabetes Obes Metab 1:285–291
Illanes A (1999) Stability of biocatalysts process biotechnology. Electronic J Biotechnol 2:7–15
Ishida Y, Kamiya T (1997) Cloning and characterization of the D-tagatose 3-epimerase gene from Pseudomonas cichorii ST-24. J Ferment Bioeng 83:529–534
Jørgensen F, Hansen O, Stougaard P (2004) Enzymatic conversion of D-galactose to D-tagatose: heterologous expression and characterisation of a thermostable L-arabinose isomerase from Thermoanaerobacter mathranii. Appl Microbiol Biotechnol 64:816–822
Kim P (2004) Current studies on biological tagatose production using L-arabinose isomerase: a review and future perspective. Appl Microbiol Biotechnol 65:243–249
Kim HJ, Oh DK (2005) Purification and characterization of an l-arabinose isomerase from an isolated strain of Geobacillus thermodenitrificans producing d-tagatose. J Biotechnol 120:162–173
Kim BC, Lee YH, Lee HS, Lee DW, Choe EA, Pyun YR (2002) Cloning, expression and characterization of L-arabinose isomerase from Thermotoga neapolitana: bioconversion of D-galactose to D-tagatose using the enzyme. FEMS Microbiol Lett 212:121–126
Kim HJ, Ryu SA, Kim P, Oh DK (2003a) A feasible enzymatic process for D-tagatose production by an immobilized thermostable L-arabinose isomerase in a packed-bed bioreactor. Biotechnol Prog 19:400–404
Kim JW, Kim YW, Roh HJ, Kim HY, Cha JH, Park KH, Park CS (2003b) Production of tagatose by a recombinant thermostable L-arabinose isomerase from Thermus sp. IM6501. Biotechnol Lett 25:963–967
Kim HJ, Hyun EK, Kim YS, Lee YJ, Oh DK (2006) Characterization of an Agrobacterium tumefaciens D-psicose-3-epimerase that converts D-fructose to D-psicose. Appl Environ Microbiol 72:981–985
Lee DW, Jang HJ, Choe EA (2004) Characterization of a thermostable L-arabinose (D-galactose) isomerase from the hyperthermophilic eubacterium Thermotoga maritima. Appl Environ Microbiol 70:1397–1404
Lee DW, Choe EA, Kim SB, Eom SH, Hong YH, Lee SJ, Lee HS, Lee DY, Pyun YR (2005a) Distinct metal dependence for catalytic and structural functions in the L-arabinose isomerase from the mesophilic Bacillus halodurans and the thermophilic Geobacillus stearothermophilus. Arch Biochem Biophys 434:333–343
Lee SJ, Lee DW, Choe EA, Hong YH, Kim SB, Kim BC, Pyun YR (2005b) Characterization of a thermoacidophilic L-arabinose isomerase from Alicyclobacillus acidocaldarius: role of Lys-269 in pH optimum. Appl Environ Microbiol 71:7888–7896
Levin GV (2002) Tagatose, the new GRAS sweetener and health product. J Med Food 5:23–36
Lowry OH, Rosenbroughn NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:256–275
Lu Y, Levin GV, Donner TW (2008) Tagatose, a new antidiabetic and obesity control drug. Diabetes Obes Metab 10:109–134
Manjasetty BA, Chance MR (2006) Crystal structure of Escherichia coli L-arabinose isomerase (ECAI), the putative target of biological tagatose production. J Mol Biol 360:297–309
Mendoza MR, Olano A, Villamiel M (2005) Chemical indicators of heat treatment in fortified and special milks. J Agric Food Chem 53:2995–2999
Mohagheghi A, Grohmann K, Himmel M, Updegraff DM (1986) Isolation and characterization of Acidothermus cellulolytics gen. nov., sp. nov., a new genus of thermophilic, acidophilic, cellulolytic bacteria. Int J Syst Bacteriol 36:435–443
Nakamatu T, Yamanaka K (1969) Crystallization and properties of L-arabinose isomerase from Lactobacillus gayonii. Biochim Biophys Acta 178:156–165
Oh DK (2007) Tagatose: properties, applications, and biotechnological processes. Appl Microbiol Biotechnol 76:1–8
Patrick JW, Lee N (1968) Purification and properties of an L-arabinose isomerase from Escherichia coli. J Biol Chem 243:4312–4318
Poonperm W, Takata G, Okada H, Morimoto K, Granström TB, Izumori K (2007) Cloning, sequencing, overexpression and characterization of L-rhamnose isomerase from Bacillus pallidus Y25 for rare sugar production. Appl Microbiol Biotechnol 76:1297–1307
Prabhu P, Tiwari MK, Jeya M, Gunasekaran P, Kim IW, Lee JK (2008) Cloning and characterization of a novel L-arabinose isomerase from Bacillus licheniformis. Appl Microbiol Biotechnol 81:283–290
Rhimi M, Bejar S (2006) Cloning, purification and biochemical characterization of metallic-ions independent and thermoactive L-arabinose isomerase from the Bacillus stearothermophilus US100 strain. Biochim Biophys Acta 1760:191–199
Rollini M, Manzoni M (2005) Bioconversion of D-galactitol to tagatose and dehydrogenase activity induction in Gluconobacter oxydans. Process Biochem 40:437–444
Rosenplenter K, Mende K (2004) Use of D-tagatose for improving aroma and flavor in foods and beverages WO patent 2004/073419
Saito H, Miura K (1963) Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim Biophys Acta 72:619–629
Troyono E, Martinez-Castro I, Olano A (1992) Kinetics of galactose and tagatose formation during heat-treatment of milk. Food Chem 45:41–43
Yoon SH, Kim P, Oh DK (2003) Properties of L-arabinose isomerase from Escherichia coli as biocatalysis for tagatose production. World J Microbiol Biotechnol 19:47–51
Acknowledgements
This work was supported by the grants from the National High Technology “863” Program of China, Project No. 2006AA10Z334, the Natural Science Foundation of China, Project No. 20906040, the Natural Science Foundation of Jiangsu Province, Project No. BK2008099 and BK2008003, the Research Program of State Key Laboratory of Food Science and Technology, Jiangnan University, Project No. SKLF-MB-200804 and SKLF-TS-200805, the Specialized Research Fund for the Doctoral Program of Higher Education, Project No. 200802951024, and the Program for Innovative Research Team of Jiangnan University, Project No. 2008CXTD01.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cheng, L., Mu, W., Zhang, T. et al. An L-arabinose isomerase from Acidothermus cellulolytics ATCC 43068: cloning, expression, purification, and characterization. Appl Microbiol Biotechnol 86, 1089–1097 (2010). https://doi.org/10.1007/s00253-009-2322-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2322-z