
Abstract
The l-rhamnose isomerase gene (L-rhi) encoding for l-rhamnose isomerase (l-RhI) from Bacillus pallidus Y25, a facultative thermophilic bacterium, was cloned and overexpressed in Escherichia coli with a cooperation of the 6×His sequence at a C-terminal of the protein. The open reading frame of L-rhi consisted of 1,236 nucleotides encoding 412 amino acid residues with a calculated molecular mass of 47,636 Da, showing a good agreement with the native enzyme. Mass-produced l-RhI was achieved in a large quantity (470 mg/l broth) as a soluble protein. The recombinant enzyme was purified to homogeneity by a single step purification using a Ni-NTA affinity column chromatography. The purified recombinant l-RhI exhibited maximum activity at 65°C (pH 7.0) under assay conditions, while 90% of the initial enzyme activity could be retained after incubation at 60°C for 60 min. The apparent affinity (K m) and catalytic efficiency (k cat/K m) for l-rhamnose (at 65°C) were 4.89 mM and 8.36 × 105 M−1 min−1, respectively. The enzyme demonstrated relatively low levels of amino acid sequence similarity (42 and 12%), higher thermostability, and different substrate specificity to those of E. coli and Pseudomonas stutzeri, respectively. The enzyme has a good catalyzing activity at 50°C, for d-allose, l-mannose, d-ribulose, and l-talose from d-psicose, l-fructose, d-ribose and l-tagatose with a conversion yield of 35, 25, 16 and 10%, respectively, without a contamination of by-products. These findings indicated that the recombinant l-RhI from B. pallidus is appropriate for use as a new source of rare sugar producing enzyme on a mass scale production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rare sugar, which has numerous applications in pharmaceutical, food, and agricultural industries, is specified as a monosaccharide and its derivatives that rarely exist in nature (Jeong and Yoo 1998; Sui et al. 2005; Lerner and Mennitt 1994; Bertelsen et al. 1999; Ly et al. 2006; Granström et al. 2004). We focus on rare sugar production using a biotransformation method, which has particular advantages over chemical method in many ways including more facilitated processing, better activity, and higher specificity under mild conditions with a low occurrence of by-products. Several enzymes involved in the biotransformation of rare sugars have been studied, i.e., d-tagatose-3-epimerase, xylitol dehydrogenase and l-arabinose isomerase (Itoh et al. 1994; Poonperm et al. 2007a; Kim and Oh 2005). In our present work, l-rhamnose isomerases (l-RhIs), which was found to have extraordinary functions in several aldoses and ketoses interconversion from new isolated bacteria, was studied.
l-RhI (E.C. 5.3.1.14), which generally occurs in the metabolism of fructose and mannose (Kanehisa and Goto 2000), is derived from various organisms such as Aerobacter aerogenes, Bacillus subtilis, Escherichia coli, Lactobacillus plantarum, Mycobacterium smegmatis, and Pseudomonas stutzeri (Noltmann 1972; Oudega et al. 1997; Badia et al. 1989; Domagk and Zech 1963; Izumori et al. 1976; Bhuiyan et al. 1997). In 1995, the gene encoding for l-RhI from E. coli was cloned and partially characterized for the first time (Garcia-Junceda et al. 1995). Later, cloning, expression, and characterization of l-RhI from P. stutzeri have been studied (Leang et al. 2004a). Leang and colleagues reported that the enzyme from E. coli showed high substrate specificity toward l-rhamnose and used to synthesize l-rhamnulose, while the enzyme from P. stutzeri exhibited a broad substrate specificity which can be applied for the catalyzing of various aldoses and ketoses into their corresponding products (Leang et al. 2004b). Moreover, the catalytic mechanism of l-RhI from these two organisms has also been elucidated through analysis of the X-ray structure of the enzyme crystal (Kornderfer et al. 2000; Yoshida et al. 2007). Applications for the production of several rare sugars using l-RhI have been implemented, i.e., d-allose from d-psicose, l-talose from l-tagatose and d-gulose from d-sorbose, mostly in the immobilizing form (Menavuvu et al. 2006; Bhuiyan et al. 1999).
A major consideration in a biotransformation application is the improvement and development of suitable biological catalysts (Illanes 1999). It has been reported that thermotolerant enzymes, which can provide a higher reaction rate and process yield, higher solubility of substrates and products, and fewer contamination problems, is a crucial variable in any biocatalytic process (Mozhaev 1993; Wasserman 1984). To attain a thermostable enzyme for the better manipulation of the processes, new sources of the thermotolerant organisms carrying the target enzyme need to be screened.
The organism used as source of the l-RhI in this study is Bacillus pallidus Y25, a facultative thermotolerant bacterium that thrives at an optimum temperature of 55°C. Generally, B. pallidus has been known as a bacterium involved in bioremediation (Bustard and Wright 2002; Cramp et al. 1997; Kuroda et al. 2004). However, we have characterized that this organism also possesses dehydrogenases that play crucial roles in the production of rare ketoses, especially for the production of l-xylulose and d-psicose from xylitol and allitol, respectively (Poonperm et al. 2007b). Coincidentally, we observed unexpected aldo-sugars, l-xylose and d-allose, which appeared during bioconversions of l-xylitol-l-xylulose and d-allitol-d-psicose. It was concluded that the enzymes catalyzing these reactions were d-arabinose isomerase and l-rhamnose isomerase, respectively.
In the present work, we describe the cloning, sequencing, and overexpression of l-rhamnose isomerase gene (L-rhi) from B. pallidus Y25. Biochemical characterization and substrate specificity of the enzyme against diverse sugars were also determined and compared to that of other microorganisms.
Materials and methods
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. B. pallidus Y25, the isolated strain, was grown at 55°C for 24 h in a yeast extract medium (0.5% yeast extract, 0.5% polypepton, and 0.5% NaCl in water, pH 8.5) supplemented with 1% sodium l-glutamate (Poonperm et al. 2007a). For the gene cloning experiments, pBluescriptII SK- (Stratagene) was used as the cloning vector and E. coli DH5α was used as the corresponding host, while pQE60 (QIAGEN) was used as the expression vector and E. coli JM109 was used as the host for protein expression. Recombinant E. coli was grown in Luria–Bertani (LB) broth medium or super broth medium (SB) supplemented with ampicillin (100 μg/ml) at 30°C with shaking (120 rpm). When required, isopropyl-β-d-thiogalactopyranoside (IPTG: 1 mM) and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal: 40 μg/ml) were added into the medium. For the induction of the lac promoter after the cell density approached OD600 = 0.7–1.0, IPTG (1 mM) was added, and cells were incubated for 4 h.
Enzymes, primers, and chemicals
Restriction enzymes, modifying enzymes, and other molecular-related reagents were purchased from Takara Shuzo. Reaction conditions were employed as recommended by the suppliers. General chemicals were obtained from Wako Pure Chemicals or Sigma Aldrich. Synthetic oligonucleotide DNA primers for polymerase chain reaction (PCR) and the sequencing reaction, as shown in Table 2, were purchased from Invitrogen. d-/l-Psicose and other rare sugars were prepared in our laboratory by a previously described method (Itoh et al. 1995; Takeshita et al. 1996).
DNA manipulations and hybridizations
Genomic DNA preparation and recombinant DNA techniques were performed according to standard procedures (Sambrook et al. 1989). DNAs were extracted from agarose gels with a Geneclean turbo kit (Q-BIO). Plasmid isolation was performed with a Mag extractor plasmid kit (Toyobo). E. coli cells were transformed according to the method of Hanahan (1983). The DNA fragment amplified by PCR was labeled with digoxigenin-11-dUTP (DIG-11-dUTP) using a DIG DNA labeling kit (Roche Diagnostics) and then utilized as probe for hybridizations. Detection was performed using DIG chemiluminescent detection kit (Roche Diagnostics) according to the attached protocol.
N-terminal and internal amino acid sequencing
To purify l-RhI, a crude extract from cultured B. pallidus was prepared by grinding washed cells with aluminum oxide and then applied onto Q-Sepharose HP (1.6 × 10 cm; GE Healthcare Bio-Science) followed by Phenyl-Sepharose HP (1.6 × 10 cm; GE) and Mono Q HR (0.5 × 5 cm; GE) columns. The purification was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) on a slab gel (12% polyacrylamide gel, 9 × 7.8 × 0.1 cm) in Tris–glycine (pH 8.3) at a current of 20 mA for 3 h by the method of Laemmli (1970). Protein bands were visualized by staining with 0.2% Coomassie brilliant blue R-250. N-terminal amino acid sequencing was carried out for the target peptide fragment. To determine the internal amino acid sequences, purified l-RhI was digested with CNBr and separated by SDS–PAGE before electroblotting onto a polyvinyldene difluoride (PVDF) membrane (FLUOROTRANS; Pall). The peptide fragments were eluted from the membrane with 70% formic acid and sequenced using an automated Edman degradation system (Model 491; Applied Biosystems).
PCR amplification of L-rhi probe
Purified chromosomal DNA was used as a template to amplify part of the L-rhi using the synthesized oligonucleotide primers derived from the N-terminal and internal peptide fragments of the enzyme (Table 2). Six pairs of synthetic primers were used for the PCR amplifications (F1-R1, F2-R1, F1-R2, F1-R3, F2-R2, and F2-R3). The amplification reactions were performed in a final volume of 20 μl containing PCR buffer, 0.25 mM of deoxyribonucleoside 5′-triphosphate mixture, 2.5 mM MgCl2, 10 pmol of each primer and 1.25 U of LA Taq DNA polymerase (Takara Shuzo). The PCR reaction was held in a thermal cycler at 95°C in for 15 min followed by 30 cycles of amplification consisting of 1 min of denaturing at 95°C, 1 min of annealing at 55°C, and 2 min of extension at 72°C, and final holding at 72°C for 5 min (iCycler thermal cycler 582BR; Bio-Rad). Under these conditions, we obtained the DNA sequence of the specific amplified 1,000-bp fragment using the pair of F1-R1. The 1,000-bp fragment amplified by PCR was labeled with digoxigenin-11-dUTP (DIG-11-dUTP) and further used as a probe for the hybridizations
Cloning of L-rhi
Genomic DNA from B. pallidus was digested with several restriction enzymes and subjected to Southern hybridization analysis with the 1,000-bp labeled probe. A 3.0-kb EcoRI–BamHI fragment was selected from several hybridized fragments and cloned into EcoRI- and BamHI-digested pBluescriptII SK- vector. The constructed plasmid was named as pLY25-I and introduced into E. coli DH5α to construct a genomic library. Transformants that could be grown on an LB plate containing ampicillin, IPTG, and X-gal were analyzed for a selection of positive insertion by colony hybridization using the 1,000-bp DNA probe. Nucleotide sequence analysis was carried out on the positive clones obtained from hybridization and were then compared with known databases using the BLAST program to find the N- and C-terminal regions of the l-RhI for the synthesis of new oligonucleotide primers of the complete sequencing (Altschul et al. 1990). The amplification was carried out using the synthetic oligonucleotide primer Forward-L1, which contains an engineered NcoI site, and the Reverse-L1, which replaced the stop codon by a BglII site to construct a C-terminal His-tag fusion protein (Table 2). The amplification condition was the same as described above. An approximately 1.3-kb NcoI–BglII fragment containing the complete coding sequence of L-rhi was obtained from agarose gel electrophoresis. To construct a cloning vector pLY25-II, the fragments were treat with Klenow enzyme and ligated with pBluescriptII SK- vector that had been previously digested with HincII. The pLY25-II vector was then transformed into E. coli DH5α. Positive clones were grown in 3-ml LB medium containing ampicillin (100 μg/ml) at 30°C for 12 h. Plasmid was prepared from the cell cultures, and the insert gene was then confirmed by nucleotide sequencing.
Nucleotide sequence analysis
DNA sequences of L-rhi derived from pLY25-I and pLY25-II were determined both directional strands by the dideoxy nucleotide chain termination method (Sanger et al. 1977) using DTCS Quick start kit (Beckman Coulter) and analyzed on a CEQ8000 system (Beckman Coulter). Homology searches were performed with nucleotide sequence and its deduced amino acid sequence available from the DDBJ databases using the BLAST program (Altschul et al. 1990). Multiple sequence alignments were assembled with the ClustalW program (Thompson et al. 1994).
Construction of the expression plasmid pLY25-III
To facilitate the purification steps, His-Tag was added to the C-terminal of the l-RhI protein. The L-rhi of pLY25-II was digested with NcoI and BglII and ligated with pQE60 predigested with the same set of restriction enzymes (named as pLY25-III) before transformed into the expression host, E. coli JM109. The transformants were grown in 3 ml SB supplemented with ampicillin (100 μg/ml) at 37°C for 12 h. The protein production was initiated by the addition of 1 mM IPTG. After cultivation, cells were collected, washed, and disrupted by sonication on ice. After centrifugation at 10,000×g, both of the supernatant and the pellet were assayed for l-RhI activity via the method described below.
Preparation of crude extract and purification of recombinant l-RhI
E. coli JM 109 harboring pLY25-III was cultivated at 30°C in 100 ml of SB medium supplemented with ampicillin (100 μg/ml) to a cell density of OD600 = 0.7–1.0. Expression was induced by the addition of 1 mM IPTG, and incubation was continued for 4 h at 30°C. After cultivation, cells were harvested by centrifugation at 10,000×g for 10 min at 4°C and washed twice with distilled water. The washed cells were disrupted by grinding with activated aluminum oxide for 30 min and resuspended in 10 mM sodium phosphate buffer (pH 7.0). Cell debris was removed by centrifugation at 10,000×g for 30 min at 4°C and the resulting crude extract (7 ml) retained for further purification. Unless otherwise noted, purification steps were carried out at room temperature. The cell-free extract was applied onto a Ni-NTA Super flow column (3.4 × 13.5 cm, QIAGEN) previously equilibrated with a binding buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0). Unbounded proteins were washed out from the column with a washing buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). Then, the l-RhI protein was eluted from the column with an elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0). The active fractions were pooled and dialyzed overnight against 10 mM sodium phosphate buffer (pH 7.0) at 4°C. Proteins pooled from each step of the purification process were analyzed by SDS–PAGE.
Enzyme assay and protein determination
l-RhI activity was measured in a 0.5-ml reaction mixture containing 50 mM sodium phosphate buffer (pH 7.0), 1 mM MnCl2, and 50 μl of appropriately diluted enzyme. The reaction was initiated by the addition of l-rhamnose (final concentration, 10 mM). The mixture was then incubated for 10 min at 65°C and the reaction terminated by the addition of 50 μl trichloroacetic acid (10%). The formation of l-rhamnulose was determined spectrophotometrically following the cystein-carbazole method (Dishe and Borenfreud 1951). One unit of activity was defined as the formation of 1 μmol l-rhamnulose within 1 min under the assay conditions. The protein concentration was measured by the method of Bradford (1979) using a protein assay kit (Bio-Rad) according to a standard procedure.
The kinetic parameters, K m (mM) and V max (μmol min−1 mg−1), and the catalytic parameters, k cat (min−1) and k cat/K m (μM−1 min−1), were calculated from Lineweaver–Burk (1/S vs 1/V) plots of the Michaelis–Menten equation.
Sugar conversion
Reactions were performed in test tubes with a final volume of 3 ml containing 50 mM sodium phosphate buffer (pH 7.0), 10 mM substrate (l-rhamnose, d-allose, l-lyxose, d-ribose, d-gulose, l-talose and l-mannose), the purified enzyme (170 U), and 1 mM MnCl2 at 50°C. Isomerization of the substrate and accumulation of the products in the reaction aqueous were determined by a colorimetric method and high-performance liquid chromatography (HPLC; Hitachi) using a separation column (GL-C611, Hitachi) at 60°C eluted with 10−4 M NaOH at a flow rate of 1.0 ml/min.
Nucleotide sequence accession number
The nucleotide sequence of the L-rhi reported in this paper has been deposited in the DDBJ nucleotide sequence database under accession number AB298546.
Results
Cloning and sequence analysis of the gene encoding L-rhi
l-RhI from wild-type B. pallidus was purified to homogeneity in three chromatographic steps as described in “Materials and methods”. The purified enzyme appeared to have a molecular mass of 47 kDa on SDS-PAGE (data not shown), and the N-terminal sequencing of the intact 47 kDa protein resulted in the sequence MVIKESFELARTVYEKKGINN. To investigate the internal amino acid, the purified enzyme was digested with CNBr, and the separation was determined by SDS–PAGE before electroblotting onto a PVDF membrane. Four fragments of 32, 23, 17, and 10 kDa were observed on SDS–PAGE, and the sequences were identified as QSIQQ, KGIYE, KENFGNF and KEFGRY, respectively. From these short amino acid sequences, degenerated oligonucleotide primers were synthesized, as shown in Table 2, and were used for the amplification of a chromosomal DNA from B. pallidus. The resultant of the 1,000-bp DNA fragment obtained from PCR with the F1–R1 pair of primers was utilized as a probe for hybridization in further steps.
The chromosomal DNA from B. pallidus was digested with several restriction enzymes and subjected to Southern hybridization. A 3.0-kb EcoRI–BamHI digested fragment, which was found to hybridize with the 1,000-bp probe, was cloned into the corresponding site of pBluescriptII SK-, generating plasmid pLY25-I, and was subsequently transformed into E. coli DH5α. More than 2,000 colonies were screened, and the positive colonies which had been hybridized with the 1,000-bp DNA probe were performed for sequencing. The deduced amino acid sequence of the gene from pLY25-I showed similarities with l-RhI from several sources, including E. coli and B. subtilis. The N- and C-terminal regions of l-RhI from theses two strains were compared with the fragment from positive clones using the BLAST program (Altschul et al. 1990). Based on the resulting homology, primers were designed to amplify the complete length of L-rhi gene. Moreover, to facilitate the cloning, the restriction sites of NcoI and BglII were combined to forward and reverse primers, respectively.
Amplification of chromosomal DNA using the synthesized primers showed about 1.3 kb on agarose gel electrophoresis. After subcloning the amplified fragment into HincII site of pBlueScriptII SK-, resulting in plasmid pLY25-II, the positive colonies that contained the complete nucleotide sequence of L-rhi were selected and performed for nucleotide sequencing. As demonstrated in Fig. 1, the sequence of the insert gene corresponded to an open reading frame of 1,236-bp encoding 412 amino acid residues with a calculated molecular mass of 47,636 Da, which was in good agreement with the molecular mass of the native purified l-RhI, 47 kDa as observed by SDS–PAGE. Ten base pairs upstream of the initiation codon was a putative 5′-AGGA-3′ ribosome binding site. Sequences similar to the −10 and −35 consensus sequences of E. coli were observed at 17 and 46-bp upstream the L-rhi.

Nucleotide sequence of L-rhi from B. pallidus Y25. The deduced amino acid sequence of L-RhI is indicated by the single-letter code below the nucleotide sequence. The putative -35 and -10 regions promoter and the possible Shine-Dalgarno sequence are shown by wave underline, double underline and underline, respectively. The stop codon is indicated by an asterisk (*)
The primary structure of the l-RhI protein exhibited homology of 58% to that from Oceanobacillus iheyensis (Swiss-Prot accession number Q8ESX0), 57% to that from l-RhI B. subtilis (Swiss-Prot accession number O05264), and 42% identical to that from E. coli (Swiss-Prot accession number P32170). However, it showed a low identity (12%) with l-RhI from P. stutzeri.
Multiple sequence alignment of l-RhI from B. pallidus with that from E. coli and P. stutzeri is displayed in Fig. 2 (Badia et al. 1989; Leang et al. 2004a). Among the nine residues involved in the active site of l-RhI from E. coli, eight residues were found to conserve in l-RhI from B. pallidus, while six residues were similar to that from P. stutzeri.

Multiple sequence alignment of L-rhi from B. pallidus Y25 (BACPL), E. coli (ECOLI) and P. stutzeri (PSEST). The alignment was performed using ClustalW program. Amino acid residues that are identical in all the displayed sequences are marked by asterisks (*), strongly conserved or weakly conserved residues are indicated by colons (:) or dots (·), respectively. The residues involved in the active site of L-rhi from E. coli are indicated with white letters in black boxes
Mass production and purification of recombinant l-RhI
To construct the expression plasmid pLY25-III, L-rhi fragment derived from pLY25-II, with an in-frame fusion His-tag sequence at the C terminus, was ligated with the pQE60 vector at multi cloning sites and was introduced into the expression host, E. coli JM109. This allowed for a generic single step purification of the recombinant l-RhI using a Ni-NTA affinity column chromatography resin.
The recombinant protein cooperating with His-tag was mass-produced in the soluble fraction with significant quantity (470 mg/l broth) and activity (36,500 U/l broth). After the purification process by Ni-NTA affinity column chromatography, the enzyme was purified to 3.87-fold with a specific activity of 77.2 U/mg and a recovery yield of 86.7%.
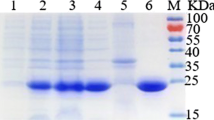
SDS–PAGE of the purified enzyme gave a single band with an apparent molecular mass of 47 kDa (Fig. 3), showing a good agreement with the observed molecular mass of the native enzyme. The enzyme presented a compatible size with several previous finding as showing in Table 3. The l-RhI activity of the purified recombinant l-RhI exhibited over 40-fold greater than that of the authentic strain (1.81 U/mg).

SDS PAGE analysis of recombinant L-rhamnose isomerase from each purification step. Lanes: 1, molecular standard marker; 2, crude extract; 3, purified L-RhI. An arrow indicates purified enzyme eluted from the Ni-NTA super flow column
Characterization of recombinant l-RhI
As summarized in Table 3, biochemical characterizations of recombinant l-RhI were determined. The recombinant enzyme was optimized at 65°C (10-min incubation) and was stable under 60°C (1-h incubation) in sodium phosphate buffer (pH 7.0). The half-life of the enzyme was 1 h at 65°C in sodium phosphate buffer (pH 7.0). The optimum pH for the enzymes was 7.0 and the enzyme was stable under alkaline conditions (pH 7–10).
The effect of metal ions on an enhancement of enzyme activity was investigated (data not shown). As a result, low activity of the enzyme was observed when the reaction lack of Mn2+ or Co2+.
Substrate specificity and kinetic properties of the recombinant l-RhI
Substrate specificity of the enzyme was studied. As shown in Table 4 and Fig. 4, the enzyme has high specificity for aldoses that have an –OH branch of C2 and C3 in the D- side by Fischer projection such as l-rhamnose, l-lyxose, d-ribose, l-mannose and d-allose. The kinetic parameters of the recombinant l-RhI from B. pallidus, for those five substrates described above, were summarized in Table 5. The enzyme represented a Michaelis–Menten constant (K m) and maximum velocity (V max) of 4.89 mM and 87.0 U/mg, respectively, against l-rhamnose. Comparisons of the K m values of l-RhI from B. pallidus with the other microorganisms, toward l-rhamnose and several aldoses, are as shown in Table 3. For l-rhamnose, l-lyxose and l-mannose, the enzyme from B. pallidus exhibited a lower affinity than the l-RhI of E. coli, but was greater than that of P. stutzeri. Although, the K m of l-RhI for d-ribose and d-allose were relatively similar in both B. pallidus and P. stutzeri, whereas no activity for these two aldoses was observed in E. coli. The catalytic efficiency (k cat/K m) values of l-RhI from B. pallidus, against l-lyxose and d-allose were 1.15 × 105, and 4.95 × 104 M−1min−1, respectively, which were determined to be significantly greater than that of P. stutzeri (Leang et al. 2004b). Meanwhile, for l-rhamnose, l-mannose, and d-ribose, the catalytic efficiency values of the enzyme from B. pallidus were 8.36 × 105, 1.59 × 105, and 2.44 × 104 M−1min−1, respectively, which were apparently similar to the values of that from P. stutzeri (Leang et al. 2004b).

Schematic representation of aldose-ketose interconversion reactions catalyzed by recombinant L-rhamnose isomerase
Analysis of isomerized products
To determine the substrate and product ratios, biotransformation of various aldoses with the recombinant l-RhI were conducted in 50 mM sodium phosphate buffer (pH 7.0) at 50°C. The products formation was then analyzed by HPLC at different times. Sugar structures in a Fischer projection and equilibrium time as well as the ratio of aldoses conversion by l-RhI are shown in Fig. 4. As the results show, l-rhamnose could be transformed into l-rhamnulose at a ratio of 55:45 within 1 h, 35% d-allose could be obtained from d-psicose within 48 h, 25% l-mannose was formed from l-fructose within 12 h, 16% d-ribulose could be produced from d-ribose in 24 h, 12% l-xylulose was obtained from l-lyxose in 24 h, 10% l-talose was formed from l-tagatose in 24 h, and 5% d-gulose could be formed from d-sorbose within 24 h.
Discussion
The l-RhI (from P. stutzeri) is wide-range utility enzyme for the mass production of various rare sugars as well as d-tagatose 3-epimerase (d-TE), which has already been applied for a commercial mass production of d-psicose from d-fructose because these two enzymes show the broad substrate specificities against various sugars and their derivatives. Although there is no doubt that d-psicose is the most important rare sugar as a starting material of all rare hexose production, d-allose, which can be prepared from d-psicose using l-RhI, is also a valuable and attractive rare sugar caused by its own physiological activities. However, productivity (82 mg/l broth), activity (7.50 U/mg for d-allose) and thermal stability (<50°C for 10 min reaction) of known recombinant l-RhI (from P. stutzeri) was still poor to apply for a continuous mass production of d-allose using immobilized enzyme (Leang et al. 2004a). In this paper, we described for the first time the cloning, sequencing, overproduction, and characterization of L-rhi from thermophilic bacterium, B. pallidus Y25, and as a result, thermostable and high activity l-RhI could be obtained.
The L-rhi was cloned into E. coli and sequenced (Fig. 1). The deduced primary structure of the l-RhI protein exhibited homology with other l-RhIs, suggesting that these genes might have evolved from a common ancestor (Badia et al. 1989; Takami et al. 2002; Moralejo et al. 1993). However, it showed a low identity (12%) with l-RhI from P. stutzeri. We suppose that the low structural relationship in these l-RhIs may cause different functions in the conversion reaction of sugars (Oudega et al. 1997; Leang et al. 2004b; Badia et al. 1991), and the diverse residues of the active site l-RhIs are possibly involved in some differentiation of enzyme properties, particularly the substrate specificity (Fig. 2).
The overexpression of the recombinant l-RhI was achieved with large quantity (470 mg/l broth) and high activity (36,500 U/l broth), but not in the inactive fraction of inclusive bodies. In previous studies, maximum enzyme productivity of the recombinant l-RhI from E. coli and P. stutzeri was observed to be 37 and 82 mg/l broth, respectively (Garcia-Junceda et al. 1995; Leang et al. 2004a). The overproduction of His-tag-fused enzyme in E. coli resulted in the simplified purification step with high yield (86.7%) and activity (77.2 U/mg). Outstandingly, the result shown here indicates a good potential of this enzyme for mass production of various rare sugars.
The molecular mass determined by SDS–PAGE indicates that the recombinant l-RhI protein is correctly expressed in E. coli and that the cloned and wild-type enzymes are really the same (Fig. 3). The recombinant l-RhI was stable up to 60°C (for 1-h incubation), suggesting that this enzyme is significantly thermostable. In previous findings, the recombinant l-RhI from E. coli and P. stutzeri was maximized at 60 and 60°C and was stable up to the temperatures of 50 and 50°C, respectively, with 10-min incubation (Table 3). Generally, sugar productions are conducted under high temperature to reduce the high viscosity of solvent and avoid microbial contamination; therefore, the biocatalyst used in biotransformation processes need to be stable, particularly at high temperature. From such reasons, the recombinant l-RhI from B. pallidus is considerably appropriate for use in bioreactor system, as it provided remarkable activity and thermostability compared to the enzyme from other strains (Leang et al. 2004a).
Crystallographic study of l-RhI from E. coli revealed that Zn2+ appeared at structural sites and required Mn2+ to catalyze ring opening (Kornderfer et al. 2000). In the case of l-RhI from P. stutzeri, although the enzyme is nearly independent of metal ions, except for Mn2+ which slightly increase the enzyme activity, recent works on crystallographic study of l-RhI from P. stutzeri have demonstrated that amino acid residues which are involved in metal-binding played the same function with those from E. coli (Kornderfer et al. 2000). This suggests a similarity of the catalytic mechanisms for l-RhI of these two strains. It is likely that the activity of the recombinant l-RhI from B. pallidus is also strictly dependent on Mn2+ or Co2+.
Meanwhile, the result of substrate specificity is different from that in other bacteria, especially l-RhI from P. stutzeri which was found to have inherent similarities with the catalytic mechanism of d-xylose isomerase (Leang et al. 2004b) and consequent broad specificity over all sugar substrates including tetroses, pentoses and hexoses. In the case of l-RhI from E. coli, the enzyme was specific for l-rhamnose, l-lyxose, and l-mannose, whereas no activity was observed with d-allose or d-ribose (Table 4). These results indicate that the enzyme from B. pallidus has the overlapped properties for the substrate specificity with that from P. stutzeri and E. coli. This finding provides an additional support for the result shown in Fig. 2 that the differentiation of the amino acid residues at the active sites may affect changes in the substrate specificity of the enzyme. Furthermore, the results of kinetic properties suggest that although the recombinant l-RhI in this study has higher activity in isomerizing of the aldose sugars, it is much more specific to the substrate, which may be cause by the rigidity of the enzyme in substrate recognition regions, compared to that of P. stutzeri (Table 5).
Noticeably, sugar conversion using the recombinant l-RhI from B. pallidus provided only a single product by HPLC, while the enzyme from P. stutzeri that had broad specificity with all kinds of aldoses resulted in multiple sugar products forming (Fig. 4). The results confirmed the kinetic parameters finding that the high specificity of its recombinant thermostable enzyme presented here is much more specific than other strains, which leads to a reduction in the number of steps for product separation and an increase in product yields. Therefore, the recombinant l-RhI from B. pallidus is demonstrated to be a good candidate for applications in the rare sugars production processes. Further studies related to the mass production of d-allose and l-talose from d-psicose and l-tagatose using this enzyme is now underway.
References
Altschul F, Gish W, Miller W, Meyers EW, Lipman Stephen DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Badia J, Baldoma L, Aguilar J, Boronat A (1989) Identification of the rhaA, rhaB and rhaD gene products from Escherichia coli K-12. FEMS Microbiol Lett 65:253–258
Badia J, Gimenez R, Baldoma L, Barnes E, Fessner WD, Aguilar J (1991) l-Lyxose metabolism employs the l-rhamnose pathway in mutant cells of Escherichia coli adapted to grow on l-lyxose. J Bacteriol 173:5144–5150
Bertelsen H, Jensen BB, Buemann B (1999) d-tagatose—a novel low-calorie bulk sweetener with prebiotic properties. World Rev Nutr Diet 85:98–109
Bhuiyan SH, Itami Y, Izumori K (1997) Isolation of an l-rhamnose isomerase-constitutive mutant of Pseudomonas sp. strain LL172: purification and characterization of the enzyme. J Ferment Bioeng 84:319–323
Bhuiyan SH, Itami Y, Takada G, Izumori K (1999) Preparation of l-talose and d-gulose from l-tagatose and d-sorbose, respectively, using immobilized l-rhamnose isomerase. J Biosci Bioeng 88:567–570
Bradford MM (1979) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Bustard MT, Wright PC (2002) Biodegradation of high concentration isopropanol by a solvent tolerant thermophile, Bacillus pallidus. Extremophiles 6:319–323
Cramp R, Gilmour M, Cowan DA (1997) Novel thermophilic bacteria producing nitrile-degrading enzymes. Microbiology 143:2313–2320
Dishe Z, Borenfreud E (1951) A new spectrophotometric method for the detection of keto sugars and trioses. J Biol Chem 192:583–587
Domagk GF, Zech R (1963) _ber den Abbau der Desoxyzucker durch Bacterienenzyme. I. l-Rhamnose-isomerase aus Lactobacillus plantarum. Biochem Z 339:145–153
Garcia-Junceda E, Shen GJ, Alajarin R, Wong CH (1995) Cloning and overexpression of rhamnose isomerase and fucose isomerase. Bioorg Med Chem 3:1349–1355
Granström TB, Takata G, Tokuda M, Izumori K (2004) Izumoring, A novel and complete strategy for bioproduction of rare sugars. J Biosci Bioeng 97:89–94
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmid. J Mol Biol 166:557–590
Illanes A (1999) Stability of biocatalysts process biotechnology. Electronic J Biotechnol 2:0717–3458
Itoh H, Okaya H, Khan AR, Tajima S, Hayakawa S, Izumori K (1994) Purification and characterization of d-tagatose 3-epimerase from Pseudomonas sp. ST-24. Biosci Biotechnol Biochem 58:2168–2171
Itoh H, Sato T, Izumori K (1995) Preparation of d-psicose from d-fructose by immobilized d-tagatose 3-epimerase. J Ferment Bioeng 80:101–103
Izumori K, Mitchell M, Elbein AD (1976) Evidence that the isomerization of d-ribose and l-rhamnose is catalyzed by the same enzyme in Mycobacterium smegmatis. J Bacteriol 126:553–555
Jeong LS, Yoo SJ (1998) Synthesis and antiviral activity of novel isodideoxy nucleosides with exocyclic methylene. Bioorg Med Chem Lett 8:847–852
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Kim HJ, Oh DK (2005) Purification and characterization of an l-arabinose isomerase from an isolated strain of Geobacillus thermodenitrificans producing d-tagatose. J Biotechnol 120:162–173
Kornderfer IP, Fessner WD, Matthews BW (2000) The structure of rhamnose isomerase from Escherichia coli and its relation with xylose isomerase illustrates a change between inter and intra-subunit complementation during evolution. J Mol Biol 300:917–933
Kuroda K, Hanajima D, Fukumoto Y, Suzuki K, Kawamoto S, Shima J, Haga K (2004) Isolation of thermophilic ammonium-tolerant bacterium and its application to reduce ammonia emission during composting of animal wastes. Biosci Biotechnol Biochem 68:286–292
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Leang K, Takada G, Ishimura A, Okita M, Izumori K (2004a) Cloning, nucleotide sequence, and overexpression of the l-rhamnose isomerase gene from Pseudomonas stutzeri in Escherichia coli. Appl Environ Microbiol 70:3298–3304
Leang K, Takada G, Fukai Y, Morimoto K, Granstrom TB, Izumori K (2004b) Novel reactions of l-rhamnose isomerase from Pseudomonas stutzeri and its relation with d-xylose isomerase via substrate specificity. Biochim Biophys Acta 1674:68–77
Lerner LM, Mennitt G (1994) A new synthesis of l-talose and preparation of its adenine nucleosides. Carbohydr Res 259:191–200
Ly KA, Milgrom P, Rothen M (2006) Xylitol, sweeteners, and dental caries. Pediatr Dent 28:192–198
Moralejo P, Egan SM, Hidalgo E, Aguilar J (1993) Sequencing and characterization of a gene cluster encoding the enzymes for l-rhamnose metabolism in Escherichia coli. J Bacteriol 175:5585–5594
Menavuvu BT, Poonperm W, Leang K, Noguchi N, Okada H, Morimoto K, Granstrom TB, Takada G, Izumori K (2006) Efficient biosynthesis of d-allose from d-psicose by cross- linked recombinant l-rhamnose isomerase: separation of product by ethanol crystallization. J Biosci Bioeng 101:340–345
Mozhaev V (1993) Mechanism-based strategies for protein thermostabilization. Trend Biotechnol 11:88–95
Noltmann EA (1972) Aldose–ketose isomerases. In: Boyer PD (ed) The enzymes, 3rd edn, vol 6. Academic, London, pp 271–354
Oudega B, Koningstein G Rodrigues L, de Sales Ramon M, Hilbert H, Dusterhoft TM, Pohl TM, Weitzenegger T (1997) Analysis of the Bacillus subtilis genome: cloning and nucleotide sequence of a 62 kb region between 275 degrees (rrnB) and 284 degrees (pai). Microbiology 143:2769–2774
Poonperm W, Takata G, Morimoto K, Granström TB, Izumori K (2007a) Production of l-xylulose from xylitol by a newly isolated strain of Bacillus pallidus Y25 and characterization of its relevant enzyme xylitol dehydrogenase. Enzyme Microb Technol 40:1206–1212
Poonperm W, Takata G, Sahachaisaree V, Lumyong P, Lumyong S, Izumori K (2007b) Efficient conversion of allitol to d-psicose by Bacillus pallidus Y25. J Biosci Bioeng 103:282–285
Sui L, Dong Y, Watanabe Y, Yamaguchi F, Hatano N, Tsukamoto I, Izumori K, Tokuda M (2005) The inhibitory effect and possible mechanisms of d-allose on cancer cell proliferation. Int J Oncol 27:907–912
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci 74:5463–5467
Takami H, Takaki Y, Uchiyama I (2002) Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res 30:3927–3935
Takeshita K, Shimonishi T, Izumori K (1996) Production of l-psicose from allitol by Gluconobacter frateurii IFO 3254. J Ferment Bioeng 3:212–215
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Wasserman B (1984) Thermostable enzyme production. Food Technol 38:78–88
Yoshida H, Yamada M, Ohyama O, Takada G, Izumori K, Kamitori S (2007) The structures of l-rhamnose isomerase from Pseudomonas stutzeri in complexes with l-rhamnose and d-allose provide insights into broad substrate specificity. J Mol Biol 365:1505–1516
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Poonperm, W., Takata, G., Okada, H. et al. Cloning, sequencing, overexpression and characterization of l-rhamnose isomerase from Bacillus pallidus Y25 for rare sugar production. Appl Microbiol Biotechnol 76, 1297–1307 (2007). https://doi.org/10.1007/s00253-007-1109-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1109-3