
Abstract
Microscopic evolution at the functional biomolecular level is an ongoing process. Leveraging functional and high-throughput assays, along with computational data mining, has led to a remarkable expansion of our understanding of multifunctional protein (and gene) families over the past few decades. Various molecular and intermolecular mechanisms are now known that collectively meet the cumulative multifunctional demands in higher organisms along an evolutionary path. This multitasking ability is attributed to a certain degree of intrinsic or adapted flexibility at the structure–function level. Evolutionary diversification of structure–function relationships in proteins highlights the functional importance of intrinsically disordered proteins/regions (IDPs/IDRs) which are highly dynamic biological soft matter. Multifunctionality is favorably supported by the fluid-like shapes of IDPs/IDRs, enabling them to undergo disorder-to-order transitions upon binding to different molecular partners. Other new malleable members of the protein superfamily, such as those involved in fold-switching, also undergo structural transitions. This new insight diverges from all traditional notions of functional singularity in enzyme classes and emphasizes a far more complex, multi-layered diversification of protein functionality. However, a thorough review in this line, focusing on flexibility and function-driven structural transitions related to evolved multifunctionality in proteins, is currently missing. This review attempts to address this gap while broadening the scope of multifunctionality beyond single protein sequences. It argues that protein intrinsic disorder is likely the most striking mechanism for expressing multifunctionality in proteins. A phenomenological analogy has also been drawn to illustrate the increasingly complex nature of modern digital life, driven by the need for multitasking, particularly involving media.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction: Protein Multifunctionality from an Evolutionary Perspective
‘My own prejudices are exactly the opposite of the functionalists’: “If you want to understand function, study structure.” ’ – Francis Crick.
One of the hallmarks of modern digital life is the need to multitask, driven by the individuality of modern times and the rapid progress of technology, particularly in media (Popławska et al. 2021). On average, American youths spend 7.5 h a day with media, with 29% of that time spent by simultaneous web-surfing on different sites (Uncapher et al. 2017). This phenomenon in the digital world mirrors a similar trend observed at the functional biomolecular level, where the concepts of protein multifunctionality (Espinosa-Cantú et al. 2020), multifunctional and multigene families (Schnoes et al. 2009) are prominent. With research over more than the past two decades, we now have a wealth of new knowledge on protein intrinsic disorder (Uversky 2013), fold-switching (Bryan and Orban 2010), moonlighting (Espinosa-Cantú et al. 2015), hub proteins (Cino et al. 2013), domain shuffling (Kawashima et al. 2009), and so on, either indirectly or directly supporting evolved multifunctionality in proteins. However different the strategies may be, a certain degree of intrinsic or adapted flexibility is found to be associated with multitasking that allows for function-driven structural transitions in proteins. This takes us far beyond over-simplistic unifunctional models limited to enzyme classes (Alberts et al. 2002) and leads us to a more sophisticated comprehension of protein functionality, both for enzymatic reactions and for non-covalent protein binding (Mannige 2014).
The evolution of binding promiscuity in IDPs/IDPs (the ability to bind to different ordered protein partners and adopt different shapes) (Jayaraman et al. 2022), moonlighting proteins, and multi-enzyme complexes (Alberts et al. 2002; Jeffery 2003) seems inevitable to fit the increasing (energy) demand in eukaryotes and higher organisms that are continuously under micro-evolutionary selection pressure. To that end, post-transcriptional mechanisms like alternative splicing and post-translational modifications are well known, giving rise to a variety of protein isoforms (Stastna and Van Eyk 2012). Gene duplication has also been extensively used in evolution (Espinosa-Cantú et al. 2015), leading to an abundance of large paralogous protein families in living systems. Paralogs within the same protein family (descended from a common evolutionary ancestor) diverge enough to provide a mutational ‘buffer’ against cross-talks with non-cognate partner proteins, typically only exhibiting ‘marginal specificity’ to constrain their evolvability (Ghose et al. 2023). Functional annotations of paralogs have further led to the concept of ‘multifunctional families’ (Zallot et al. 2016), explicated by comparative genomics, phylogeny, metabolic reconstruction, and signature motifs. Subgroups within large multifunctional families have also been disambiguated by careful manual curation. Hence, attempts have even been made to further backtrack the collective functionality of enzyme superfamilies to the level of multigene families, defined as having at least two members of the family in a given genome (Schnoes et al. 2009). Strolling along the same line, this review explores the various evolutionary strategies used to achieve functional diversification in proteins, resulting in both ‘direct’ and ‘indirect’ means of evolved protein multifunctionality. These strategies range from an array of naturally evolved genetic editing mechanisms that eventually lead to functional variation in ortho-/paralogs (indirect) (Zallot et al. 2016) to moonlighting proteins and other conformationally flexible ‘single protein’ candidates (e.g., fold-switching proteins, disordered proteins, etc.) that directly perform more than one function (direct). The review also argues in favor of protein intrinsic disorder as probably the most powerful and demanded mechanism.
Regarding ‘indirect’ means of evolved protein multifunctionality, protein families often harbor multifunctional ortho-/paralogs, adapting to diverse biological roles beyond their original functions by the process of neofunctionalization (He and Zhang 2005). In the Zinc Finger protein family (IPR043359),Footnote 1 distinct roles have evolved within vertebrates: GLI1 enhances transcription, while GLI2 variably acts as an activator or repressor, and GLI3 acts primarily as a repressor in the Hedgehog pathway, demonstrating functional diversification from a single ancestral gene (Laity et al. 2001). Meanwhile, the Opsin (IPR001760) protein family across different species illustrates evolutionary adaptation to diverse light environments, with primates developing trichromatic vision and insects, like bees, evolving sensitivity to ultraviolet light (Yokoyama 2000). On the other hand, there are protein families that are functionally conserved and only perform a single core function (i.e., unifunctional), similar to the function of the ancestral organism, without acquiring any new functionalities in their evolutionary descendants. In other words, a unifunctional protein family refers to a group of proteins that share a common core functionality, typically enzymatic, with potential differences in isoforms, subcellular localization, or substrate specificity, but without any additional evolved secondary functions. For enzymes and multi-enzyme complexes (Patel et al. 2014), the core protein functionality includes enzymatic regulation, allosterism, and all other necessary steps to maintain the enzyme homeostasis over evolution. Urease (EC 3.5.1.5)Footnote 2 (Mazzei et al. 2020), Cytochrome c oxidase (EC 7.1.1.9) (Wikström and Krab 1979), and Carbonic Anhydrase (EC 4.2.1.1) (Imtaiyaz Hassan et al. 2013) are just to name a few. A more detailed (non-exhaustive) list is presented in the Supplementary Materials (Data S1). It is important to note that protein isoforms and/or isozymes with a single, conserved core function that differ in their subcellular localization (e.g., carbonic anhydrase (Imtaiyaz Hassan et al. 2013)), and consequently in their biophysical properties (such as enzyme kinetics, pH, etc.) (Lynch and Conery 2000), are still part of one unifunctional protein family. Similarly, large protein superfamilies (e.g., serine proteases, EC 3.4.21. – (Rawlings et al. 2012)) that perform the same core functionality using a characteristic catalytic mechanism on a variety of similar substrates (Hedstrom 2002) would also fall under unifunctional families. This characteristic catalytic mechanism is what defines their classification in the EC system.
In contrast, ‘direct’ means of evolved protein multifunctionality frequently lead to protein moonlighting (a single protein performing more than one function), ample evidence of which is present in the MoonProt 3.0 database (Chen et al. 2021). These proteins retain their primary enzymatic function while acquiring additional secondary functions (non-enzymatic). For instance, Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) (EC 1.2. 1.12), primarily known for its role in glycolysis (D-glyceraldehyde 3-phosphate → 1,3-bis-phosphoglycerate), also participates in the GAIT complex, which inhibits translation of ceruloplasmin mRNA in response to interferon (IFN)-gamma (Mazumder et al. 2010). Another notable example is Enolase (EC 4.2.1.11), also in glycolysis (2-phosphoglycerate → phosphoenolpyruvate) which further acts as a plasminogen receptor on the surface of various cell types, facilitating plasminogen activation and thereby playing a role in tissue remodeling and cell migration (Wygrecka et al. 2009). A detailed list (100 examples, culled from MoonProt 3.0 (Chen et al. 2021)) of moonlighting proteins along with their primary (enzymatic) and secondary (non-enzymatic) functions is presented in the Supplementary Materials (Table S2).
Proteins can serve multiple functions due to evolutionary pressures (Jayaraman et al. 2022) that lead to adaptations in their structure and function. The fundamental evolutionary basis of functional diversification in proteins is rooted in an activity–stability trade-off between conservation of the core protein function (and corresponding key residue positions in the structure) and variation at other mutable peripheral positions allowing the scope for novel functions to evolve (Tokuriki et al. 2008; Sikosek and Chan 2014). This trade-off can be attained by various evolutionary mechanisms. One indirect mechanism is gene duplication (Espinosa-Cantú et al. 2015) where redundant copies of a gene can get differentially mutated or domain-shuffled (Kawashima et al. 2009) to evolve into homologous proteins with novel functionalities crucial for an organism's survival and adaptation.
Another more direct mechanism to express multifunctionality in proteins is intrinsic disorder which can be manifested throughout the whole protein chain (Intrinsically Disordered Proteins or IDPs) or in patches (Intrinsically Disordered Regions or IDRs). Interestingly, eukaryotic proteins have more intrinsic disorder compared to prokaryotes. In fact, more than 30% of all eukaryotic proteins consist of long disordered regions comprising ≥ 50 consecutive amino acid residues (Dunker et al. 2001). Viruses can alter their host cells through short linear motifs in their IDRs that lead to potential binding promiscuity with a direct impact at the functional level (Davey et al. 2011). In comparative genomics studies, it has been observed that bacteriophages and their host bacteria co-evolve on the chromosomal level (Brüssow et al. 2004). Evolutionary pathways of multifunctional proteins, when viewed together with intrinsic disorder, unravel the complex interplay between evolving life forms.
Biophysical Basis of Multifunctionality in IDPs
Unlike well-folded globular proteins, IDPs lack a stable fold. Instead, they exist as conformational ensembles rather than a single structure, which gives them structural plasticity and flexibility. This, in turn, allows them to exhibit their characteristic binding promiscuity when interacting with different partners, enabling multifunctionality (Fersht 2009). The phenomenon of ‘coupled folding and binding’ (Sugase et al. 2007) allows their transitions from disordered (unstructured) to ordered (structured) states upon binding to different partners. Their non-disjoint flexible backbone trajectory enables them to accommodate different combinations of side-chain rotamers, consistent with different befitting surfaces of ordered protein partners (Dunker et al. 2001).
In contrast to well-folded proteins, IDPs continuously search for suitable intra- and/or intermolecular interactions to stabilize themselves (Tsai et al. 1999; Levy et al. 2005). This makes them hover across different self-similar (Bandyopadhyay and Basu 2020) local minima across their rugged energy landscape. Eukaryotic IDPs remain disordered under normal conditions and only fold into ordered structures when they come into contact with their cellular targets (Wright and Dyson 1999; Dyson and Wright 2002, 2005; Uversky 2002). It has been theorized that disordered proteins bind weakly and non-specifically to the target and align structurally to a befitting surface to become structured when approaching the cognate binding sites (Shoemaker et al. 2000). Most of these interactions, especially those in signal transduction pathways, are transient (meta-stable).
IDPs can also undergo a co-translational folding mechanism involving the ribosomal surface and molecular chaperons (Simister et al. 2011; Waudby et al. 2019) to escape protein degradation. These adaptabilities enable IDPs to engage in numerous cellular processes despite lacking a defined structure. Importantly, IDRs (in hybrid or partially disordered proteins) also serve as promising (fuzzy) drug targets, offering a new approach for drug development (Kamagata et al. 2019, p. 53; Saurabh et al. 2023). In distinct contrast to well-demarcated drug-binding pockets of the folded proteins, such an approach accounts for an acceptable representation of the conformational ensemble of an IDR (Saurabh et al. 2023) as the receptor surface, thereby increasing the interaction cross-section for the ligands (drugs).
The promiscuous binding nature of IDRs makes them potential drug targets. One example is in the case of castration-resistant prostate cancer, where the disordered N-terminal domain of androgen receptors is targeted to overcome existing drug resistance (Yi et al. 2023). The formation of binding-competent transient structures induced by molecular crowding in the close vicinity of IDPs/IDRs is another unique mechanism that demonstrates binding promiscuity and multifunctionality (Gruber et al. 2022). These results can potentially lead us to decipher and understand the mechanism of assembly of very large and distinct signal transduction protein complexes (viz., ‘signalosomes’ that are stimulus specific) in response to certain stimuli in a short period of time (Simister et al. 2011).
Arsenal of Direct and Indirect Means of Multifunctionality in Proteins
Multifunctionality in proteins is harnessed by several molecular evolutionary strategies or mechanisms, both indirect and direct (Fig. 1). While some mechanisms accommodate swapping between distinct well-defined structural states (domains, folds), leading to the emergence of protein isoforms and ortho-/paralogs, others render more flexibility and allow transitions among multiple conformations adapted by the same protein sequence in response to environmental cues (e.g., vicinity of a binding partner, change in pH, etc.). Following is a comparative discussion of these evolutionary mechanisms.

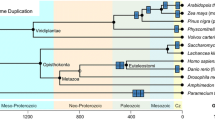
The composite figure portrays different indirect and direct evolutionary mechanisms to express evolved protein multifunctionality. The represented repertoire includes as indirect tools: (i) Domain Shuffling: human aggrecan core peptide (highlighted as dots) presented by class II major histocompatibility complexes (7RDV) (Kawashima et al. 2009), (ii) Gene Duplication: Two in-del mutations (Q211 → D231, D299 → T365) in Actins (tested structurally in yeasts, camelidaes, insects) that seem to inhibit filament formation of ARP4 (PDB ID: 5NBM, left), which instead heteromerizes with its paralog ACT1 (6BNO, right) (Mallik et al. 2022), (iii) Adaptive Evolution: the Spike protein with highly mutable FLCSSpike (highlighted as dots) from SARS-CoV-2 (6XR8); and as direct tools: (iv) Hub Proteins: Canine GDP-Ran (monomer: 1QG4 ↔ dimer: 1BYU) with its interactome consisting of Ran binding protein (RBP): 1RRP, Ran GAP: 1K5D, karyopherin β2: 1QBK, Nuclear Transport Factor 2 (NTF-2): 5BXQ, etc. (Higurashi et al. 2008), (v) Intrinsic Disorder: human alpha synuclein (a conformational ensemble picked up from its MD-simulated trajectory (Bandyopadhyay and Basu 2020)) with its two cognate globular binding partners: Tubulin (4YRL) and β-neurexin 1 (3MW2), (vi) Moonlighting: Yeast Heat shock protein Hsp70 bound with ADP (3QFU) (Jeffery 2018), (vii) Fold-switching: human lymphotactin (1J8I ↔ 2JP1) (Bryan and Orban 2010), and (viii) Multi-domain Proteins: carboxylate transfer in pyruvate carboxylase (PC) (Maurice et al. 2007) using its three main domains: an N-terminal Biotin Carboxylation domain, an internal Biotin carrier domain, and a C-terminal Carboxyltransferase domain to eventually produce oxaloacetate
Indirect Mechanisms
These mechanisms involve the evolution of new protein functions through modifications at the genetic level, resulting in protein families with diverse functions. While individual members of these families may only have one function, the overall group exhibits multifunctionality due to the divergence of their ancestral genes.
Gene Duplication and Functional Divergence
Gene duplications create redundant gene copies, allowing one copy to retain the original function while the paralog (often varying at their oligomeric states (Mallik et al. 2022)) accumulates mutations at a higher rate. The paralog is often fixed in the population by acquiring an adaptive function according to the classical model of divergence by neofunctionalization (Ohno 1970). To that end, accelerated evolution in retained paralogs (e.g., Rck1/Rck2, Ptc2/Ptc3, Sim1/Sun4, Ktr5/Ktr6 (Hughes and Friedman 2003)) has been observed through evolving post-translational regulation mechanisms, utilizing diversified short linear motif like sequences (Nguyen Ba et al. 2014). At the other end, there is the more flexible model of subfunctionalization, where after gene duplication and divergence, the biological functions of the ancestor get partitioned between two paralogs (Fig. 1). Furthermore, subfunctionalization may be qualitative or quantitative. Qualitative subfunctionalization refers to the molecular functions that trade-off with each other in the ancestral gene. Each paralog may then evolve toward the optimization of the retained function. Alternatively, quantitative subfunctionalization occurs when neutral evolution results in complementary loss-of-function mutations between the paralogs. In this model, both duplicates become indispensable as they together provide the ancestral functional requirements (Fewell and Woolford 1999; Lynch and Force 2000; He and Zhang 2005).
Domain Shuffling
Reorganization of protein domains can create multifunctional homologous proteins through the combination of existing functional units in new ways. It may come through horizontal gene transfer (e.g., from prokaryotes to eukaryotes) or by insertion–deletion (in-del) mutations of genes, post duplication. One common way in which domain shuffling (Fig. 1) leads to novel functions is by the shuffling of exons (exon shuffling, analogous to alternative splicing at the mRNA level), followed by in-del mutations. Usually, this is established by a mapping of exons and domains (e.g., a single exon coding for a single complete domain) (Kawashima et al. 2009). Additionally, insertion of a ‘nested’ domain may also interrupt the linear sequence of a structural domain. Such insertions often map to disordered loops in the parent structure. For example, in phospholipase-γ (EC 3.1.4.3)(PLCγ), an insert of ~ 300 residues (comprising one SH3 and two SH2 domains) separates one of its two Pleckstrin Homology (PH) domains (Bill and Vines 2020). The C-terminal PH domain is believed to bind to calcium channels, resulting in agonist-induced calcium entry into the cell, while interactions at the SH3/SH2 domains help stabilize the recruitment of PLCγ to the plasma membrane, crucial for its functions as a signal transducer. The structural integration of the SH2, SH3, and PH domains ensures that PLCγ regulates proliferation (e.g., through the SH3 domain’s recruitment of a Ras exchange factor, SOS1) independently of its lipase activity (PIP2 + H2O → IP3 + DAG) (Bill and Vines 2020). Certain domains (e.g., the Xlink domain) of the protein aggrecan (P16112)Footnote 3 (Kawashima et al. 2009), the most abundant non-collagenous protein in cartilage, are also said to have been created by domain shuffling in ancestral vertebrates.
Adaptive Evolution
Environmental pressures drive proteins to adapt, acquiring new functions that enhance an organism’s survival fitness. Adaptive mutations are largely amino acid substitutions that occur at the protein’s surface. A high degree of solvent accessibility of the exposed residues at the protein surface makes them most prone to mutations. Population genomics studies in model systems like Drosophila and Arabidopsis have surveyed a variety of genomic, structural, and functional descriptors. These studies have revealed that the rate of adaptive substitutions differs for various functional classes, with the fastest rates of adaptation observed in proteins involved in translation, degradation, and signaling (Moutinho et al. 2019). The studies also suggest that intermolecular interactions, such as host–pathogen co-evolution, play a significant role in adaptive evolution (Moutinho et al. 2019). Multifunctional viral proteins are classic examples of adaptive evolution (Hasiów-Jaroszewska et al. 2014). The most prominent candidate in recent times is perhaps the Spike protein of the Coronavirus (Fig. 1), rapidly undergoing mutations from SARS-CoV-2 → Omicron (Araf et al. 2022), Deltacron (Maulud et al. 2022), and so on. Again, one of the key mutational hotspots in the Coronavirus Spike protein is the ‘solvent exposed’ disordered loop containing the ‘Furin like cleavage site’ or FLCSSpike (Balaram 2021; Roy et al. 2022). The mutational patterns in FLCSSpike have contributed much to the COVID-origin debate (Balaram 2021)—further emphasizing the importance of solvent exposed surface residues in adaptive evolution. Significant patterns of co-occurrence of adaptive events have also been identified in the RNA binding domains with functional overlapping of the HC-Pro of the potyvirus (established by covariation analyses) (Hasiów-Jaroszewska et al. 2014).
Direct Mechanisms
These mechanisms enable a single protein to directly perform multiple functions through inherent structural features or conformational changes, without relying on gene duplication or other genetic modifications as seen in indirect mechanisms.
Protein Moonlighting
In contrast to gene-fusion, alternative splicing, or functional peptides resulting from multiple proteolysis, protein moonlighting (Jeffery 2003, 2018) refers to the multifunctionality evolved in proteins (especially enzymes) without requiring any change in their primary sequence. It is therefore a more direct expression of multifunctionality derived from a single protein sequence. Moonlighting (Fig. 1) is typically expressed via alternative sites to that of the primary active site (usually a catalytic pocket) (Jeffery 1999; Piatigorsky 2009). In these proteins, both classic and non-classic type protein functions co-exist, wherein the former refers to enzymatic activities (i.e., involving covalent bond breaking and making), while the latter refers to protein–protein interactions (PPI) via an alternative part of the protein’s surface. These alternative sites may also include allosteric relay of conformational changes, for example, in moonlighting kinase guanylate cyclase (EC 4.6.1.2) (Turek and Irving 2021). The structure of a moonlighting protein can get altered by its secondary (non-classic PPI type) function, thereby putting constraints on its structural flexibility with the primary function (enzymatic activity) somewhat compromised, as observed in ƞ-crystallin (Jeffery 2004). Another related example from the same protein family is ε-crystalline which serves as a major component of the lens of the eye in ducks while retaining a basal level of its primary function as the ubiquitous enzyme lactate dehydrogenase (EC 1.1. 1.27) (pyruvate ↔ lactate) (Jeffery 2018). Moonlighting has also been found to evolve in heat shock proteins (HSPs) part of ATP-dependent molecular chaperones (e.g., HSP10, HSP70, HSP90, etc.) enabling them to work in ATP-poor conditions (Jeffery 2018).
Fold-Switching Proteins
Fold-switching proteins (Bryan and Orban 2010), a newly emerging class of proteins, undergo a distinct switching of their folds by remodeling their secondary structures upon change in environmental (physiological) conditions (Fig. 1), for example, a change in pH (Baruah and Biswas 2015). Upon fold-switching, they respond to altered cellular stimuli, enabling them to perform important alternative regulatory (e.g., transcriptional regulation) functions of the cell (demonstrated in proteins like RfaH, KaiB, etc.) (Bernhardt and Hansmann 2018; Kim and Porter 2021). Another dramatic example is lymphotactin, which undergoes conformational switching (Bryan and Orban 2010). The conformational switching from a ‘lying down’ to a ‘standing up’ position of the receptor binding domain of the SARS-CoV-2 Spike protein (Mercurio et al. 2020; Cai et al. 2020) may also be envisaged analogously in the sense that a close vicinity of the ACE-2 host cell receptor serves as the environmental (physiological) trigger in this switching.
Intrinsically Disordered Proteins
IDPs are biological soft matters (Bandyopadhyay and Basu 2020) that are highly dynamic and biologically active (Uversky 2016a). Unlike globular proteins, they do not have enough hydrophobic residues to trigger a hydrophobic collapse. Instead, they have high amounts of polar and charged residues (Sun et al. 2013; Uversky 2016a; Basu and Biswas 2018; Már et al. 2023) which contribute to less sequence complexity in the absence of folding (Uversky 2016a; Már et al. 2023). This results in partial temporal order by hydrogen bonding, water-mediated contacts (indirect readouts) (Reid et al. 2023), and the formation of transient interchangeable salt-bridges (Basu and Biswas 2018). Unlike globular proteins, IDPs lack a characteristic deep well in their energy landscapes as they do not conform to a lone stable 3D structure under physiological conditions, and, rather, have an affinity to undergo transition from disorder to order and back to disorder (Sun et al. 2013; Basu and Biswas 2018). This makes them highly flexible and adaptable. Partially disordered (or hybrid) proteins contain a varying degree of IDRs, enabling them with both ordered and disordered regions (Sun et al. 2013; Uversky 2016a). A classic example of hybrid protein is p53 (IPR002117) (Xue et al. 2013).
Recurrent salt-bridges (especially, those with short-range contact orders) impart local temporal structural rigidity in IDPs. Studies (Basu and Biswas 2018; Bandyopadhyay and Basu 2020; Roy et al. 2022) have demonstrated that salt-bridges in IDPs are typically not stable (or persistent) and tend to dissolve and reform frequently with various interchangeable counter-ionic partners. This phenomenon is referred to as ‘transient salt-bridge dynamics.’ This is a necessary mechanism to accommodate an abundance of oppositely charged residues and to allow for sampling of different conformations, leading to conformational ensembles (Fig. 1). These conformations are not random but revolve around a finite number of structurally degenerate conformational clusters (Bandyopadhyay and Basu 2020). Phase transitions among these clusters are often triggered by the switching of transient salt-bridges, demonstrating critical behavior similar to a sand-pile model. The presence of these transient or flitting salt-bridges may stabilize the IDP in a conformationally dependent manner, locked by the befitting surfaces of its globular partners. Such function-guided conformational dynamics is especially relevant in the case of cell signaling, e.g., in suppressors of cytokine signaling (SOCs) (Bandyopadhyay and Basu 2020), where IDRs in eukaryotic transcription factors (Már et al. 2023) are evolving with high sequence heterogeneity and demonstrated dynamic multifunctionality by means of their characteristic binding promiscuity. This way, IDP/IDRs can remain potentially multifunctional in a non-random conformational ensemble, while structural proximity of a befitting binding partner shapes the conformational dynamics to a specific ordered (bound) conformation. Salt-bridge dynamics and criticality in phase transitions in disordered loops (IDRs) have also been found to be associated with proteolytic priming in host–pathogen interactions, wherein the host and the pathogenic molecular partners seem to have co-evolved (e.g., Spike—Furin in SARS-CoV-2) from an evolutionary perspective (Roy et al. 2022).
IDPs, lacking a fixed structure or folding code, exist as highly dynamic ‘dancing protein clouds’ (Uversky 2016a) that can adopt different shapes depending on their local environment. When IDPs interact with ordered proteins, their binding contributes to at least partial folding, depending on the binding partner. Different binding partners can induce different folds (Wright and Dyson 1999; Uversky 2016a), making them highly adaptable. Additionally, IDPs exhibit fractals and heterogeneity, meaning that they neither converge to a steady state nor diverge to infinity but rather stay within a ‘chaotically defined region’ (Uversky 2016a).
Hub Proteins
Hub proteins (Higurashi et al. 2008) are proteins with a (hub-like) high degree in a protein–protein interaction (PPI) network. They can interact with multiple partners, even those associated with very different protein networks, leading to diverse biological processes. Hub proteins can further be differentiated into stable or static hubs (also known as date hubs, intra-module) and dynamic hubs (party hubs, inter-module) (Ekman et al. 2006; Kenley et al. 2011). Stable hubs maintain their high connectivity over time, while dynamic hubs are often involved in transient interactions (Han et al. 2004), with different interaction patterns depending on cellular conditions. Stable hubs are often involved in specific protein complexes or pathways rather than having broad interaction capabilities. More often than not, stable hubs are found to be hybrid proteins (e.g., p53 (Eriksson et al. 2019)) containing disordered regions (IDRs)—which sets them apart from dynamic hubs and proteins with low connectivity (non-hubs). Dynamic hubs, being involved mostly in transient interactions, have a tendency to interact with disordered partners (IDPs/IDRs) (Cino et al. 2013), broadening the array of their interactome (Sun et al. 2013). Stable hubs, on the other hand, having both ordered and disordered regions in them, bear the structural potential to interact with both ordered and disordered partners based on specific binding motifs and structural features. These specific binding motifs, often found within intrinsically disordered regions (IDRs) of partner proteins, are known as Molecular Recognition Features (MoRFs) (Mohan et al. 2006). MoRFs are short linear stretches of amino acids that are typically disordered in their unbound state but undergo a disorder-to-order transition upon binding to their target proteins, such as stable hubs (Oldfield et al. 2005). This transition often involves the formation of secondary structures like alpha-helices or beta-sheets, which are stabilized by interactions with the hub protein (Vacic et al. 2007). The inherent flexibility of MoRFs allows them to adopt different conformations upon binding to diverse partners, significantly contributing to the hub’s ability to engage in a wide range of protein–protein interactions, enhancing its multifunctionality (Dunker et al. 2005). Hub proteins, due to their strategic high-degree multifunctional importance, constitute an important component in drug design (Fu et al. 2015; Eriksson et al. 2019). One example of hub proteins is the canine GDP-Ran (Fig. 1), the active monomeric form of which interacts with RBP, Ran GAP, karyopherin β2, NTF-2, etc. apart from forming a biological dimer (Higurashi et al. 2008). Other more well-known hub proteins in biological systems are p53 and β-Catenin—whose interaction partners, roles of each interaction, and functional mechanistic details are enlisted in Table S1 in the Supplementary Materials. For p53, partners include MDM2 (negative regulation) (El-Deiry et al. 1993), p21 (transcriptional activation) (Wang et al. 2023), BAX (apoptosis promotion) (Miyashita and Reed 1995), p300/CBP (transcription co-activation) (El-Deiry et al. 1993), and DNA repair proteins (Seoane et al. 2002). For β-Catenin, the partners are E-Cadherin (cellular adhesion) (Huber and Weis 2001), TCF/LEF (transcriptional activation) (Metcalfe and Bienz 2011), and APC (tumor suppression and signaling regulation) (Parker and Neufeld 2020).
Multi-domain Proteins
Multi-domain proteins (Vishwanath et al. 2018) are also useful tools to express direct multifunctionality in proteins. Besides, they provide folding benefits and structural stability. Often, multi-domain proteins map to multifunctional enzymes with allosteric regulations and activated intermediates shuttled across different domains till the formation of the desired final product. For example, pyruvate carboxylase (EC 6.4.1.1) (PC) is an ATP-dependent multi-domain ligase belonging to the family of biotin-dependent multifunctional enzymes (Maurice et al. 2007). PC uses its three major domains (Fig. 1) to shuttle the carboxyl transfer through biotin (prosthetic group) to eventually form oxaloacetate.
Necessity of IDPs as SOS (ad hoc) Tools for Multifunctionality in Higher Organisms
The oversimplified ‘one gene–one enzyme’ hypothesis (Beadle and Tatum 1941) has long been outdated with an evolving definition of ‘gene’ (Portin and Wilkins 2017), and, perhaps even more so, with the growing knowledge of IDPs in recent times. In a human cell, there are approximately 104 protein coding genes, giving rise to ~ 106 different proteins. This genetic efficiency of an organism, leading to a surplus of effective proteins further, expands the multifunctional potential of the proteome in a higher organism (Sun et al. 2013; Uversky 2016b). Alternative splicing serves as a common post-transcriptional mechanism that enables the generation of multiple transcripts (leading to alternatively spliced variants) from a single gene, thereby expanding the phenotypic diversity (Keren et al. 2010; Wright et al. 2022) of the organism. Intrinsic disorder can also contribute to the phenotypic diversity of a genome, as disordered segments (IDRs) within hybrid or partially disordered proteins can get alternatively spliced at the mRNA level, eventually leading to diverse functionality exhibited by disordered proteins (Sickmeier et al. 2007; Clark et al. 2015). These variants can reshape signaling and participate in regulatory networks across different cell types during development, thereby enhancing the functional versatility of proteins, expanding interaction networks in various tissue types (Babu 2016). Furthermore, the flexibility associated with IDRs and alternative splicing may contribute to the emergence of novel phenotypes and increase the complexity of protein families for an organism’s microevolution. The acquired multifunctionality in IDPs/hybrid proteins is structurally supported by their fluid-like flexibility, serving to their conformational dynamics (Basu and Biswas 2018; Bandyopadhyay and Basu 2020) and binding promiscuity (Morris et al. 2021). IDPs are highly flexible and can undergo conformational changes to fit the surfaces of their binding partners. They also exhibit high binding plasticity and a low affinity–high specificity trade-off due to their conformational flexibility that enables them to become almost tailor made for their globular partners (Sun et al. 2013). IDPs, or IDRs, due to their physical flexibility to adopt different shapes upon binding to different protein surfaces, complement the repertoire of ordered (largely globular) proteins by providing transient SOS multifunctionality when required (e.g., in signaling cascades), while the ordered proteins carry out their routine functions (Uversky 2016a). In particular, IDPs are essential for cell signaling pathways, as they allow for high specificity, transitory (switch-like, fuzzy), and reversible (Wright and Dyson 2015) interactions that are not possible with ordered proteins alone. Rather, ordered and disordered proteins work together in a complementary manner to bring about cellular functions efficiently. The complexity of an organism is directly proportional to the demand for IDRs, as higher organisms often require more cellular signaling that relies on these protein interactions (Gao et al. 2021). Hence, it is no wonder that the presence of long IDRs is more common in eukaryotes than in Archaea and Bacteria (Wright and Dyson 2015). Also, ~ 1/4th of eukaryotic proteins have intrinsic disorder (Basile et al. 2019; Zamora-Briseño et al. 2021), while about 70% of signaling proteins are disordered.
Computational Tools to Unravel the World of IDPs
A great deal of effort has been invested in the past two decades to unravel the world of IDPs, once their existence was revealed (Wright and Dyson 1999). The growing sequence data, facilitated by the advent of modern-day sequencing techniques, necessitated the development of bioinformatic and computational tools to address specific queries regarding IDPs/IDRs. The rapid progress of machine learning (ML) techniques and artificial intelligence (AI) methods contributed substantially to the fast growth of such computational tools. At one end, there are sequence-based disorder predictors with gradually increasing prediction accuracies, namely, IUPred (Dosztányi et al. 2005), PrDOS (Ishida and Kinoshita 2007), DISOPRED3 (Jones and Cozzetto 2015), SPOT-Disorder2 (Hanson 2020), Metapredict (Emenecker et al. 2021), and so on. At the other end, there are predictors of disorder-to-order transitioning residues/regions (known as ‘protean’ segments) such as ANCHOR (Mészáros et al. 2009), MoRFpred (Disfani et al. 2012), MFSPSSMpred (Fang et al. 2013), Proteus (Basu et al. 2017), SPOT-MoRF (Hanson et al. 2020), flDPnn (Hu et al. 2021), as well as predictors of DNA, RNA, and protein-binding IDRs, namely, DeepDISOBind—developed through deep multitask learning (Zhang et al. 2022). At the other end, there are important databases of structure-functional annotations for IDPs such as DisProt (Sickmeier et al. 2007), disorder annotations based on the literature such as MobiDB 3.0 (Piovesan et al. 2023), and consensus-based prediction of long disorder in proteins (The UniProt Consortium 2021). Further, continually expanded structural ensembles have also been generated for IDPs (Ghafouri et al. 2024) with or without explicit experimental (electron paramagnetic resonance or circular dichroism) data compiled through novel ML techniques. Among the structural modeling tools, MODELLER (Eswar et al. 2006) can be effectively used to model whole IDPs (Baruah et al. 2015; Rani and Biswas 2015; Basu and Biswas 2018) as well as missing disordered loops (Roy et al. 2022) in hybrid proteins, using its ‘loop model’ module. AlphaFold (Ruff and Pappu 2021), when used to model disordered proteins, highlights the importance of IDRs (in quantitative sequence–ensemble relationships) and leaves room for misprediction and improvement in functional annotations from the predicted structures. However, AlphaFold2 (Bret et al. 2024), which has revolutionized the state-of-the-art in unraveling the complexity of PPI networks, performs comprehensive scanning of IDRs (short linear motifs, disordered stretches, etc.) across PPI networks and protein interfaces, with an improved prediction accuracy of their functional annotations. The improvement can be rationalized by considering different evolutionary rates of rewiring of the different IDRs in the AlphaFold2 algorithm. For example, under negative selection to maintain function, most domain–motif interactions evolve faster than stable protein complexes. AlphaFold2 has demonstrated the use of PPI network analyses to effectively probe the non-enzymatic functions (binding) of IDRs. To study the interactome of given IDPs computationally, as a next step to experimental binding data (non-structural), often molecular docking is performed, either in a blind mode using ClusPro 2.0 (Kozakov et al. 2017) or in a guided mode (Chen et al. 2003) using ZDock, depending upon the availability of the binding site information. HADDOCK (High Ambiguity Driven biomolecular DOCKing) is a docking tool that allows for the flexible docking of IDPs by integrating experimental data, such as NMR-derived interfacial contacts. This capability makes HADDOCK well-suited for exploring the interactions of IDPs with various partners (Honorato et al. 2024). Since IDPs exist as conformational ensembles, ensemble modeling is often necessarily followed by molecular dynamic simulations. The AWSEM molecular dynamics simulation package (Davtyan et al. 2012) includes a specialized force field for IDPs that accounts for their unique properties. AWSEM simulations can be used to study the conformational dynamics of IDPs, their interactions with other molecules, and their aggregation behavior. To that end, even force fields such as ff14IDPSFF have been developed as part of molecular dynamic packages to specifically deal with IDPs (Song et al. 2017) with improved conformational sampling. The IDP-specific force field ff19SB IDP (Tian et al. 2020) is an updated version of the ff14IDPSFF (Song et al. 2017) force field that has been optimized for IDPs. It has been shown to improve the accuracy of molecular dynamics simulations of IDPs, particularly for their conformational sampling and dynamics. Furthermore, the growing interest in targeting IDPs for drug discovery has spurred the development of computational approaches to identify potential druggable pockets and design small-molecule inhibitors (Joshi and Vendruscolo 2015). Ensemble docking for fuzzy complexes involving IDPs/IDRs (Saurabh et al. 2023) is yet another new computational structural endeavor in the realm of drug discovery. Algorithms have also been developed to study the evolutionary dynamics of disordered regions and that of disorder-to-order transitions in proteins (Nunez-Castilla and Siltberg-Liberles 2020), with the construction of phylogenetic trees and the investigation of site-specific conservation of disorder.
p53: Example of a Unique Idiosyncratic Multifunctional Hybrid Protein with Functionally Crucial IDRs
Hybrid proteins contain structured regions that are connected by disordered loops (i.e., IDRs). IDRs are directly correlated with sequence diversity, making them robust for their regulatory functions. A prime example of this is p53, a protein found in both vertebrates and invertebrates, which has a unique structure-functional mapping. Its primary function is to suppress tumors by regulating cell cycle and control. However, it also has many other related non-enzymatic biological functions, such as PPI and DNA-binding. It can form different biologically active multimers, like homo-tetramers and isoform-based hetero-tetramers. Additionally, it undergoes alternative splicing and has many preferentially localized pre- and post-translational modifications that lead to various isoforms known as ‘proteoforms.’ These combinations, along with the presence of multiple disorder-based protein-binding sites, allow p53 to adopt meta-stable states upon interacting with many binding partners in a switch-like transient manner, characteristic of signal transducers and eukaryotic transcription factors (Uversky 2016b; Már et al. 2023). This is possible due to the flexibility and sequence diversity offered by its IDRs. While acting as a tumor suppressor, it binds to DNA via its highly conserved, well-structured DNA-binding domains. The flanking and interconnecting IDRs often promote these bindings to different partners transiently (Xue et al. 2013). These IDRs situated amidst structured domains in hybrid proteins have high amino acid substitution rates, leading to high sequence heterogeneity. The resultant expressed structural heterogeneity can be categorized into foldons (independently folding units) (Panchenko et al. 1997), inducible foldons (IDRs capable of at least partial folding promoted by interactions with their binding partners), semi-foldons (partially folded regions), non-foldons (non-foldable regions), and unfoldons (ordered regions that require order-to-disorder transition to become functional) (Uversky 2013, 2016b; Kulkarni et al. 2022), underscoring their promiscuous binding capabilities, their presence in PPI networks, and signaling pathways. With over 1000 binding partners, p53’s intrinsic disorder is essential for its functionality. This intricate interplay between protein variation, intrinsic disorder, and functionality underscores the complexity of the biological machinery, with implications for understanding disease pathogenesis and the regulation of cellular processes.
Multifunctional IDPs Involved in Neurodegenerative Disorders and Cancer
The aggregation of the pre-synaptic protein α-synuclein (α-Syn) (IPR002460) as oligomers, protofibrils, and insoluble fibrils within the brain is the pathological hallmark of Parkinson’s disease. It is a 140-amino acid containing small acidic protein comprising three domains—N-terminal lipid-binding domain (NTD), non-amyloid core domain (NAC), and C-terminal acidic tail (CTD) (Emamzadeh 2016). Computational models suggest that α-Syn has ~ 30–80% disordered region, which could convert to α helix or β sheet upon oligomerization or binding to different partners (Piovesan et al. 2023). This conformational flexibility allows α-Syn to engage in multiple functions, such as synaptic vesicle trafficking, regulation of neurotransmitter release, and lipid metabolism (Bendor et al. 2013). Specifically, the disordered NAC region has been shown to be essential for α-Syn’s interactions with lipid membranes and its ability to modulate synaptic vesicle fusion (Burré et al. 2010). Moreover, the flexibility of the CTD enables α-Syn to interact with various protein partners, including chaperones and other synaptic proteins, thereby influencing diverse cellular processes (Emamzadeh 2016). Under normal physiological conditions, it exists in a dynamic equilibrium between unfolded monomers and α-helical tetramers with a low tendency to form aggregates. The tetramer:monomer ratio governed by the binding of molecular chaperones to the NTD reduces monomeric α-Syn, which, in turn, inhibits aggregate formation (Gómez-Benito et al. 2020). Post-translation modification of the NTD and the existence of multiple highly conserved KTKEGV hexameric motif induces helicity important for the protein–lipid interaction. The CTD exists as a random coil due to the abundance of negatively charged amino acids and could be used in different ways, viz., Ca2+ binding, proteolytic cleavage to transition between aggregated and non-aggregated states. Under various cellular stressed conditions, the percentage of free α-Syn increases, leading to cytotoxicity and neurodegenerative disorders (Aspholm et al. 2020).
In line with α-Syn toxicity leading to synucleinopathies, recent reports suggest that it is impacted by a class of evolutionarily conserved disordered proteins, known as small EDRK-rich factor (SERF) (Liu et al. 2024). Computational models estimate this protein to be > 90% disordered (Piovesan et al. 2023). The NMR structure of SERF2 from human shows a disordered N-terminal region wobbling near the C-terminal region (Sahoo et al. 2024). The polar C-terminal, on the other hand, interacts with the acidic tail of monomeric α-Syn promoting aggregation without rendering any ordered structural state of SERF (Falsone et al. 2012). The predominating unstructured SERF fails to recognize the correct binding partner, and its interaction with free, monomeric α-Syn leads to pathological conditions under cellular stress (Nh et al. 2020). This disordered nature of SERF, particularly its flexibility and adaptability, allows it to interact with both α-Syn and RNA, suggesting a potential role in both protein aggregation and RNA regulation. Specifically, the disordered N-terminal region of SERF has been implicated in its interaction with RNA, while the disordered C-terminal region is responsible for its interaction with α-Syn and promotion of aggregation (Liu et al. 2024).
Retinoblastoma protein (pRb) (IPR028309) is an example of a hybrid protein comprising several domains interconnected by disordered loops or IDRs. It also has a disordered CTD, which houses kinases and phosphatases, and flexible linker regions connecting the NTD with one of the subdomains containing a pocket. The intrinsically disordered regions of pRb, particularly the flexible linkers and the disordered CTD with its phosphorylation sites, are central to its multifunctionality. These regions enable pRb to interact with a vast array of over 100 binding partners, including viral oncoproteins and cell cycle regulators (Dick and Rubin 2013). The disordered CTD, for example, acts as a hub for interactions with chromatin remodeling complexes, modulating transcriptional activity (Longworth and Dyson 2010). The flexible linkers allow pRb to adopt diverse conformations upon binding to different partners, enabling it to fine-tune its regulatory roles in cell cycle progression, DNA damage response, and apoptosis (Morris and Dyson 2001). Competitive binding events by multiple partners and misregulation of de-phosphorylation in the IDRs make pRb malfunctional, leading to uncontrolled cellular growth and progression to tumor growth (Dick and Rubin 2013).
Another example of IDP associated with neurodegenerative disorder is the Tau protein (IPR002955) in Alzheimer’s disease (AD), which exists in six different isoforms in the brain and regulates microtubule growth, each containing IDRs contributing to its multifunctionality (Levine et al. 2015; Avila et al. 2016). Under normal physiological conditions, these IDRs can adopt transient secondary structures like α-helix or β-sheet due to post-translational modifications (PTMs), enabling Tau to regulate microtubule growth (Levine et al. 2015). The disordered regions of Tau have been shown to mediate interactions with various other proteins, such as kinases and phosphatases, and these interactions can modulate Tau’s function and aggregation propensity (Mandelkow and Mandelkow 2012). But upon losing the structure and activity, it fails to bind to the microtubules and adopts rigid cross-β structures. This aggregation process is a pathological hallmark of Alzheimer’s disease and gives rise to tauopathies, where highly soluble, disordered Tau protein becomes highly insoluble filaments (Skrabana et al. 2006; Levine et al. 2015). The Tau pathology followed by neuronal death in AD is known to be triggered by the increase of yet another 42 residue long IDP, amyloid-β (Aβ42) (IPR037071), which has served the “Amyloid Cascade Hypothesis” for the last three decades. Relatively newer findings show that both Aβ42 and Tau oligomers bind to amyloid-β protein precursor (AβPP) (P05067) and enter neurons (Gulisano et al. 2018) to induce abnormal synaptic function and memory. Thus, extracellular oligomers of Aβ42 and Tau act in parallel and upstream of AβPP. This has brought about a reconsideration of therapeutic approaches, with an increased interest toward AβPP. Overall, the biomedical relevance of IDPs and their multifunctionality appears to be ever increasing.
Conclusion
The long-established unifunctional model of enzyme classes in proteins was questioned by the increasing evidences of multifunctionality of proteins, for which evolutionary pressure is believed to be one of the guiding forces. This adaptation is also reflected in function-driven structural transitions where IDRs and/or IDPs play a crucial role. Structurally, these are rich in secondary structure-breaking residues such as glycine and proline, as well as other polar and charged residues. They lack hydrophobic and aromatic residues, which are essential core components of a well-folded globular protein. The IDRs and/or IDPs may adopt energetically favorable complexes upon interacting with a diverse range of binding partners. These high specificity–low affinity binding events, along with different structural mosaic patterns, label the proteins as promiscuous and reinforce multitasking capabilities. Although the IDRs and/or IDPs increase the working efficiency in the cellular milieu, the structural flexibility and binding promiscuity often lead to pathological conditions. The problem of aggregate formation (α-Syn, Tau), competitive binding of different partners (SERF), and being the hub nodes of PPI (p53, pRb) attribute to neurodegenerative diseases and different types of cancer. So, these are now being treated as potential drug targets with significant biomedical relevance. The drug-development route to address the diseases requires the knowledge of protein structures to design appropriate molecules. But the structural flexibility of IDPs often hinders the understanding (correlation) of the structure–function relationship experimentally. With increasing numbers of computational tools (AlphaFold, DeepMind, etc.), modeling hypothetical, uncharacterized, and putative proteins with/without IDRs is becoming easier. Software and database like PocketFinder, MobiDB, IUPred, PrDOS, MD simulation, etc. are instrumental in assessing the approximate percentage of disordered regions in hybrid proteins, predicting their probable binding sites, regions undergoing disorder-to-order transitions, as well as the thermodynamic parameters of binding. Understanding how these disordered regions have evolved to adapt and work in such ways is crucial for uncovering the full extent of IDRs and/or IDPs—which would be more insightful in the context of evolved multifunctionality in proteins.
Data Availability
Not applicable.
Notes
INTERPRO ID.
EC number (for enzymes).
UNIPROT ID.
References
Alberts B, Johnson A, Lewis J et al (2002) Protein function. Molecular biology of the cell, 4th edn. Garland Science, New York
Araf Y, Akter F, Tang Y-D et al (2022) Omicron variant of SARS-CoV-2: genomics, transmissibility, and responses to current COVID-19 vaccines. J Med Virol 94:1825–1832. https://doi.org/10.1002/jmv.27588
Aspholm EE, Matečko-Burmann I, Burmann BM (2020) Keeping α-synuclein at bay: a more active role of molecular chaperones in preventing mitochondrial interactions and transition to pathological states? Life (Basel) 10:289. https://doi.org/10.3390/life10110289
Avila J, Jiménez JS, Sayas CL et al (2016) Tau structures. Front Aging Neurosci. https://doi.org/10.3389/fnagi.2016.00262
Babu MM (2016) The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans 44:1185–1200. https://doi.org/10.1042/BST20160172
Balaram P (2021) The murky origins of the coronavirus SARS-CoV-2, the causative agent of the COVID-19 pandemic. Curr Sci 120:4
Bandyopadhyay A, Basu S (2020) Criticality in the conformational phase transition among self-similar groups in intrinsically disordered proteins: probed by salt-bridge dynamics. Biochim Biophys Acta 1868:140474. https://doi.org/10.1016/j.bbapap.2020.140474
Baruah A, Biswas P (2015) Designing pH induced fold switch in proteins. J Chem Phys 142:185102. https://doi.org/10.1063/1.4920938
Baruah A, Rani P, Biswas P (2015) Conformational entropy of intrinsically disordered proteins from amino acid triads. Sci Rep 5:srep11740. https://doi.org/10.1038/srep11740
Basile W, Salvatore M, Bassot C, Elofsson A (2019) Why do eukaryotic proteins contain more intrinsically disordered regions? PLoS Comput Biol 15:e1007186. https://doi.org/10.1371/journal.pcbi.1007186
Basu S, Biswas P (2018) Salt-bridge dynamics in intrinsically disordered proteins: a trade-off between electrostatic interactions and structural flexibility. Biochim Biophys Acta 1866:624–641. https://doi.org/10.1016/j.bbapap.2018.03.002
Basu S, Söderquist F, Wallner B (2017) Proteus: a random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J Comput Aided Mol Des. https://doi.org/10.1007/s10822-017-0020-y
Beadle GW, Tatum EL (1941) Genetic control of biochemical reactions in neurospora*. Proc Natl Acad Sci 27:499–506. https://doi.org/10.1073/pnas.27.11.499
Bendor JT, Logan TP, Edwards RH (2013) The function of α-synuclein. Neuron 79:1044–1066. https://doi.org/10.1016/j.neuron.2013.09.004
Bernhardt NA, Hansmann UHE (2018) Multifunnel landscape of the fold-switching protein RfaH-CTD. J Phys Chem B 122:1600–1607. https://doi.org/10.1021/acs.jpcb.7b11352
Bill CA, Vines CM (2020) Phospholipase C. Adv Exp Med Biol 1131:215–242. https://doi.org/10.1007/978-3-030-12457-1_9
Bret H, Gao J, Zea DJ et al (2024) From interaction networks to interfaces, scanning intrinsically disordered regions using AlphaFold2. Nat Commun 15:597. https://doi.org/10.1038/s41467-023-44288-7
Brüssow H, Canchaya C, Hardt W-D (2004) Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev 68:560–602. https://doi.org/10.1128/MMBR.68.3.560-602.2004
Bryan PN, Orban J (2010) Proteins that switch folds. Curr Opin Struct Biol 20:482–488. https://doi.org/10.1016/j.sbi.2010.06.002
Burré J, Sharma M, Tsetsenis T et al (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. https://doi.org/10.1126/science.1195227
Cai Y, Zhang J, Xiao T et al (2020) Distinct conformational states of SARS-CoV-2 spike protein. Science. https://doi.org/10.1126/science.abd4251
Chen R, Li L, Weng Z (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52:80–87. https://doi.org/10.1002/prot.10389
Chen C, Liu H, Zabad S et al (2021) MoonProt 3.0: an update of the moonlighting proteins database. Nucleic Acids Res 49:D368–D372. https://doi.org/10.1093/nar/gkaa1101
Cino EA, Killoran RC, Karttunen M, Choy W-Y (2013) Binding of disordered proteins to a protein hub. Sci Rep 3:2305. https://doi.org/10.1038/srep02305
Clark SA, Jespersen N, Woodward C, Barbar E (2015) Multivalent IDP assemblies: unique properties of LC8-associated, IDP duplex scaffolds. FEBS Lett 589:2543–2551. https://doi.org/10.1016/j.febslet.2015.07.032
Davey NE, Travé G, Gibson TJ (2011) How viruses hijack cell regulation. Trends Biochem Sci 36:159–169. https://doi.org/10.1016/j.tibs.2010.10.002
Davtyan A, Schafer NP, Zheng W et al (2012) AWSEM-MD: protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B 116:8494–8503. https://doi.org/10.1021/jp212541y
Dick FA, Rubin SM (2013) Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14:297–306. https://doi.org/10.1038/nrm3567
Disfani FM, Hsu W-L, Mizianty MJ et al (2012) MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 28:i75–i83. https://doi.org/10.1093/bioinformatics/bts209
Dosztányi Z, Csizmok V, Tompa P, Simon I (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21:3433–3434. https://doi.org/10.1093/bioinformatics/bti541
Dunker AK, Lawson JD, Brown CJ et al (2001) Intrinsically disordered protein. J Mol Graph Model 19:26–59. https://doi.org/10.1016/S1093-3263(00)00138-8
Dunker AK, Cortese MS, Romero P et al (2005) Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 272:5129–5148. https://doi.org/10.1111/j.1742-4658.2005.04948.x
Dyson HJ, Wright PE (2002) Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 12:54–60. https://doi.org/10.1016/s0959-440x(02)00289-0
Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6:197–208. https://doi.org/10.1038/nrm1589
Ekman D, Light S, Björklund ÅK, Elofsson A (2006) What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol 7:R45. https://doi.org/10.1186/gb-2006-7-6-r45
El-Deiry WS, Tokino T, Velculescu VE et al (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825. https://doi.org/10.1016/0092-8674(93)90500-P
Emamzadeh FN (2016) Alpha-synuclein structure, functions, and interactions. J Res Med Sci. https://doi.org/10.4103/1735-1995.181989
Emenecker RJ, Griffith D, Holehouse AS (2021) Metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys J 120:4312–4319. https://doi.org/10.1016/j.bpj.2021.08.039
Eriksson SE, Ceder S, Bykov VJN, Wiman KG (2019) p53 as a hub in cellular redox regulation and therapeutic target in cancer. J Mol Cell Biol 11:330–341. https://doi.org/10.1093/jmcb/mjz005
Espinosa-Cantú A, Ascencio D, Barona-Gómez F, DeLuna A (2015) Gene duplication and the evolution of moonlighting proteins. Front Genet 6:227
Espinosa-Cantú A, Cruz-Bonilla E, Noda-Garcia L, DeLuna A (2020) Multiple forms of multifunctional proteins in health and disease. Front Cell Dev Biol 8:451. https://doi.org/10.3389/fcell.2020.00451
Eswar N, Webb B, Marti-Renom MA et al (2006) Comparative protein structure modeling using modeller. Curr Protoc Bioinform. https://doi.org/10.1002/0471250953.bi0506s15
Falsone SF, Meyer NH, Schrank E et al (2012) SERF protein is a direct modifier of amyloid fiber assembly. Cell Rep 2:358–371. https://doi.org/10.1016/j.celrep.2012.06.012
Fang C, Noguchi T, Tominaga D, Yamana H (2013) MFSPSSMpred: identifying short disorder-to-order binding regions in disordered proteins based on contextual local evolutionary conservation. BMC Bioinform 14:300. https://doi.org/10.1186/1471-2105-14-300
Fersht PTA (2009) Structure and function of intrinsically disordered proteins. Chapman and Hall/CRC, New York
Fewell SW, Woolford JL (1999) Ribosomal protein S14 of Saccharomyces cerevisiae regulates its expression by binding to RPS14B pre-mRNA and to 18S rRNA. Mol Cell Biol 19:826–834. https://doi.org/10.1128/MCB.19.1.826
Fu Y, Guo Y, Wang Y et al (2015) Exploring the relationship between hub proteins and drug targets based on GO and intrinsic disorder. Comput Biol Chem 56:41–48. https://doi.org/10.1016/j.compbiolchem.2015.03.003
Gao C, Ma C, Wang H et al (2021) Intrinsic disorder in protein domains contributes to both organism complexity and clade-specific functions. Sci Rep 11:2985. https://doi.org/10.1038/s41598-021-82656-9
Ghafouri H, Lazar T, Del Conte A et al (2024) PED in 2024: improving the community deposition of structural ensembles for intrinsically disordered proteins. Nucleic Acids Res 52:D536–D544. https://doi.org/10.1093/nar/gkad947
Ghose DA, Przydzial KE, Mahoney EM et al (2023) Marginal specificity in protein interactions constrains evolution of a paralogous family. Proc Natl Acad Sci 120:e2221163120. https://doi.org/10.1073/pnas.2221163120
Gómez-Benito M, Granado N, García-Sanz P et al (2020) Modeling Parkinson’s disease with the alpha-synuclein protein. Front Pharmacol 11:356. https://doi.org/10.3389/fphar.2020.00356
Gruber T, Lewitzky M, Machner L et al (2022) Macromolecular crowding induces a binding competent transient structure in intrinsically disordered Gab1. J Mol Biol 434:167407. https://doi.org/10.1016/j.jmb.2021.167407
Gulisano W, Maugeri D, Baltrons MA et al (2018) Role of amyloid-β and Tau proteins in Alzheimer’s disease: confuting the amyloid cascade. J Alzheimers Dis 64:S611–S631. https://doi.org/10.3233/JAD-179935
Han J-DJ, Bertin N, Hao T et al (2004) Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430:88–93. https://doi.org/10.1038/nature02555
Hanson J, Litfin T, Paliwal K, Zhou Y (2020) Identifying molecular recognition features in intrinsically disordered regions of proteins by transfer learning. Bioinformatics 36:1107–1113. https://doi.org/10.1093/bioinformatics/btz691
Hanson J (2020) SPOT-Disorder2: protein disorder prediction Jack Hanson. In: Sparks Lab. https://sparks-lab.org/server/spot-disorder2/ Accessed 19 Jul 2024
Hasiów-Jaroszewska B, Fares MA, Elena SF (2014) Molecular evolution of viral multifunctional proteins: the case of potyvirus HC-Pro. J Mol Evol 78:75–86. https://doi.org/10.1007/s00239-013-9601-0
He X, Zhang J (2005) Rapid subfunctionalization accompanied by prolonged and substantial neofunctionalization in duplicate gene evolution. Genetics 169:1157–1164. https://doi.org/10.1534/genetics.104.037051
Hedstrom L (2002) Serine protease mechanism and specificity. Chem Rev 102:4501–4524. https://doi.org/10.1021/cr000033x
Higurashi M, Ishida T, Kinoshita K (2008) Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci 17:72–78. https://doi.org/10.1110/ps.073196308
Honorato RV, Trellet ME, Jiménez-García B et al (2024) The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat Protoc. https://doi.org/10.1038/s41596-024-01011-0
Hu G, Katuwawala A, Wang K et al (2021) flDPnn: accurate intrinsic disorder prediction with putative propensities of disorder functions. Nat Commun 12:4438. https://doi.org/10.1038/s41467-021-24773-7
Huber AH, Weis WI (2001) The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell 105:391–402. https://doi.org/10.1016/s0092-8674(01)00330-0
Hughes AL, Friedman R (2003) Parallel evolution by gene duplication in the genomes of two unicellular fungi. Genome Res 13:794–799. https://doi.org/10.1101/gr.714603
Imtaiyaz Hassan M, Shajee B, Waheed A et al (2013) Structure, function and applications of carbonic anhydrase isozymes. Bioorg Med Chem 21:1570–1582. https://doi.org/10.1016/j.bmc.2012.04.044
Ishida T, Kinoshita K (2007) PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res 35:W460-464. https://doi.org/10.1093/nar/gkm363
Jayaraman V, Toledo-Patiño S, Noda-García L, Laurino P (2022) Mechanisms of protein evolution. Protein Sci 31:e4362. https://doi.org/10.1002/pro.4362
Jeffery CJ (1999) Moonlighting proteins. Trends Biochem Sci 24:8–11. https://doi.org/10.1016/s0968-0004(98)01335-8
Jeffery CJ (2003) Multifunctional proteins: examples of gene sharing. Ann Med 35:28–35. https://doi.org/10.1080/07853890310004101
Jeffery CJ (2004) Molecular mechanisms for multitasking: recent crystal structures of moonlighting proteins. Curr Opin Struct Biol 14:663–668. https://doi.org/10.1016/j.sbi.2004.10.001
Jeffery CJ (2018) Protein moonlighting: what is it, and why is it important? Philos Trans R Soc Lond B 373:20160523. https://doi.org/10.1098/rstb.2016.0523
Jones DT, Cozzetto D (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31:857–863. https://doi.org/10.1093/bioinformatics/btu744
Joshi P, Vendruscolo M (2015) Druggability of intrinsically disordered proteins. Adv Exp Med Biol 870:383–400. https://doi.org/10.1007/978-3-319-20164-1_13
Kamagata K, Mano E, Itoh Y et al (2019) Rational design using sequence information only produces a peptide that binds to the intrinsically disordered region of p53. Sci Rep 9:8584. https://doi.org/10.1038/s41598-019-44688-0
Kawashima T, Kawashima S, Tanaka C et al (2009) Domain shuffling and the evolution of vertebrates. Genome Res 19:1393–1403. https://doi.org/10.1101/gr.087072.108
Kenley EC, Kirk L, Cho Y-R (2011) Differentiating party and date hubs in protein interaction networks using semantic similarity measures. In: Proceedings of the 2nd ACM conference on bioinformatics, computational biology and biomedicine. Association for Computing Machinery, New York, NY, USA, pp 641–645
Keren H, Lev-Maor G, Ast G (2010) Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11:345–355. https://doi.org/10.1038/nrg2776
Kim AK, Porter LL (2021) Functional and regulatory roles of fold-switching proteins. Structure 29:6–14. https://doi.org/10.1016/j.str.2020.10.006
Kozakov D, Hall DR, Xia B et al (2017) The ClusPro web server for protein-protein docking. Nat Protoc 12:255–278. https://doi.org/10.1038/nprot.2016.169
Kulkarni P, Behal A, Mohanty A et al (2022) Co-opting disorder into order: intrinsically disordered proteins and the early evolution of complex multicellularity. Int J Biol Macromol 201:29–36. https://doi.org/10.1016/j.ijbiomac.2021.12.182
Laity JH, Lee BM, Wright PE (2001) Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol 11:39–46. https://doi.org/10.1016/s0959-440x(00)00167-6
Levine ZA, Larini L, LaPointe NE et al (2015) Regulation and aggregation of intrinsically disordered peptides. Proc Natl Acad Sci USA 112:2758. https://doi.org/10.1073/pnas.1418155112
Levy Y, Cho SS, Onuchic JN, Wolynes PG (2005) A survey of flexible protein binding mechanisms and their transition states using native topology based energy landscapes. J Mol Biol 346:1121–1145. https://doi.org/10.1016/j.jmb.2004.12.021
Liu H-N, Wang T, Hu J-J et al (2024) The disordered protein SERF promotes α-Synuclein aggregation through liquid-liquid phase separation. J Biol Chem 300:105667. https://doi.org/10.1016/j.jbc.2024.105667
Longworth MS, Dyson NJ (2010) pRb, a local chromatin organizer with global possibilities. Chromosoma 119:1–11. https://doi.org/10.1007/s00412-009-0238-0
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290:1151–1155. https://doi.org/10.1126/science.290.5494.1151
Lynch M, Force A (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459–473. https://doi.org/10.1093/genetics/154.1.459
Mallik S, Tawfik DS, Levy ED (2022) How gene duplication diversifies the landscape of protein oligomeric state and function. Curr Opin Genet Dev. https://doi.org/10.1016/j.gde.2022.101966
Mandelkow E-M, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2:a006247. https://doi.org/10.1101/cshperspect.a006247
Mannige RV (2014) Dynamic new world: refining our view of protein structure, function and evolution. Proteomes 2:128–153. https://doi.org/10.3390/proteomes2010128
Már M, Nitsenko K, Heidarsson PO (2023) Multifunctional intrinsically disordered regions in transcription factors. Chemistry 29:e202203369. https://doi.org/10.1002/chem.202203369
Maulud SQ, Hasan DA, Ali RK et al (2022) Deltacron: apprehending a new phase of the COVID-19 pandemic. Int J Surg 102:106654. https://doi.org/10.1016/j.ijsu.2022.106654
Maurice MSt, Reinhardt L, Surinya KH, et al (2007) Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science 317:1076–1079. https://doi.org/10.1126/science.1144504
Mazumder B, Li X, Barik S (2010) Translation control: a multifaceted regulator of inflammatory response. J Immunol 184:3311–3319. https://doi.org/10.4049/jimmunol.0903778
Mazzei L, Musiani F, Ciurli S (2020) The structure-based reaction mechanism of urease, a nickel dependent enzyme: tale of a long debate. J Biol Inorg Chem 25:829–845. https://doi.org/10.1007/s00775-020-01808-w
Mercurio I, Tragni V, Busto F et al (2020) Protein structure analysis of the interactions between SARS-CoV-2 spike protein and the human ACE2 receptor: from conformational changes to novel neutralizing antibodies. Cell Mol Life Sci. https://doi.org/10.1007/s00018-020-03580-1
Mészáros B, Simon I, Dosztányi Z (2009) Prediction of protein binding regions in disordered proteins. PLOS Comput Biol 5:e1000376. https://doi.org/10.1371/journal.pcbi.1000376
Metcalfe C, Bienz M (2011) Inhibition of GSK3 by Wnt signaling—two contrasting models. J Cell Sci 124:3537–3544. https://doi.org/10.1242/jcs.091991
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299. https://doi.org/10.1016/0092-8674(95)90412-3
Mohan A, Oldfield CJ, Radivojac P et al (2006) Analysis of molecular recognition features (MoRFs). J Mol Biol 362:1043–1059. https://doi.org/10.1016/j.jmb.2006.07.087
Morris EJ, Dyson NJ (2001) Retinoblastoma protein partners. Adv Cancer Res 82:1–54. https://doi.org/10.1016/s0065-230x(01)82001-7
Morris OM, Torpey JH, Isaacson RL (2021) Intrinsically disordered proteins: modes of binding with emphasis on disordered domains. Open Biol 11:210222. https://doi.org/10.1098/rsob.210222
Moutinho AF, Trancoso FF, Dutheil JY (2019) The impact of protein architecture on adaptive evolution. Mol Biol Evol 36:2013–2028. https://doi.org/10.1093/molbev/msz134
Nguyen Ba AN, Strome B, Hua JJ et al (2014) Detecting functional divergence after gene duplication through evolutionary changes in posttranslational regulatory sequences. PLoS Comput Biol 10:e1003977. https://doi.org/10.1371/journal.pcbi.1003977
Nh M, H D, C T-A, et al (2020) Structural fuzziness of the RNA-organizing protein SERF determines a toxic gain-of-interaction. J Mol Biol 432:930–951
Nunez-Castilla J, Siltberg-Liberles J (2020) An easy protocol for evolutionary analysis of intrinsically disordered proteins. Methods Mol Biol 2141:147–177. https://doi.org/10.1007/978-1-0716-0524-0_7
Ohno S (1970) Evolution by gene duplication. Springer, Heidelberg
Oldfield CJ, Cheng Y, Cortese MS et al (2005) Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 44:12454–12470. https://doi.org/10.1021/bi050736e
Panchenko AR, Luthey-Schulten Z, Cole R, Wolynes PG (1997) The foldon universe: a survey of structural similarity and self-recognition of independently folding units 1. J Mol Biol 272:95–105. https://doi.org/10.1006/jmbi.1997.1205
Parker TW, Neufeld KL (2020) APC controls Wnt-induced β-catenin destruction complex recruitment in human colonocytes. Sci Rep 10:2957. https://doi.org/10.1038/s41598-020-59899-z
Patel MS, Nemeria NS, Furey W, Jordan F (2014) The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem 289:16615–16623. https://doi.org/10.1074/jbc.R114.563148
Piatigorsky J (2009) Gene sharing and evolution: the diversity of protein functions. Gene sharing and evolution. Harvard University Press, Cambridge
Piovesan D, Del Conte A, Clementel D et al (2023) MobiDB: 10 years of intrinsically disordered proteins. Nucleic Acids Res 51:D438–D444. https://doi.org/10.1093/nar/gkac1065
Popławska A, Szumowska E, Kuś J (2021) Why do we need media multitasking? A self-regulatory perspective. Front Psychol 12:624649. https://doi.org/10.3389/fpsyg.2021.624649
Portin P, Wilkins A (2017) The evolving definition of the term “gene.” Genetics 205:1353–1364. https://doi.org/10.1534/genetics.116.196956
Rani P, Biswas P (2015) Local structure and dynamics of hydration water in intrinsically disordered proteins. J Phys Chem B 119:10858–10867. https://doi.org/10.1021/jp511961c
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343–D350. https://doi.org/10.1093/nar/gkr987
Reid KM, Poudel H, Leitner DM (2023) Dynamics of hydrogen bonds between water and intrinsically disordered and structured regions of proteins. J Phys Chem B 127:7839–7847. https://doi.org/10.1021/acs.jpcb.3c03102
Roy S, Ghosh P, Bandyopadhyay A, Basu S (2022) Capturing a crucial ‘disorder-to-order transition’ at the heart of the coronavirus molecular pathology—triggered by highly persistent, interchangeable salt-bridges. Vaccines 10:301. https://doi.org/10.3390/vaccines10020301
Ruff KM, Pappu RV (2021) AlphaFold and implications for intrinsically disordered proteins. J Mol Biol 433:167208. https://doi.org/10.1016/j.jmb.2021.167208
Sahoo BR, Subramanian V, Bardwell JCA (2024) Backbone 1H, 13C, and 15N chemical shift assignments for human SERF2. Biomol NMR Assign 18:51–57. https://doi.org/10.1007/s12104-024-10167-5
Saurabh S, Nadendla K, Purohit SS et al (2023) Fuzzy drug targets: disordered proteins in the drug-discovery realm. ACS Omega 8:9729–9747. https://doi.org/10.1021/acsomega.2c07708
Schnoes AM, Brown SD, Dodevski I, Babbitt PC (2009) Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol 5:e1000605. https://doi.org/10.1371/journal.pcbi.1000605
Seoane J, Le H-V, Massagué J (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419:729–734. https://doi.org/10.1038/nature01119
Shoemaker BA, Portman JJ, Wolynes PG (2000) Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Natl Acad Sci USA 97:8868–8873. https://doi.org/10.1073/pnas.160259697
Sickmeier M, Hamilton JA, LeGall T et al (2007) DisProt: the database of disordered proteins. Nucleic Acids Res 35:D786–D793. https://doi.org/10.1093/nar/gkl893
Sikosek T, Chan HS (2014) Biophysics of protein evolution and evolutionary protein biophysics. J R Soc Interface 11:20140419. https://doi.org/10.1098/rsif.2014.0419
Simister PC, Schaper F, O’Reilly N et al (2011) Self-organization and regulation of intrinsically disordered proteins with folded N-termini. PLoS Biol 9:e1000591. https://doi.org/10.1371/journal.pbio.1000591
Skrabana R, Skrabanova M, Csokova N et al (2006) Intrinsically disordered tau protein in Alzheimer’s tangles: a coincidence or a rule? Bratisl Lek Listy 107:354–358
Song D, Luo R, Chen H-F (2017) The IDP-specific force field ff14IDPSFF improves the conformer sampling of intrinsically disordered proteins. J Chem Inf Model 57:1166–1178. https://doi.org/10.1021/acs.jcim.7b00135
Stastna M, Van Eyk JE (2012) Analysis of protein isoforms: can we do it better? Proteomics 12:2937–2948. https://doi.org/10.1002/pmic.201200161
Sugase K, Dyson HJ, Wright PE (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447:1021–1025. https://doi.org/10.1038/nature05858
Sun X, Rikkerink EHA, Jones WT, Uversky VN (2013) Multifarious roles of intrinsic disorder in proteins illustrate its broad impact on plant biology. Plant Cell 25:38–55
The UniProt Consortium (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49:D480–D489. https://doi.org/10.1093/nar/gkaa1100
Tian C, Kasavajhala K, Belfon KAA et al (2020) ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J Chem Theory Comput 16:528–552. https://doi.org/10.1021/acs.jctc.9b00591
Tokuriki N, Stricher F, Serrano L, Tawfik DS (2008) How protein stability and new functions trade off. PLOS Comput Biol 4:e1000002. https://doi.org/10.1371/journal.pcbi.1000002
Tsai CJ, Kumar S, Ma B, Nussinov R (1999) Folding funnels, binding funnels, and protein function. Protein Sci 8:1181–1190. https://doi.org/10.1110/ps.8.6.1181
Turek I, Irving H (2021) Moonlighting proteins shine new light on molecular signaling niches. Int J Mol Sci 22:1367. https://doi.org/10.3390/ijms22031367
Uncapher MR, Lin L, Rosen LD et al (2017) Media multitasking and cognitive, psychological, neural, and learning differences. Pediatrics 140:S62–S66. https://doi.org/10.1542/peds.2016-1758D
Uversky VN (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci 11:739–756. https://doi.org/10.1110/ps.4210102
Uversky VN (2013) Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 1834:932–951. https://doi.org/10.1016/j.bbapap.2012.12.008
Uversky VN (2016a) Dancing protein clouds: the strange biology and chaotic physics of intrinsically disordered proteins. J Biol Chem 291:6681–6688. https://doi.org/10.1074/jbc.R115.685859
Uversky VN (2016b) p53 Proteoforms and intrinsic disorder: an illustration of the protein structure-function continuum concept. Int J Mol Sci 17:1874. https://doi.org/10.3390/ijms17111874
Vacic V, Oldfield CJ, Mohan A et al (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res 6:2351–2366. https://doi.org/10.1021/pr0701411
Vishwanath S, de Brevern AG, Srinivasan N (2018) Same but not alike: structure, flexibility and energetics of domains in multi-domain proteins are influenced by the presence of other domains. PLoS Comput Biol 14:e1006008. https://doi.org/10.1371/journal.pcbi.1006008
Wang H, Guo M, Wei H, Chen Y (2023) Targeting p53 pathways: mechanisms, structures, and advances in therapy. Sig Transduct Target Ther 8:1–35. https://doi.org/10.1038/s41392-023-01347-1
Waudby CA, Dobson CM, Christodoulou J (2019) Nature and regulation of protein folding on the ribosome. Trends Biochem Sci 44:914–926. https://doi.org/10.1016/j.tibs.2019.06.008
Wikström M, Krab K (1979) Proton-pumping cytochrome c oxidase. Biochim Biophys Acta 549:177–222. https://doi.org/10.1016/0304-4173(79)90014-4
Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 293:321–331. https://doi.org/10.1006/jmbi.1999.3110
Wright PE, Dyson HJ (2015) Intrinsically disordered proteins in cellular signaling and regulation. Nat Rev Mol Cell Biol 16:18–29. https://doi.org/10.1038/nrm3920
Wright CJ, Smith CWJ, Jiggins CD (2022) Alternative splicing as a source of phenotypic diversity. Nat Rev Genet 23:697–710. https://doi.org/10.1038/s41576-022-00514-4
Wygrecka M, Marsh LM, Morty RE et al (2009) Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood 113:5588–5598. https://doi.org/10.1182/blood-2008-08-170837
Xue B, Brown CJ, Dunker AK, Uversky VN (2013) Intrinsically disordered regions of p53 family are highly diversified in evolution. Biochim Biophys Acta 1834:725–738. https://doi.org/10.1016/j.bbapap.2013.01.012
Yi Q, Liu W, Seo JH et al (2023) Discovery of a small-molecule inhibitor targeting the androgen receptor N-terminal domain for castration-resistant prostate cancer. Mol Cancer Ther 22:570–582. https://doi.org/10.1158/1535-7163.MCT-22-0237
Yokoyama S (2000) Molecular evolution of vertebrate visual pigments. Prog Retin Eye Res 19:385–419. https://doi.org/10.1016/s1350-9462(00)00002-1
Zallot R, Harrison KJ, Kolaczkowski B, de Crécy-Lagard V (2016) Functional annotations of paralogs: a blessing and a curse. Life (Basel) 6:39. https://doi.org/10.3390/life6030039
Zamora-Briseño JA, Pereira-Santana A, Reyes-Hernández SJ et al (2021) Towards an understanding of the role of intrinsic protein disorder on plant adaptation to environmental challenges. Cell Stress Chaperones 26:141–150. https://doi.org/10.1007/s12192-020-01162-5
Zhang F, Zhao B, Shi W et al (2022) DeepDISOBind: accurate prediction of RNA-, DNA- and protein-binding intrinsically disordered residues with deep multi-task learning. Brief Bioinform 23:bbab521. https://doi.org/10.1093/bib/bbab521
Acknowledgements
We thank Dr. Ritobrita Chakraborty, Post Doctoral Researcher, University of Pennsylvania, Philadelphia, USA and Dr. Kuntal Kanti Goswami, Assistant Professor, Department of Microbiology, Asutosh College for crucial discussions on the topic and wise terminological inputs. We also take the opportunity to sincerely acknowledge the contribution of the two anonymous reviewers reviewing this manuscript, whose insightful comments and constructive criticism have undoubtedly raised the quality of the paper substantially.
Funding
The project was self-funded.
Author information
Authors and Affiliations
Contributions
SB conceptualized the problem. AA wrote the first draft assisted by AB. Everybody participated in the literature survey. SB made the figure and edited the manuscript rigorously with crucial inputs from SN. SN wrote the conclusion and other sections during revision. SB, AA, and SN did the first major revisions. SS did bulk of the second major revision alongside SB with help from AB. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Handling editor: Erich Bornberg-Bauer.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aftab, A., Sil, S., Nath, S. et al. Intrinsic Disorder and Other Malleable Arsenals of Evolved Protein Multifunctionality. J Mol Evol (2024). https://doi.org/10.1007/s00239-024-10196-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00239-024-10196-7