
Abstract
Although the proteins in all the current major classes considered to be druggable are folded in their native states, intrinsically disordered proteins (IDPs) are becoming attractive candidates for therapeutic intervention by small drug-like molecules. IDPs are challenging targets because they exist as ensembles of structures, thereby making them unsuitable for standard rational drug design approaches, which require the knowledge of the three-dimensional structure of the proteins to be drugged. As we review in this chapter, several different small molecule strategies are currently under investigation to target IDPs, including: (i) to stabilise IDPs in their natively disordered states, (ii) to inhibit interactions with ordered or disordered protein partners, and (iii) to induce allosteric inhibition. In this context, biophysical techniques, including in particular nuclear magnetic resonance (NMR) spectroscopy and small-angle X-ray scattering (SAXS) coupled with molecular dynamics simulations and chemoinformatics approaches, are increasingly used to characterize the structural ensembles of IDPs and the specific interactions that they make with their binding partners. By analysing the results of recent studies, we describe the main structural features that may render IDPs druggable, and describe techniques that can be used for drug discovery programs focused on IDPs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Intrinsically disordered proteins
- Drug design
- Nuclear magnetic resonance spectroscopy
- Small molecule library
- Drug discovery
1 Introduction: IDPs as Therapeutic Targets
IDP Function and Dysfunction
As described in earlier chapters of this book, intrinsically disordered proteins (IDPs) play major roles in a wide range of biochemical processes in living organisms. A range of recent studies has revealed that the functional diversity provided by disordered regions complements that of ordered regions of proteins (Iakoucheva et al. 2002; Dyson and Wright 2005; Tompa 2012; Uversky 2013; Dunker et al. 2002; van der Lee et al. 2014; Babu et al. 2012), in particular in terms of key cellular functions such as signaling and regulation (Iakoucheva et al. 2002; Dyson and Wright 2005; Tompa 2012; Uversky 2013; Dunker et al. 2002; Uversky et al. 2008, 2009; Knowles et al. 2014; Babu et al. 2012; van der Lee et al. 2014). The high flexibility and lack of stable secondary and tertiary structures allow IDPs to interact with multiple partners, often placing them at the hubs of protein-protein interaction networks (Mészáros et al. 2011; Malaney et al. 2013; Dunker et al. 2005). It has also been realized that the failure of the regulatory processes responsible for the correct behaviour of IDPs, which can be referred to as ‘IDP homeostasis’, is associated with a variety of pathological conditions (Uversky et al. 2008, 2009; Knowles et al. 2014). Indeed, intrinsic disorder is often observed in peptides and proteins implicated in a series of human conditions including cancer, cardiovascular diseases and neurodegenerative disorders (Uversky et al. 2008, 2009; Knowles et al. 2014) (Fig. 13.1).

Presence of intrinsic disorder in disease-associated proteins. The histogram presents the percentage of disease-associated proteins with more than 30 to more than 100 consecutive residues predicted to be disordered, with error bars representing 95 % confidence intervals; for comparison, data for signalling and ordered proteins are also shown. Reprinted from (Uversky et al. 2008)
IDPs Are Implicated in Misfolding Diseases
Among all the diseases associated with IDPs, great attention has recently been devoted to misfolding diseases (Chiti and Dobson 2006; Dobson 2001; Knowles et al. 2014). Most of these diseases originate from the conversion of specific proteins from their soluble functional states into stable, highly ordered filamentous protein aggregates, known as amyloid fibrils, which accumulate in a variety of organs and tissues (Chiti and Dobson 2006; Dobson 2001; Knowles et al. 2014). Amyloid fibrils display common properties including a characteristic cross-β structure in which continuous β-sheets are formed with β-strands running perpendicularly to the long axis of the fibrils (Fitzpatrick et al. 2013; Eisenberg and Jucker 2012; Knowles et al. 2014). Although amyloid fibrils from different diseases are structurally and morphologically similar to each other, the polypeptide chains that constitute the fibrils are diverse and the native conformation may be rich in β-sheets and α-helices, or they may be natively unfolded (Fitzpatrick et al. 2013; Eisenberg and Jucker 2012; Knowles et al. 2014). The conformational diversity of the native states of amyloidogenic proteins, as opposed to the close structural similarity of the resultant amyloid fibrils, implies that substantial structural rearrangements need to occur for fibril formation to happen. As IDPs are devoid of stable structure, the primary step of their fibrillogenesis requires the stabilization of monomeric or oligomeric partially folded conformations. Such partially folded conformations are characterised by the presence of specific intermolecular interactions, including electrostatic attraction, hydrogen bonding, and hydrophobic contacts, which promote the assembly process. Therefore, a possible strategy to target aggregation-prone IDPs implicated in misfolding diseases is to stabilize the protein in conformations that do not readily enable the formation of higher order species. Indeed, recent studies are beginning to suggest that such approaches may be promising (Zhu et al. 2013; Toth et al. 2014; Dunker and Uversky 2010; Metallo 2010; Rezaei-Ghaleh et al. 2012; Uversky 2012).
Therapeutic Targeting of IDPs
The foremost reason to target IDPs pharmacologically is that they are closely associated with a wide range of diseases. Therapeutic intervention strategies aimed at maintaining the normal functionality of these proteins require the characterization of their structures and functional mechanisms, as indeed is beginning to be done (Iakoucheva et al. 2002; Dyson and Wright 2005; Tompa 2012; Uversky 2013; Dunker et al. 2002; Uversky et al. 2008, 2009; Knowles et al. 2014). This objective may be achieved by a range of different strategies, including: (i) the stabilization of IDPs in their natively disordered states, for instance in the case of aggregation-prone proteins, (ii) the inhibition of interactions with molecular partners, in cases where a small molecule perturbs the binding interface, and (iii) the regulation of the behaviour of IDPs by allosteric effects, for instance when the binding of a small molecule to an ordered region causes the disordered region to become ordered.
Most drugs target enzymes or cell surface receptors by modulating their functions, where small molecules can mimic the interactions made by their natural substrates (Imming et al. 2006). Even though enzymes possess a certain degree of flexibility to be able to act on a variety of substrates, their structures tend to fluctuate around equilibrium positions, making it easier to identify binding pockets and subsequently tailor drugs to fit in them. By contrast, IDPs explore large ensembles of structures, which exhitbit large conformational fluctuations and no evidence of permanent binding pockets. This type of conformational features does not present suitable cavities for small drug-like molecules to form stable interactions (Dunker and Uversky 2010; Metallo 2010; Toth et al. 2014; Zhu et al. 2013).
Quite generally, the binding of a small molecule to a disordered protein may seem counterintuitive because of the large difference expected between the entropic loss and the enthalpic gain upon binding (Metallo 2010; Toth et al. 2014; Zhu et al. 2013). As we will describe in more detail in the remainder of this chapter, however, some intrinsically disordered proteins have been shown to be capable of forming adaptable, specific interfaces for small molecule binding (Metallo 2010; Toth et al. 2014; Zhu et al. 2013).
Druggability of IDPs
The ability to find compounds that bind to a target protein does not by itself imply that this protein is fully druggable, as the compounds should also be orally bioavailable (Hopkins and Groom 2002). Lipinski proposed a set of five criteria to readily estimate bioavailability, the so-called ‘rule of five’, that includes restrictions on the molecular weight (< 500 Da), number of hydrogen bond acceptors (< 10), number of hydrogen bond donors (< 5) and the octanol-water partition coefficient (< 5) of the potential lead compound (Lipinski 2004). These rules should be in some way combined with the primary demand for the design of small molecule drugs, that they should be able to form relatively strong interactions with the protein to overcome the entropy loss upon binding. IDPs are in this context particularly challenging targets, as their mobility implies that, compared to structured proteins, their interactions with small molecules are weaker and more transient, and the entropic loss greater.
As noted above, the high abundance and peculiar roles of IDPs in protein-protein interactions (PPIs) makes them desirable drug targets. However, attempts at developing small molecule drugs that block protein-protein interactions have generally been challenging (Arkin and Wells 2004; Wells and McClendon 2007; Hopkins and Groom 2002). Investigations on the reasons that make PPIs difficult drug targets are beginning to reveal some of the molecular mechanisms responsible for these problems. It has been shown that PPIs display complex binding surfaces, have discontinuous epitopes or multiple continuous epitopes, are devoid of groves or pockets, exhibit interaction regions that are often as large as 1500–3000 Å2, and have energies that are not evenly distributed over a large contact area but over smaller regions called hotspots (Jones and Thornton 1996; Conte et al. 1999; Wells and McClendon 2007).
Specialized libraries for IDPs have not been extensively adopted to date, and the properties of compounds that bind IDPs have not been intensively investigated. Compounds that bind IDPs tend to have properties that make them different from those from more conventional target classes, as in particular they are larger and more three-dimensional. In the absence of a more complete understanding of these properties, the question regarding the set of compounds that could adequately provide coverage of the chemical space specifically for IDPs is still open. To this end, the construction of libraries of compounds specific for IDPs would be highly useful, as it would lead to a better and faster design of inhibitor libraries and promote more effective efforts towards drug discovery programs for IDPs.
2 Strategies for Drug Discovery for IDPs
Strategies for therapeutic intervention for IDPs involve a wide range of objectives, including:
-
I.
To maintain IDPs in their natively disordered states;
-
II.
To inhibit interactions with ordered or disordered partners;
-
III.
To induce allosteric inhibition.
Below we discuss some examples that illustrate how these strategies have begun to be implemented in recent studies.
2.1 Case Study 1. Targeting the p53-Mdm2 Interaction
p53 protein is a transcription factor at the centre of a large signalling network that targets genes involved in cell cycle regulation, apoptosis and DNA repair (Muller and Vousden 2014). For this reason, p53 dysregulation is a major factor in cancer development (Muller and Vousden 2014). The interactions made by p53 can involve: (i) the N-terminal domain (the transcription domain), (ii) the C-terminal domain (the regulatory domain), and (iii) the DNA binding domain (DBD). Of these domains only the DBD is structured, while the N- and C-terminal domains are intrinsically disordered (Dawson et al. 2003; Oldfield et al. 2008). Thus, p53 extensively uses disordered regions to mediate and modulate interactions with other proteins, as about 70 % of the interactions of p53 are of this type (Oldfield et al. 2008; Uversky et al. 2008).
Among the proteins that regulate p53, Mdm2 plays a major role by inactivating it through binding to its transcription activation domain (Kubbutat et al. 1997). X-ray and NMR studies have revealed that the Mdm2 binding region of p53 near the N-terminus, which is highly flexible when unbound, forms a α-helical structure that binds into a deep groove on the surface of Mdm2 (Chène 2004; Michelsen et al. 2012). Several peptides and small molecules have been designed to inhibit this interaction (Wang et al. 2011; Fry et al. 2013). Predictions made using the molecular recognition features (MoRFs) method (van der Lee et al. 2014), as well as the presence of hydrophobic clusters and other distinctive structural features of the p53-Mdm2 complex, have rationalised why this region appears to be a promising drug target (Cheng et al. 2006), thus providing support to the notion that a protein-protein interaction involving one disordered and one structured partner is likely to be druggable (Uversky 2012). Analysis of the MoRF dataset (Habchi et al. 2014; Cheng et al. 2006) has suggested that for example in some cases the disordered region of a protein can form an α-helix with a hydrophobic face that fits into the groove of the ordered protein. In the disorder-to-order transition, the binding energy should balance off the high entropy of the unfolded state. This interaction may thus be weaker than that between two ordered proteins, and thus potentially more easily targeted by small molecule inhibitors.
2.2 Case Study 2. Targeting the c-Myc-Max Interaction
The oncoprotein c-Myc is a transcription factor implicated in a broad range of human cancers (Dang 1999). Its activity is dependent on heterodimerisation with the disordered protein Max (Blackwood and Eisenman 1991). Both c-Myc and Max are highly disordered in their free forms and undergo mutual coupled binding and folding to form the heterodimer complex via a basic helix-loop-helix leucine zipper domain present in both proteins (Hammoudeh et al. 2009; Harvey et al. 2012; Michel and Cuchillo 2012). This interaction has been reported to be druggable by small molecules that bind to the monomeric and disordered c-Myc (Hammoudeh et al. 2009; Harvey et al. 2012; Michel and Cuchillo 2012). NMR spectroscopy revealed that these molecules bind to a disordered site in the c-Myc monomer located at the interface between the helix-loop-helix and leucine zipper in the c-Myc-Max complex (Hammoudeh et al. 2009). c-Myc is flexible in both the free and small molecule-bound states with only secondary structural elements being transiently populated, thus representing a clear example of the strategy of using small molecules to inhibit interactions between disordered partners. This case study highlights the importance of the specificity of the interactions between disordered proteins and small molecules, and provide insights into the types of small molecules that could be capable of binding highly dynamic targets.
2.3 Case Study 3. Targeting the Aβ Peptide
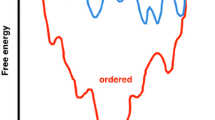
The aggregation of the Aβ peptide into oligomeric assemblies and ordered fibrils is associated with Alzheimer’s disease (Chiti and Dobson 2006; Haass and Selkoe 2007; Knowles et al. 2014). For this reason, this peptide has been the subject of intense research and an increasing number of small molecules have been reported to interfere with its aggregation process (Necula et al. 2007; McKoy et al. 2012; Hawkes et al. 2009; Porat et al. 2006; Wang et al. 2014; Yamin et al. 2009). The existence of various metastable structures of this peptide, however, poses tremendous challenges in developing strategies to avoid its aggregation. Its two main isoforms, Aβ40 and Aβ42, are highly disordered, with the latter having a greater pathological relevance due to its more aggressive aggregation behaviour. Although no predominant binding modes have been identified, many of the small molecules that have been reported to interact with the Aβ peptide appear to do so at the central portion of the peptide (residues 16–22). A recent mapping of the free energy landscape of this peptide (Zhu et al. 2013) (Fig. 13.2) has suggested that the presence of these small molecules may affect the populations of the conformational substates (Zhu et al. 2013). These findings suggest the intriguing possibility that the binding of small molecules to the Aβ peptide may change its behaviour by shifting the statistical weights of multiple substates.

Representative free energy landscape of an IDP. The free energy landscape of the Aβ peptide is shown as a function of the number of hydrogen bonds (backbone-backbone, backbone-sidechain, and sidechain-sidechain) and of the solvent-exposed surface area of hydrophobic residues. The most populated clusters of structures, of which some examples are shown, are found in different regions of the free energy landscape. Reprinted from (Zhu et al. 2013)
3 Fragment-based Drug Discovery for IDPs
So far, owing to the heterogeneous structural characteristics of IDPs, it has been challenging to identify possible initial drug candidates (or ‘hits’) through high-throughput screening of compounds. A promising approach to overcome this problem is based on fragment-based drug design (FBDD), an approach that allows smaller starting structures to be identified and then subsequently grown into small-drug like molecules (Warr 2009; Hajduk and Greer 2007; Congreve et al. 2008; Murray and Rees 2009; Murray and Blundell 2010). Druggable epitopes are identified in an unbound protein structure using such fragments. This approach is based on the observation that a few adjacent residues often make a significant contribution to the binding free energy. These hotspot regions (Wells and McClendon 2007; Clackson and Well 1995) consist in large part of polar and conserved residues, which is consistent with their roles in binding. Experimental fragment screens have confirmed that the druggable hotspots of proteins are characterized by their ability to bind a variety of fragments and that the number of different probe molecules observed to bind to a particular site predicts the potential importance of the site, as well as its overall druggability (Kozakov et al. 2011). A set of fragment probes containing diverse functional groups and shapes, which enable them to bind to a variety of hotspots and binding sites, has recently been used to identify druggable sites in Aβ and α-synuclein (Zhu et al. 2013; Toth et al. 2014). In the case of Aβ, the identification of hotspots and binding pockets was carried out using a computational fragment probe mapping methodology (Zhu et al. 2013).
Unlike the case of structured proteins, as for example G protein-coupled receptors (GPCRs) and kinases, dedicated small molecule libraries are not yet available for IDPs. The development of such libraries may be based on the idea that IDP sequences present peculiar features that enable them to assume heterogeneous structures. Therefore, for individual IDPs, privileged small molecule scaffolds can in principle be identified for drug discovery. The term ‘privileged scaffold’ stems from the notion that multiple molecules of similar scaffolds have similar bioactivities (Welsch et al. 2010). Further, these privileged scaffolds can be used to identify compounds that are biologically active towards IDPs by building chemical libraries. This ligand-based approach could be used together with structure-based methods where an ensemble of structures are probed by multiple fragments to identify hotspots comprised of conserved residues across the ensemble. Subsequently, the fragment ligands could be linked to achieve greater binding (Fig. 13.3). In our group we are currently pursuing a strategy in which fragments from known inhibitors of Aβ are used to identify new molecules that could be potential inhibitors of aggregation. This approach could also be used to identify those scaffolds that contribute significantly towards the free energy of binding to the monomeric peptide, then further expanding them with appropriate linkers and functional groups to optimize binding and bioavailability.

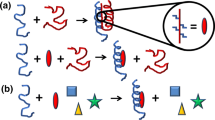
Schematic illustration of the fragment-based drug discovery strategy. A small molecule made up of three fragments (orange square, blue triangle and pink circle) is bound weakly to an IDP (a) since it does not fit very well into a transient binding pocket (c). The fragments are considered separately and some of them bind well to distinct binding pockets in different conformations (b, d). The fragments that bind well are bonded together by a linker whose length and properties match the binding mode of the individual fragments (e)
Quite generally, the FBDD approach is most promising for drug design, given that a few fragments can sample vast amounts of chemical space, reducing the number of compounds for screening (Sun et al. 2011). Fragments bound at various regions of IDPs can be linked together via an appropriate linker (Fig. 13.3). In most cases, fragment-sized compounds need hydrogen bonds to achieve detectable binding. This enthalpic gain compensates for the entropic loss upon binding of the small molecules, and overall lowers the free energy of the protein upon binding. This effect is similar to the mimicking of the interacting partners of IDPs by these molecules.
4 Techniques Used to Study IDP-Small Molecule Interactions
Any drug discovery programme should include a quantitative assessment of the interactions of therapeutic targets with drug candidates. In the case of IDPs such interactions can be characterized in great detail by a wide range of experimental techniques, including NMR spectroscopy and small-angle X-ray scattering (SAXS), particularly if complemented with advanced computational methods.
4.1 Nuclear Magnetic Resonance (NMR) Spectroscopy
4.1.1 NMR Spectroscopy for Drug Development for IDPs
As described in earlier chapters of this book (Chaps. 3–5), NMR spectroscopy is a particularly suitable technique to study the structure and dynamics of IDPs. In the context of drug development, already in the early 1970s NMR spectroscopy was used to study ligand-protein transient interactions, mainly to obtain information from the ligand point of view, by observing perturbations of different NMR parameters including nuclear relaxation time, chemical shifts, inter and intra-molecular nuclear Overhauser effects (NOEs) and intermolecular spin diffusion (Pellecchia et al. 2002). NMR spectroscopy has thus played a major role in the development of rational drug design approaches based on the knowledge of the three-dimensional structures of potential pharmacological targets and their complexes with drug-like molecules (Pellecchia et al. 2008). In the last 25 years, an extended arsenal of NMR methodologies has become available at different stages of the drug discovery process, including target identification, screening compounds, hit validation and lead-compound characterization, structural and mechanistic characterization of ligand-receptor interactions to design and assess target druggability, pharmacophore identification, and potentially structure based drug design (Hajduk et al. 1999; Pellecchia et al. 2002; Renaud and Delsuc 2009). More recently, NMR spectroscopy has demonstrated its utility in FBDD strategies, thus providing an alternative to conventional high-throughput screening, or to support hit-to-lead optimization for a particular drug target (Klages et al. 2007). NMR spectroscopy can also be used to determine low-resolution structures of target-ligand complexes for IDPs or membrane proteins that are not amenable to crystallographic approaches (Pellecchia et al. 2008). Several NMR-based strategies have been developed for FBDD applications, ranging from the traditional chemical shift mapping to ligand-based techniques that monitor changes in ligand nuclear spin relaxation properties upon binding, to measurements of diffusion (Klages et al. 2007). Some of these approaches are better suited to screen and/or to validate hits coming from high-throughput screening (HTS) campaigns, whereas others are better suited to guide hit optimization into more potent, selective drug-like compounds.
4.1.2 Using Chemical-Shift Mapping for Ligand Binding, Screening and Validation
Applications of NMR-based approaches have been extended to ‘non-traditional’ targets such as protein-protein interactions and IDPs. Chemical-shift mapping is one of the most robust, reliable and reproducible ligand binding assays available, and is thus the most utilized approach for ligand binding studies (Hajduk et al. 1999; Pellecchia et al. 2002). This technique exploits the differences in chemical shifts between free and bound protein targets in 15N-1H and 13C-1H two-dimensional correlation spectra of the target upon titration of a ligand or a mixture of ligands. When combined with resonance assignments, at least for proteins with a molecular weight under 30–40 kDa, this approach can give valuable structural information on the site of binding. This method can be extended to larger macromolecular targets in which an amino acid type has been selectively labelled to reduce spectral complexity, thus extending its applicability to targets greater than 100 kDa (Pellecchia et al. 2002). Similar to ordered targets, when the structures of the IDPs have been previously determined and characterized by NMR spectroscopy, in some cases it should be possible to rapidly derive ligand-protein distances via NOE-type experiments that allow sufficiently more precise determination of the ligand binding mode. One of the advantages of this method is that compounds that bind to a given protein can be found and characterized without the need to develop more complex assays (Pellecchia et al. 2008). This aspect is particularly useful in the case of IDPs, where assay development is challenging given the heterogeneity that exists among the different species. Unlike X-ray crystallography, however, this approach does not readily provide information on the dissociation constants of the complexes, nor can it be easily used to monitor ligand binding. Moreover, the amount of protein that is required for individual NMR experiments is relatively high as compared to other techniques, making it expensive for the testing of large libraries of compounds. Hence, these assays have found widespread use in hit validation and in FBDD, in which smaller libraries are used. Taken together, the results obtained thus far indicate that strategies based on the use of FBDD, NMR and computational methods provide an attractive strategy for targeting IDPs for drug design.
Instead of studying the protein-ligand complex upon binding, ligand-based methods could be used to observe the perturbations induced by sub-stoichiometric amounts of targets on the NMR spectra of the ligands. Examples of these approaches include the saturation transfer difference (STD) or T1ρ measurements. For IDPs, whose structures are difficult to calculate (as explained in Chaps. 2–5), these ‘transferred’ ligand-based methods could prove valuable. These approaches, however, are less informative than chemical-shift mapping and are used primarily for screening and for validating ligand binding. This information could be very useful in pharmacophore-based design (Pellecchia et al. 2008).
Hybrid approaches based on the combination of NMR computational docking and screening methods could also be used. In these methods, NMR restraints such as internuclear distance information, chemical shift mapping and residual dipolar couplings are used to calculate an ensemble of conformations of IDPs through molecular dynamics simulations (Vendruscolo 2007), even without full structure determination. Selective conformations based on low energy, compaction, or other structural indicators can then be subjected to virtual screening via docking wherein the top ranked compounds are experimentally verified using NMR spectroscopy. Introducing the docking approach reduces the number of test compounds in a library and saves on running an expensive and time-consuming high-throughput screening experiment. Similarly, a fragment-based library based on these docked compounds could also be used via the ligand-based methods to identify pharmacophores for IDPs.
4.1.3 Using NMR Spectroscopy for Hit and Lead Optimization
Some of the NMR approaches, such as the structure-activity relationships (‘SAR by NMR’) (Shuker et al. 1996), have proven very useful in deriving high-affinity ligands for challenging targets for which other approaches have failed to produce viable leads. In targets that have been previously classified as challenging and ‘undruggable’ or for which there are few alternatives-such as IDPs-NMR information is clearly very useful. The observation that some targets yield more free energy of binding per atom for the initial binding fragments provides a good indicator for assessing the druggability of the target and identifying potential hotspots on the surface of the protein (Pellecchia et al. 2008). These hotspots can also be identified using computational algorithms, such as for example FTMap (Ivetac and McCammon 2012), as shown in a recent study on the Aβ peptide (Zhu et al. 2013).
4.2 Small-Angle X-ray Scattering
As described earlier in this book (Chap. 8), SAXS methods can be effectively employed to study systems with conformational polydispersity, i.e. completely or partially disordered macromolecules, including multi-domain proteins with flexible linkers and IDPs. SAXS is a label-free biophysical method that is particularly suitable in the study of the overall structure and structural transitions of biological macromolecules in solution (Chap. 8). It is a powerful, although low-resolution, tool for the quantitative analysis of flexible systems such as IDPs, and is highly complementary to the high-resolution methods X-ray crystallography and NMR spectroscopy.
Advances in instrumentation at synchrotron facilities, including data collection times within seconds, charge-coupled device detectors, automated sample changers, and integrated robotic control software, have enabled the collection of scattering profiles for high-throughput studies within seconds, making data collection on IDPs relatively accessible (Bernado and Svergun 2012). This progress is very significant for drug discovery programs aimed at IDPs, as structural effects induced by small molecules can be studied using a medium-throughput screen. Analysis of the conformationally heterogeneous ensembles provides quantitative information about flexibility and insights into the structural features (Bernadó et al. 2007). This type of information is extremely useful for drug discovery pipelines, where after an in silico hit identification, the goal is to identify lead compounds. As SAXS methods give relatively low-resolution information (Blobel et al. 2009) but well-defined evidence for overall structural changes, they are very useful tools for studying the effects of small molecules on IDPs.
In peptide and protein systems that undergo aggregation, such as for example the Aβ peptide, possible therapeutic interventions can be aimed at targeting the flexible monomeric species to stabilize them without forming higher order oligomers (Toth et al. 2014; Zhu et al. 2013). Compounds interacting with peptides and proteins at the monomeric level are expected to have three kinds of effects: (i) to inhibit aggregation, (ii) to accelerate aggregation (iii) or to leave aggregation unaltered. These effects can be monitored through time-dependent scattering measurements. In particular, Kratky plots collected at different stages of the aggregation process give the scattering profiles characteristic of either an ordered protein or a disordered protein (Bernado and Svergun 2012). The effects of the molecule on the protein can be observed by studying the Kratky plots, which are characteristically different for ordered, disordered and multidomain proteins. Alternatively, for an IDP, the radius of gyration could be monitored as a function of time. Since flexible proteins normally have larger radius of gyration values than fully folded ones, the addition of an effector compound often leads to a decrease in the radius of gyration. More generally, any deviation from the normal trend of the aggregation behaviour of an IDP suggests that the molecule has an effect. Such data can be used to build low-resolution models of the structural species in cases where structural species are not very well described, as for example the Aβ peptide, where structures are challenging to obtain. Additionally, SAXS data could also be used in combination with molecular simulations to study the dynamics of these interactions or to obtain models of these structural species.
4.3 Chemical Kinetics
Chemical kinetics is a powerful approach for the quantitative analysis of the rates at which biochemical reactions take place (Knowles et al. 2009). This method offers the possibility of detecting and analysing even very weak binding events and their effects, and thus shows promise for studying the interactions of small molecules with IDPs (Arosio et al. 2014). The inhibitory effects of different compounds on protein aggregation have been evaluated by monitoring the kinetics of aggregation in vitro, in particular by means of thioflavin T (ThT) fluorescence based assays (Cohen et al. 2013; Arosio et al. 2014; Meisl et al. 2014). Potential drugs are based on their capacity to delay or arrest fibril formation as monitored in this way (Arosio et al. 2014). This approach has the advantage of rapidly providing potential information on specific compounds, and it can define the specific mechanism by which fibril formation is inhibited by enabling the characterization of the individual microscopic processes underlying the aggregation process, including primary nucleation, elongation, fragmentation and secondary nucleation (Arosio et al. 2014). By using this method, it has recently been possible to reveal that the aggregation of Aβ42 into toxic oligomers and fibrils is primarily caused by secondary nucleation events in which the presence of existing aggregates catalyses the formation of new ones (Cohen et al. 2013). Compounds that prevent toxic oligomers from forming are thus likely to be promising drug candidates in this case (Cohen et al. 2013; Arosio et al. 2014). For IDPs, it is clear that the understanding of the mechanism of inhibition of any potential drug is a key requirement to achieve the desired targeting (Arosio et al. 2014).
4.4 Molecular Dynamics Simulations
As discussed in Chap. 2 of this book, molecular dynamics simulations, in particular when used in combination with advanced sampling techniques and information from experimental data, can provide highly quantitative information on the dynamics of a small molecule upon binding. When the objective is to stabilize the monomeric native states of IDPs, the major goal of these efforts should be to map their free energy landscapes (Vendruscolo and Dobson 2005). Unlike ordered proteins, which have fully-folded native structures that populate well-defined free energy minima, IDPs are characterized by numerous local free energy minima without any stable well-folded conformations. The target of the structure-based drug discovery strategy, therefore, rather than being an individual conformation, should be the variety of low free energy states populated by IDPs. The interaction with a particular binding partner is likely to affect the whole free energy landscape of an IDP, making some minima deeper and some barriers higher (Fig. 13.4). One possibility is to follow a ‘double-hit’ strategy in which the first step is to identify a small molecule that causes a conformational change in the target IDP by creating a binding pocket, and the second step is to find a molecule capable of recognizing this binding pocket. To implement this strategy, it would be very helpful to characterize the free energy landscape of the target IDP in order to gauge the effects of the lead compounds of interest. From an ensemble of conformations, structures of low free energies could be further stabilized by exploiting chemical features suitable to form significant enthalpic interactions with the target IDP.

Schematic illustration of the binding of small molecules to target IDPs. In ordered proteins, the free energy landscape is characterised by a well-defined global minimum, which contains structured binding pockets that can bind small molecules with high affinity. In IDPs, the free energy landscape exhibits multiple minima populated by conformations with transient binding pockets and generally low affinity for small molecules
4.5 Chemoinformatics
Chemoinformatic approaches exploit the knowledge base of the existing chemical space (Dobson 2004) of small molecules. Although the chemical space for IDPs has not yet been fully mapped, the existence of specific physicochemical properties of IDPs suggests that there may be chemical and functional groups particularly favourable for IDP binding. Following this approach, databases such as ChEMBL, PubChem, DrugBank, and ZINC (Gaulton et al. 2012; Wishart et al. 2006; Irwin and Shoichet 2004; Wang et al. 2009) can be exploited to search for compounds that could be repurposed towards targeting IDP ensembles (Varadi et al. 2014). It will be exciting to see whether advances will be made in the future through this strategy.
5 Conclusions
In this chapter we have described how IDPs represent highly challenging, and yet crucial, drug targets. Traditional drug discovery strategies are not ideally suited for these peptides and proteins, as they are structurally heterogeneous and usually devoid of clear binding sites, and it is therefore difficult to design small molecules that bind IDPs with high affinity and high specificity. Despite these problems, we have discussed how recent developments in biophysical techniques, including NMR spectroscopy, SAXS, chemical kinetics and molecular dynamics simulations, have provided initial evidence that IDPs may be druggable. In perspective, given the small but fair amount of success stories so far, we can anticipate that IDPs will be included in the list of targets in drug discovery initiatives in the future.
References
Arkin MR, Wells JA (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 3(4):301–317
Arosio P, Vendruscolo M, Dobson CM et al (2014) Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol Sci 35(3):127–135
Babu MM, Kriwacki RW, Pappu RV (2012) Versatility from protein disorder. Science 337(6101):1460–1461
Bernadό P, Svergun DI (2012) Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol BioSys 8(1):151–167
Bernadó P, Mylonas E, Petoukhov MV et al (2007) Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc 129(17):5656–5664
Blackwood EM, Eisenman RN (1991) Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 251(4998):1211–1217
Blobel J, Bernadό P, Svergun DI et al (2009) Low-resolution structures of transient protein-protein complexes using small-angle X-ray scattering. J Am Chem Soc 131(12):4378–4386
Chène P (2004) Inhibition of the p53-MDM2 interaction: targeting a protein-protein interface. Mol Cancer Res 2(1):20–28
Cheng Y, LeGall T, Oldfield CJ et al (2006) Rational drug design via intrinsically disordered protein. Trends Biotechnol 24(10):435–442
Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 75:333–366
Clackson T, Well JA (1995) A hotspot of binding energy in a hormone-receptor interface. Science 267(5196):383–386
Cohen SIA, Linse S, Luheshi LM et al (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110(24):9758–9763
Congreve M, Chessari G, Tisi D et al (2008) Recent developments in fragment-based drug discovery. J Med Chem 51(13):3661–3680
Conte LL, Chothia C, Janin J (1999) The atomic structure of protein-protein recognition sites. J Mol Biol 285(5):2177–2198
Dang CV (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19(1):1–11
Dawson R, Müller L, Dehner A et al (2003) The N-terminal domain of p53 is natively unfolded. J Mol Biol 332(5):1131–1141
Dobson CM (2001) The structural basis of protein folding and its links with human disease. Philos Trans R Soc B 356(1406):133–145
Dobson CM (2004) Chemical space and biology. Nature 432(7019):824–828
Dunker AK, Uversky VN (2010) Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr Op Pharmacol 10(6):782–788
Dunker AK, Brown CJ, Lawson JD et al (2002) Intrinsic disorder and protein function. Biochemistry 41(21):6573–6582
Dunker AK, Cortese MS, Romero P et al (2005) Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 272(20):5129–5148
Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6(3):197–208
Eisenberg D, Jucker M (2012) The amyloid state of proteins in human diseases. Cell 148(6):1188–1203
Fitzpatrick AWP, Debelouchina GT, Bayro MJ et al (2013) Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc Natl Acad Sci U S A 110(14):5468–5473
Fry DC, Wartchow C, Graves B et al (2013) Deconstruction of a nutlin: dissecting the binding determinants of a potent protein-protein interaction inhibitor. ACS Med Chem Lett 4(7):660–665
Gaulton A, Bellis LJ, Bento AP et al (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucl Acids Res 40(Database issue):D1100–D1107
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol 8(2):101–112
Habchi J, Tompa P, Longhi S et al (2014) Introducing protein intrinsic disorder. Chem Rev 114(13):6561–6588
Hajduk PJ, Greer J (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat Rev Drug Discov 6(3):211–219
Hajduk PJ, Meadows RP, Fesik SW (1999) NMR-based screening in drug discovery. Q Rev Bioph 32(03):211–240
Hammoudeh DI, Follis AV, Prochownik EV et al (2009) Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc 131(21):7390–7401
Harvey SR, Porrini M, Stachl C et al (2012) Small-molecule inhibition of c-MYC: MAX leucine zipper formation is revealed by ion mobility mass spectrometry. J Am Chem Soc 134(47):19384–19392
Hawkes CA, Ng V, McLaurin J (2009) Small molecule inhibitors of Aβ aggregation and neurotoxicity. Drug Develop Res 70(2):111–124
Hopkins AL, Groom CR (2002) The druggable genome. Nat Rev Drug Discov 1(9):727–730
Iakoucheva LM, Brown CJ, Lawson JD et al (2002) Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol 323(3):573–584
Imming P, Sinning C, Meyer A (2006) Drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov 5:821–834
Irwin JJ, Shoichet BK (2004) ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model 45(1):177–182
Ivetac A, McCammon JA (2012) A molecular dynamics ensemble-based approach for the mapping of druggable binding sites. In: Baron R (ed) Computational drug discovery and design, vol 819. Methods in molecular biology. Springer New York, New York, pp 3–12
Jones S, Thornton JM (1996) Principles of protein-protein interactions. Proc Natl Acad Sci U S A 93(1):13–20
Klages J, Coles M, Kessler H (2007) NMR-based screening: a powerful tool in fragment-based drug discovery. Analyst 132(7):692
Knowles TP, Waudby CA, Devlin GL et al (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326(5959):1533–1537
Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15(6):384–396
Kozakov D, Hall DR, Chuang GY et al (2011) Structural conservation of druggable hotspots in protein-protein interfaces. Proc Natl Acad Sci U S A 108(33):13528–13533
Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387(6630):299–303
Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today 1(4):337–341
Malaney P, Pathak RR, Xue B et al (2013) Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci Rep 3:2035
McKoy AF, Chen J, Schupbach T et al (2012) A novel inhibitor of amyloid β (Aβ) peptide aggregation: from high throughput screening to efficacy in an animal model of Alzheimer disease. J Biol Chem 287(46):38992–39000
Meisl G, Yang X, Hellstrand E et al (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Aβ40 and Aβ42 peptides. Proc Natl Acad Sci U S A 111(26):9384–9389
Mészáros B, Simon I, Dosztányi Z (2011) The expanding view of protein–protein interactions: complexes involving intrinsically disordered proteins. Phys Biol 8(3):035003
Metallo SJ (2010) Intrinsically disordered proteins are potential drug targets. Curr Op Chem Biol 14(4):481–488
Michel J, Cuchillo R (2012) The impact of small molecule binding on the energy landscape of the intrinsically disordered protein c-Myc. PloS ONE 7(7):e41070
Michelsen K, Jordan JB, Lewis J et al (2012) Ordering of the N-terminus of human MDM2 by small molecule inhibitors. J Am Chem Soc 134(41):17059–17067
Muller PA, Vousden KH (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25(3):304–317
Murray CW, Blundell TL (2010) Structural biology in fragment-based drug design. Curr Op Struct Biol 20(4):497–507
Murray CW, Rees DC (2009) The rise of fragment-based drug discovery. Nat Chem 1(3):187–192
Necula M, Kayed R, Milton S et al (2007) Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem 282(14):10311–10324
Oldfield CJ, Meng J, Yang JY et al (2008) Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9(Suppl 1):S1
Pellecchia M, Sem DS, Wuthrich K (2002) NMR in drug discovery. Nat Rev Drug Discov 1(3):211–219
Pellecchia M, Bertini I, Cowburn D et al (2008) Perspectives on NMR in drug discovery: a technique comes of age. Nat Rev Drug Discov 7(9):738–745
Porat Y, Abramowitz A, Gazit E (2006) Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des 67(1):27–37
Renaud JP, Delsuc MA (2009) Biophysical techniques for ligand screening and drug design. Curr Op Pharmacol 9(5):622–628
Rezaei-Ghaleh N, Blackledge M, Zweckstetter M (2012) Intrinsically disordered proteins: from sequence and conformational properties toward drug discovery. ChemBioChem 13(7):930–950
Shuker SB, Hajduk PJ, Meadows RP et al (1996) Discovering high-affinity ligands for proteins: SAR by NMR. Science 274(5292):1531–1534
Sun C, Petros AM, Hajduk PJ (2011) Fragment-based lead discovery: challenges and opportunities. J Comput-Aided Mol Des 25(7):607–610
Tompa P (2012) Intrinsically disordered proteins: a 10-year recap. Trends Biochem Sci 37(12):509–516
Toth G, Gardai SJ, Zago W et al (2014) Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PloS ONE 9(2):e87133
Uversky VN (2012) Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discov 7(6):475–488
Uversky VN (2013) Unusual biophysics of intrinsically disordered proteins. Biochim Bioph Acta 1834(5):932–951
Uversky VN, Oldfield CJ, Dunker AK (2008) Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Bioph 37:215–246
Uversky VN, Oldfield CJ, Midic U et al (2009) Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics 10(Suppl 1):S7
van der Lee R, Buljan M, Lang B et al (2014) Classification of intrinsically disordered regions and proteins. Chem Rev 114:6589–6631
Varadi M, Kosol S, Lebrun P et al (2014) pE-DB: a database of structural ensembles of intrinsically disordered and of unfolded proteins. Nucl Acids Res 42(D1):D326–D335
Vendruscolo M (2007) Determination of conformationally heterogeneous states of proteins. Curr Op Struct Biol 17(1):15–20
Vendruscolo M, Dobson CM (2005) Towards complete descriptions of the free–energy landscapes of proteins. Philos Trans R Soc, A 363(1827):433–452
Wang Y, Xiao J, Suzek TO et al (2009) PubChem: a public information system for analyzing bioactivities of small molecules. Nucl Acids Res 37(suppl 2):W623–W633
Wang J, Cao Z, Zhao L et al (2011) Novel strategies for drug discovery based on Intrinsically Disordered Proteins (IDPs). Int J Mol Sci 12(5):3205–3219
Wang Q, Liang G, Zhang M et al (2014) De novo design of self-assembled hexapeptides as β-Amyloid (Aβ) peptide inhibitors. ACS Chem Neurosci 5(10):972–81
Warr WA (2009) Fragment-based drug discovery. J Comput-Aided Mol Des 23(8):453–458
Wells JA, McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450(7172):1001–1009
Welsch ME, Snyder SA, Stockwell BR (2010) Privileged scaffolds for library design and drug discovery. Curr Op Chem Biol 14(3):347–361
Wishart DS, Knox C, Guo AC et al (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucl Acids Res 34(Database issue):D668–D672
Yamin G, Ruchala P, Teplow DB (2009) A peptide hairpin inhibitor of amyloid βprotein oligomerization and fibrillogenesis. BioChemistry 48(48):11329–11331
Zhu M, De Simone A, Schenk D et al (2013) Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J Chem Phys 139(3):035101
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Joshi, P., Vendruscolo, M. (2015). Druggability of Intrinsically Disordered Proteins. In: Felli, I., Pierattelli, R. (eds) Intrinsically Disordered Proteins Studied by NMR Spectroscopy. Advances in Experimental Medicine and Biology, vol 870. Springer, Cham. https://doi.org/10.1007/978-3-319-20164-1_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-20164-1_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20163-4
Online ISBN: 978-3-319-20164-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)