
Abstract
Our knowledge on the mode of evolution of the multifunctional viral proteins remains incomplete. To tackle this problem, here, we have investigated the evolutionary dynamics of the potyvirus multifunctional protein HC-Pro, with particular focus on its functional domains. The protein was partitioned into the three previously described functional domains, and each domain was analyzed separately and assembled. We searched for signatures of adaptive evolution and evolutionary dependencies of amino acid sites within and between the three domains using the entire set of available potyvirus sequences in GenBank. Interestingly, we identified strongly significant patterns of co-occurrence of adaptive events along the phylogenetic tree in the three domains. These patterns suggest that Domain I, whose main function is to mediate aphid transmission, has likely been coevolving with the other two domains, which are involved in different functions but all requiring the capacity to bind RNA. By contrast, episodes of positive selection on Domains II and III did not correlate, reflecting a trade-off between their evolvability and their evolutionary dependency likely resulting from their functional overlap. Covariation analyses have identified several groups of amino acids with evidence of concerted variation within each domain, but interdomain significant covariations were only found for Domains II and III, further reflecting their functional overlapping.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Potyviridae family, named after its prototypical member Potato virus Y (PVY), is one of the largest plant virus families currently recognized by the International Committee on Taxonomy of Viruses (Ward and Shukla 1991; Gibbs and Ohshima 2010). The family is further divided into eight genera, being the Potyvirus genus the most populated one. This genus contains at least 30 % of all known plant viruses, which cause significant losses in agriculture (Ward and Shukla 1991). Despite its importance, little is known about the evolutionary dynamics of this group of viruses. In particular, the dynamics of selection driving their diversification into new species with potential to infect new hosts remain poorly explored (although see Hughes (2009) for an exception). The RNA genome of potyviruses (ca. 9.5 kb) contains a single long open reading frame encoding a ca. 350 kDa polyprotein precursor (Riechmann et al. 1992). The polyprotein is proteolytically processed by three viral proteinases (Riechmann et al. 1992; Urcuqui-Inchima et al. 2001; Adams et al. 2005) to yield ten mature proteins denoted as P1, HC-Pro, P3, 6K1, CI, 6K2, VPg, NIaPro, NIb, and CP. An additional peptide, PIPO, is translated in the +2 reading frame relative to P3 via ribosomal frameshifting or transcriptional slippage at a highly conserved motif at the 5′ end of PIPO (Chung et al. 2008). Most potyviral proteins are multifunctional, and thus expected to be under strong selective constraints imposed by the trade-off between alternative functions.
One of the most intriguing proteins encoded in potyvirus genome is HC-Pro, the helper-component proteinase (Fig. 1). Broadly speaking, HC-Pro is involved in at least five different functions (Maia et al. 1996; Urcuqui-Inchima et al. 2001): aphid-mediated virus transmission, RNA amplification, systemic movement, suppression of RNA silencing, and as a proteinase. Structural and functional analyses of HC-Pro from Tobacco etch virus (TEV) (Kasschau and Carrington 1995, 2001; Kasschau et al. 1997), Lettuce mosaic virus (LMV) (Plisson et al. 2003), and Plum pox virus (PPV) (Varrelmann et al. 2007) have shown that different biologic functions can be assigned to three different domains of this protein (Fig. 1). The N-terminus, hereafter referred to as Domain I, which contains a cysteine-rich region with a Zn finger-like metal-binding motif, controls virus transmission by aphids, symptoms severity, genome amplification, systemic infection, and virus accumulation (Atreya et al. 1992; Cantó et al. 1995; Revers et al. 1999; Urcuqui-Inchima et al. 2001; Yap et al. 2009). The conserved KITC motif, together with the PTK motif located in the central part of the protein, is both essential for aphid transmission (Fig. 1) (Blanc et al. 1997, 1998; Peng et al. 1998). Indeed, a direct interaction between the PTK motif and a DAG motif in the N-terminus of the coat protein (CP) is necessary to facilitate the binding of the two proteins (Blanc et al. 1998). The central region of HC-Pro, to which we will refer as Domain II, is also involved in long-distance movement and replication-maintenance functions (Kasschau et al. 1997). Genetic evidence supports the role of the highly conserved motif of Domain II IGN in the amplification process (Fig. 1) (Cronin et al. 1995; Kasschau et al. 1997). In addition, alanine-scanning mutation studies have pointed the existence of a central C(C,S)C sequence motif necessary for the systemic spread of the virus within host and for genome amplification (Fig. 1) (Cronin et al. 1995; Kasschau et al. 1997). Last, Domain II also contains the FRNK box essential for the RNA silencing suppressor (RSS) activity of the protein (Fig. 1) (Shiboleth et al. 2007). The C-terminal region of HC-Pro, or Domain III, is a cysteine-type proteinase that catalyzes the autoproteolytic cleavage from the polyprotein in a GG dipeptide at its own C-terminus (Guo et al. 2011). In addition, Domain III also plays a role in cell-to-cell movement (Carrington et al. 1989). So far, only the cysteine proteinase domain (Domain III) has been crystalized and its structure in solution solved at 2.0 Å resolution, showing an α/β-fold (Guo et al. 2011). A hypervariable region of six amino acids at the C-terminus of Domain II, which shows high variability among potyvirus species as well as among isolates of Potato virus A (PVA) (Haikonen et al. 2013), serves as boundary between Domains II and III. This hypervariable region has been shown to interact with microtubule-associated protein HIP2 of potato and tobacco (Haikonen et al. 2013).

Schematic representation of HC-Pro divided into three modules. The N-terminal Domain I is involved in transmission, whereas the central Domain II is involved in genome amplification, suppression of RNA silencing, and cell-to-cell movement. The C-terminal Domain III is involved in movement and has the proteinase activity. On the lower panel, the functions of different regions and positions of conserved functional motifs are indicated: KITC and PTK are involved in aphid transmission; FRNK in suppression of RNA silencing; IGN in cell-to-cell movement and amplification; C(C,S)C in movement; the C in box GYCY along with H147 in the proteolytic activity; and YNVG is the proteolytic site. Modified from Plisson et al. (2003)
As mentioned above, HC-Pro has also been shown to be an RSS and to interfere with microRNA function (Llave et al. 2000; Kasschau and Carrington 2001; Shiboleth et al. 2007). Moreover, mounting evidence shows that potyvirus HC-Pro interferes with RNA silencing by siRNA binding (Lakatos et al. 2006; Torres-Barceló et al. 2010b). A number of reports have suggested that the Zn finger-like motifs in the N-terminus of HC-Pro might contain a self-interaction domain (Guo et al. 1999; Urcuqui-Inchima et al. 1999, 2001). However, Zheng et al. (2010) showed that the C-terminus was also necessary for self-interaction in Turnip mosaic virus (TuMV). Similar finding was made for PVA (Guo et al. 1999). Early studies suggested that HC-Pro functional form was a dimer (Plisson et al. 2003), however, structural analyses by ultracentrifugation and single-particle electron microscopy three-dimensional (3D) reconstructions of TEV HC-Pro suggested that the protein in solution exists as a mixture of multimers of dimers (e.g., dimers, tetramers, hexamers, and octamers) (Ruiz-Ferrer et al. 2005).
The multifunctionality of HC-Pro shall be strongly constrained by selection. The protein-coding sequences encoding the different HC-Pro functions overlap and, thus, their evolution is not independent from one another. That is, selective constraints acting on one function will unavoidably constrain other functions. To explore the selective constraints operating upon HC-Pro, Torres-Barceló et al. (2008) generated a collection of single amino acid substitution mutants of TEV scattered along HC-Pro and evaluated their activity as RSS. Most mutations either had no quantitative effect or completely abolished RNA silencing suppression, but a number of mutations induced quantitative increases or reductions in RSS activity. This observation, and in particular, the existence of alleles with increased RSS activity, suggests that this trait can evolve at the cost of the alternative functions. Indeed, in a subsequent experiment, some of the mutants with significant effect on RNA silencing suppression were subjected to a compensatory evolution experiment (Torres-Barceló et al. 2010a). Accumulation of second-site compensatory mutations drove the evolved lineages back to the wild-type RSS activity, suggesting that under biologically realistic conditions, the enzyme is under stabilizing selection, likely resulting from the orthogonality of the different functions.
It is well known that a protein function results from structural and non-structural communications between amino acid sites—that is, amino acid sites exercise reciprocal constraints upon one another, becoming hence linked through a coadaptation dynamic (Maia et al. 1996; Moroni et al. 2012). The strong selective constraints operating in such a multifunctional protein and the link between the different functions mean that advantageous mutations fixed at some sites or functional domains must be followed by coadaptive mutations at functionally or structurally linked sites or domains in the protein. Identifying such coadaptation events may illuminate the functional importance of amino acid sites or regions within HC-Pro. Here, we aimed at identifying such selective constraints to help drawing a site-specific evolutionary profile for HC-Pro. We investigated several questions: how do the selective constraints on multiple HC-Pro functions trade-off? Did functions evolve independent of one another and in a modular manner? Does HC-Pro functional plasticity correspond to an evolutionary plasticity?
Materials and Methods
Nucleotide Sequences
The analyses reported here involved ca. 300 complete potyviruses HC-Pro sequences. However, we performed further phylogenetic and evolutionary analyses using only representative sequences from each of the species (e.g., if they infected different hosts or strong differences in symptoms were described), thereby avoiding representation biases. For example, identical sequences within species clades or sequences placed at the root of such clades were removed from downstream analyses. The selection of sequences was made also to minimize the possible effect of recombination on subsequent evolutionary analyses. For example, sequences that did not present sufficient phylogenetic robustness were removed from the analyses. The assumption was that genomes affected by recombination at regions neighboring HC-Pro would lead to conflicting phylogenetic signals when using HC-Pro sequences. The final set comprised 76 complete HC-Pro sequences; GenBank accession numbers for all sequences used in the study are available upon request. We first conducted multiple protein sequence alignments using MUSCLE (Edgar 2004) as implemented in MEGA5 (Tamura et al. 2011), and then, we built protein-coding nucleotide sequence alignments by concatenating nucleotide triplets according to the protein sequence alignments. To gain further insights on the evolutionary dynamics of functional regions within the protein, we divided the full-protein alignment into the three functional domains previously described (Kasschau et al. 1997; Kasschau and Carrington 2001; Plisson et al. 2003; Syller 2006; Varrelmann et al. 2007). All alignments are available upon request.
While we have discarded intergenomic recombination in regions neighboring HC-Pro, we have examined intragenic recombination; as this could distort the result of selection analyses, it is important to detect them before any further study (Kosakovsky Pond et al. 2006). Accordingly, we first analyzed the HC-Pro sequences searching for evidence of recombination events using the RDP3 package (Martin et al. 2010). RDP3 incorporates several recombination detection methods into a single suite of tools: RDP, GENECONV, BOOTSCAN, MAXCHI, CHIMAERA, SISCAN, and 3SEQ. We ran the program with the default parameters and considered as true recombination events only those detected by more than half of the methods.
Other sequence manipulations were done with BIOEDIT version 7.1.3.0 (Hall 1999).
Phylogenetic Reconstruction
Maximum likelihood trees were inferred for the full-protein alignment and each of the three functional domains using MEGA5. In each case, MODELTEST (Posada and Crandall 1998), as implemented in MEGA5, was used to determine the best fitting model of nucleotide substitution. We assessed confidence of branching patterns in the phylogenetic tree by the bootstrap method, with 1,000 pseudo-random replicates. The congruency of the four trees was checked using the hierarchical likelihood-ratio test for congruence in multilocus phylogenies implemented in the CONCATERPILLAR version 1.5 software (Leigh et al. 2008).
Tests of selection
The ratio between the nonsynonymous (dN) and synonymous (dS) substitutions rates ω = dN/dS was used as a proxy to evaluate the intensity and sign of selection operating on HC-Pro. Values of ω = 1, ω < 1, and ω > 1 indicate neutral evolution, purifying or negative selection, and diversifying or positive selection, respectively. First, we conducted per-codon analyses using the following methodologies implemented in DATAMONKEY (Kosakovsky Pond and Frost 2005a, b): SLAC (single-likelihood ancestor counting), fixed effects likelihood (FEL), and random effects likelihood (REL). A consensus scoring approach was applied to determine the sites experiencing positive selection.
Next, to analyze a broader range of selective constraints in protein-coding genes, we applied the sliding window analysis procedure implemented in SWAPSC (Fares et al. 2002; Fares 2004). SWAPSC uses a statistically optimized window size to detect selective constraints in specific codon regions of the alignment at a particular branch of phylogenetic tree. In each sliding step, SWAPSC identifies signatures of purifying and positive selection as well as neutral evolution. The performance of the method relies on the use of sets of simulated alignments to generate a null distribution of dS and dN using Li (1993) method against which data from the real alignment can be compared. A statistically optimum window size is then estimated that makes the detection of adaptive evolution independent of the window size. Simulated sequences were obtained with the program EVOLVER from the PAML package version 4.44 (Yang and Bielawski 2000) with parameters estimated from the true sequence alignments after running M0 codon-based model in PAML.
Testing Coevolution Between Amino Acid Sites
To identify correlated variation among amino acid sites, in particular, those with evidence of selection pressures, we performed analyses of coevolution within HC-Pro. Coevolution analyses using proteins compare the correlated variance of the evolutionary rates at two amino acid sites, identifying those pairs with significant evidence of coevolution. We identified coevolution using the program CAPS version 1.0 (Fares and McNally 2006). The algorithm implemented in CAPS has been shown to outperform other coevolution-detection methods (Fares and Travers 2006). Briefly, this program identifies covariation between pairs of sites in the multiple sequence alignment by calculating the correlation in the amino acid patterns variation between both sites. The BLOSUM amino acid substitution matrix is then used to score the strength of the amino acid variation for a particular amino acid site, and these scores are corrected taken into account the divergence time between the sequences of the multiple sequence alignment (measured as the estimated number of synonymous substitutions). The significance of the correlation coefficients was tested using 10,000 pseudo-random pairs of amino acid sites and a confidence value α = 0.001. We also tested whether coevolving amino acids can be used to predict protein–protein contact interfaces. Both intra- and interdomains analyses were performed.
Since the identification of coevolution relies on the variability at a pair of amino acid sites, we quantified the intradomain coevolution using the equation:
Here, the coevolution coefficient (CC) is calculated as the sum of the correlation coefficients (ρ) for all pairs of amino acid sites (i = 1, …, K) from a domain showing coevolution normalized by the number of pairs of variable sites (V) potentially coevolving. Variable sites were here defined as those amino acid sites from the multiple sequence alignments with at least two amino acid transitions (three amino acid states). Therefore, Eq. 1 takes into account both, the number of sites potentially involved in a coevolutionary relationship and the strength of the correlated variation of amino acid sites, being a measure of coevolution per site.
To further quantify the amount of coevolution, we modified slightly Eq. 1 to calculate the interdomain CC:
In this equation, V n and V m refer to the number of variable sites in domains n and m, respectively. K refers to the total number of pairs of sites reported as significantly coevolving.
Networks of interacting sites were drawn and analyzed using CYTOSCAPE version 2.8 (Smoot et al. 2011).
Analysis of the Distribution of Coevolving Amino Acid Sites on a Model Structure
Structural clustering of coevolving sites can shed light on their functional and structural reciprocal selective constraints. HC-Pro structure has not been experimentally determined before in full; only the C-terminus containing the cysteine proteinase domain was crystalized and solved (Guo et al. 2011). Following the modeling approach described in Haikonen et al. (2013) with PVA HC-Pro, we modeled the three-dimensional structure of TEV protein using the platform I-TASSER (Roy et al. 2010). I-TASSER is a program that iteratively conducts threading assembly refinement starting with a single amino acid and generating three-dimensional atomic models. The modeling is performed in three stages. First, the query sequence is PSI-blasted against a non-redundant sequence database and secondary structures predicted with PSIPRED (Jones 1999). Then, the sequence and the predicted secondary structures are launched against a PDB structure library using a suit of seven threading programs, all compiled in LOMETS (Wu and Zhang 2007). Second, continuous fragments are excised from threading alignments and assembled to build structural conformations, with the structure of non-aligned regions being ab initio modeled. Third, a consensus set of models, those that are the closest to the centroid of the simulations, are used to refine the models. The final stage of the modeling provides a set of models and their corresponding scores (TM scores), with the highest such score referring to the best model.
Results
Basic evolutionary parameters and phylogenetic inference
The entire alignment, including gaps, comprised 1,407 nucleotides and 469 amino acids, respectively. The identity between 76 studied sequences ranged from 46.7 to 99.5 % (average identity ± 1 SEM: 54.14 ± 0.12 %) at the nucleotide level and from 32.3 to 99.0 % (average identity ± 1 SEM: 48.6 % ± 0.18 %) at the amino acid level. Aligned Domain I consists of 315 nucleotides and 105 amino acids, aligned Domain II of 618 nucleotides and 206 amino acids, and aligned Domain III 474 nucleotides and 158 amino acids including gaps. In addition to previously described conserved motifs (e.g., 57-KITC-60, 188-FRNK-191, and 321-PTK-323; numbering according to the amino acid alignment), the consensus sequence generated from the amino acid alignment shows the existence of three other highly conserved motifs. The first one, spanning amino acids 202-IxCDNQLDxN-211, is located in the middle of Domain II and alternated positively and negatively charged lateral chains, with an overall negative charge. The second motif, 355-CYxNIF(L,F)A-362 located in Domain III, is rich in positively polar lateral chains at the N-terminal side and rich in nonpolar chains in the C-terminal side, but with an overall neutral charge. Finally, the third conserved motif, in the C-terminal side of Domain III, 419-LVDH-422, does not generate any obvious physical context, being the overall charge neutral.
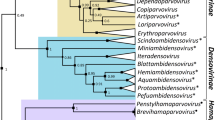
The maximum-likelihood phylogenetic trees inferred for each of the domains shown in Fig. 1 were congruent with the tree inferred for the entire protein (Weibull-smoothed P value for the CONCATERPILLAR congruency test, P ≥ 0.123), and, therefore, we will use the tree inferred for the full alignment hereafter. The GTR+I+Γ 5 model of nucleotide substitutions was the one among the 24 evaluated that had the smallest BIC statistic, which indeed corresponded to an Akaike weight of 100 %. Model parameters were I = 0.087, indicating that very few sites in the alignment were invariable, and with a shape parameter α = 0.761 for the Γ distribution of rates per site, illustrating the fact that the distribution has a highly skewed L-shape, with most sites having very low rates of substitution, or are nearly invariable, but some substitution hotspots with high rates exist. The transitions to transversions bias was R = 2.01, consistent with the principle that transitions are biochemically more likely than transversions.
Figure 2 shows the resulting phylogenetic tree. Overall, the tree topology we have obtained for HC-Pro is remarkably similar to the one obtained by Gibbs and Ohshima (2010) using full genomes; thus, we refer those readers interested in the taxonomic implications of this topology to Gibbs and Ohshima (2010) review article. The only noticeable exception to this similarity is that the HC-Pro of TEV isolates forms a monophyletic group with Bean yellow mosaic virus (BYMV) and Clover yellow vein virus (ClYVV), whereas in Gibbs and Ohshima (2010), TEV genome was forming a monophyletic group with those of PVA and Tobacco vein mottling virus (TVMV).

Maximum-likelihood phylogenetic unrooted tree obtained for the entire HC-Pro using the GTR+I+Γ 5 model of nucleotide substitution. Numbers over the branches represent the bootstrap support values. Significant nodes (P ≥ 0.75) are indicated in red (Color figure online)
Detection of positive selection events
The average values for ω were 0.456 ± 0.019 (± 1 SD based on 1,000 bootstrap samples), 0.248 ± 0.004, and 0.146 ± 0.002, for Domains I, II, and III, respectively, indicating that HC-Pro has been subjected to strong purifying selection (in all cases z test P < 0.001). Both SLAC and FEL methods showed no signatures of positively selected sites. Only the IFEL method found codon 5 in Domain I to be under positive selection with ω = 2.16. It is known that identification of punctual adaptive evolution is often precluded by strong purifying selection under which a protein evolves most of its evolutionary time. This is particularly true when the sequences being compared belong to distantly related organisms. To overcome this problem, we used SWAPSC to screen the alignments at given protein domains in each of the tree branches. We identified positively selected regions (ω > 1) at 1.34, 1.02, and 0.64 % of the codons in Domains I, II, and III, respectively. In all cases, accelerated rates of amino acid substitution (that is, cases in which dN was larger than expected under neutrality but where dS was not reliably estimated to infer ω) were also observed in 2.12, 1.62, and 1.25 in Domains I, II, and III, respectively. Different domains of proteins are likely to be subjected to distinct selective constraints and thus to evolve at different rates. Positively selected regions were randomly distributed in Domain II (Wald-Wolfowitz runs test, P = 0.642), whereas these positively selected regions were pervasive in the 3′ regions of Domains I and II along several branches of the tree (Wald-Wolfowitz runs test, P ≤ 0.021 in both cases).
Next, we sought to analyze whether episodes of positive selection along the phylogenetic tree occurred in a coordinated manner for all domains or, alternatively, different domains evolved independently. That is, if an episode of positive selection is observed in a tree branch for a given domain, how likely is that we observe an event of positive selection in the same branch for other domain? To answer this question, we estimated the probability of the correlation coefficients in the phylogenetic profiles of positively selected regions in the three domains. This probability was calculated by comparing the correlation coefficients of positive selection between domains to a null distribution of such coefficients (Fig. 3). We generated the null distribution by shuffling regions identified under positive selection from the three domains, recalculating the correlations between two domains, and repeating these steps 106 times. Clear differences in the correlation of the evolutionary patterns among domains were observed. The strongest correlation was identified between Domains I and II and Domains I and III (Fig. 3). The weakest and barely significant correlation was observed between Domains II and III (Fig. 3) and can be explained by these domains being mutually exclusive regarding their evolution patterns -that is, strong constraints in one domain involve relaxed constraints in the other- as their sequences overlap.

Probability of congruent coevolution between functional HC-Pro domains. We have compared the correlation coefficients in the phylogenetic profile of positively selected regions in each of the domains between domains. To determine the probability of these correlations, we drew a null distribution of such correlations by randomly shuffling 106 times positively selected regions between domains and calculating the correlations of positive selection phylogenetic profiles between pairs of domains. The position of the real correlation coefficients for a Domains I and II, b Domains I and III, and c Domains II and III indicates that domains significantly correlate in their phylogenetic profiles
Covariation within and between HC-Pro domains
We identified signatures of evolutionary dependencies among amino acids from the same and different functional HC-Pro domains. In absolute numbers, Domain III showed the largest number of sites undergoing coevolution (Fig. 4c) followed by Domain II (Fig. 4b) and Domain I (Fig. 4a).

Intradomain coevolution networks in HC-Pro protein for Domain I (a), Domain II (b), and Domain III (c). Coevolutionary relationships are represented as networks, with amino acid sites represented as nodes (or circles) with the position of the site in the multiple sequence alignment indicated within the circle and coevolutionary links between pairs of sites represented as edges
We identified 100, 180, and 120 variable sites for Domains I, II, and III, respectively. The CCs values computed using Eq. 1 were 0.0023, 0.0027, and 0.0045 for Domains I, II, and III, respectively, suggesting that Domain III was bearing the strongest signature of coevolution (Fig. 4c). Remarkably, Domain III also showed the most complex network of coevolving residues, with three main subnetworks and a number of pairs of sites coevolving (Fig. 4c). In contrast to Domain III, Domains I and II showed a single coevolution network (Fig. 4a and Fig. 4b, respectively). Centrality measures applied to all three domains showed no difference between amino acid sites for Domains I and II and only slight differences between amino acid sites of Domain III. Since most covarying sites were neighbors in the protein sequence (e.g., sites 100–105 in Domain I, sites 296–301 in Domain II, and sites 459–468 in Domain III), these sites were assumed to form clusters in the protein structure. However, this is not always the case. For example, the second cluster found in Domain II (Fig. 4b) comprises sites distributed all over the domain. Similarly, the main cluster found in Domain III (Fig. 4c) in addition to neighboring sites in the region 459–468 contains sites closer to the N-terminal part (site 355) and expands down to site 443. The second and third clusters found in Domain III also include non-contiguous sites, as it is the case for all the pairwise covariations found within this domain (Fig. 4c).
To determine whether functional correlation between domains overlaps with their covariation, we also performed interdomain coevolution analyses. Interestingly, we detected very little coevolution between Domains I and II (only one pair of amino acid sites) and Domains I and III (two pairs of sites), while we detected a strong signature of coevolution between Domains II and III (Fig. 5). The coefficients of interdomain coevolution (Eq. 2) for the three pairwise comparisons were 2.88 × 10−5, 9.25 × 10−5, and 5.8 × 10−3, for the comparisons of Domains I and II, I and III, and II and III, respectively. Coevolution between Domains II and III was two orders of magnitude stronger than that between Domains I and II or I and III. This weak covariation between Domain I and the other two domains is in concert with the different functionality of Domain I, involved in aphid transmission, in comparison with Domains II and III. Conversely, the remarkable covariation between Domains II and III reflects the evolutionary dependency of their functions, with sites coevolving between these domains being involved in RNA silencing and cell-to-cell movement, respectively.

Network of coevolving residues between Domains II (blue) and III (orange). Coevolutionary relationships are represented as networks, with amino acid sites represented as nodes (or circles) with the position of the site in the multiple sequence alignment indicated within the circle and coevolutionary links between pairs of sites represented as edges (Color figure online)
To gain further insights onto the distribution of important sites in the structure, we also generated five 3D structure models of TEV HC-Pro using I-TASSER. The TM-score for the models ranged between 0.608 and 0.878. We used the model with the highest score as the most representative structure of TEV HC-Pro. This model shows three main domains, all of which were enriched with alpha helices (Fig. 6). The hinge beta sheet structure between Domains II and III was remarkable, as it allows for a better packing of the structure, making feasible the interaction among amino acids from both these domains. We mapped on the predicted structure the amino acid sites identified by our coevolution analyses. Sites identified in intradomain coevolutionary analyses formed clear clusters in the structure (Fig. 6). Indeed, the Euclidean distance between all pairs of coevolving amino acid sites within each of the domains (Fig. 3a–c) was significantly low when compared to a 1,000 replicates-based null distribution of mean distances for randomly clustered groups of amino acids, supporting their physical interaction. Domain I coevolution group (yellow spheres in Fig. 6) showed an average Euclidean distance of 2.68 Å ± 0.27 (z test, P = 0.005). Likewise, Domain II presented the main coevolution group (blue spheres in Fig. 6) with a distance of 10.60 Å ± 1.18 (P = 0.044). Domain III comprised several coevolution groups, with the mean distances being 8.55 Å ± 1.31 (P = 0.035) for the main group (green spheres), 7.89 Å ± 1.14 (P = 0.03) for the second group (turquoise spheres), and 3.20 Å for the third and minor group (yellow pairs of amino acids in Fig. 6).

Three-dimensional (3D) structural model of TEV HC-Pro. The model was generated using the I-TASSER platform. Amino acids found as relevant in our selection analyses or as part of covariation groups are mapped into the structure. The blue spheres represent the larger coevolution group in Domain II (those of Fig. 4b). The green spheres represent the largest coevolution group in Domain III (those of Fig. 4c), the turquoise spheres represent the second largest coevolution group in Domain III (as in Fig. 4c), and the yellow spheres represent pairs of coevolving amino acids in Domain III (Fig. 4c) (Color figure online)
Discussion
Revealing important regions in proteins has largely relied on the identification of signatures of positive selection. A remarkable result of our study, however, is that sites do not evolve in isolation but form complex co-evolutionary networks that may involve functional sites and their neighbors in the protein structure. The reciprocal constraints between coevolving amino acid sites of a protein mean that regions evolving under apparent neutral evolution may exercise positive and adaptive effects on other structurally or functionally linked regions of the protein.
There are two major forces driving the evolution of virus proteins: recombination and positive selection. We found no evidence of recombination in our data and found evidence pointing to strong purifying selection governing the evolution of HC-Pro sequences, in general. However, we have also identified positive selection events at some phylogenetic lineages in all three domains of HC-Pro. In the absence of a 3D structural model, it is difficult to unravel the evolutionary constraints that are responsible for these events. The N-terminal (amino acids 1–105) and central parts of HC-Pro (amino acids 106–311) are examples of the multifunctionality of viral proteins. Both parts of HC-Pro are likely to be subjected to stronger action of diversifying selection than C-terminal part of protein (amino acids 312–469). In Domain II, positively selected sites were randomly distributed, while these were clustered around the C-terminal region in Domains I and III. Despite these differences, the patterns of diversifying selection were congruent for the three domains across the phylogeny, hinting their evolutionary dependencies.
HC-Pro N-terminus (Domain I) controls virus transmission by aphids, symptom severity, and virus accumulation (Zheng et al. 2010). In Domain I, several regions have undergone positive selection, mostly clustered in the C-terminal amino acid part of the domain, suggesting that this region may play an important role in the function of the protein. In good agreement, a stretch of coevolving amino acid residues have been also detected in C-terminal end of Domain I. A number of sites were close in primary sequence, which suggests that they may be nearby located in the 3D protein structure. In Domain II, regions under positive selection were randomly distributed along the lineages. The central part of Domain II (amino acids 100–225) of helix-rich region 1 corresponds to the RNA-binding site A (Urcuqui-Inchima et al. 2000) and contains parts of the regions involved in viral movement, genome amplification, and suppression of RNA silencing. It has been suggested that the region comprising amino acids 170–176 is probably exposed and not highly structured (Plisson et al. 2003). This is in line with our results, which pointed to the action of diversifying selection on several branches of this region’s phylogeny. It is plausible that the regions identified are ligand-binding domains and that positive selection reflects gains or losses of affinity for these ligands. In this respect, interactions with plant or vector factors as well as interactions with other viral proteins can be the driving forces of this evolution. It is interesting to note that the highly conserved FRNK motif (positions 188–191 in the alignment) is located downstream of these unstructured (170–176) regions. The hinge region, suggested by secondary structure prediction and identified in the projection maps as a constriction between Domains I and II, was remarkably resistant to trypsin digestion, implying that it is probably well structured (Plisson et al. 2003). Prediction programs indicate that this region is composed mainly of beta sheets. Consequently, a region under significant negative selection was found in this part of the protein. However, several regions within Domain II were identified as being under positive selection. Interestingly, Urcuqui-Inchima et al. (2000) have mapped the RNA-binding capacity of PVY HC-Pro to two independent regions that are located in Domain II, amino acids 161-IGNLV-165 and 296-DGNYVY-301. This illustrates that amino acid substitutions in these regions can affect the fitness of potyviruses quantitatively and can consequently be the target of selection. Two amino acids sequences 57-KITC-60 and 321-PTK-323 located in N- and C-terminal parts of protein, respectively, have been proposed to be involved in protein–protein interactions during vector transmission of potyviruses. Both regions are conserved in the majority of potyviruses, although the KITC motif was found to be under positive selection in only three branches of phylogenetic tree. In Domain III, the action of positive selection is mostly concentrated on the C-terminus part of the protein, and a stretch of coevolving sites have been found there. It has been also shown that C-terminal was necessary for self-interactions in TuMV, which is similar to the finding in PVA (Zheng et al. 2010). The proteinase domain has been mapped to the C-terminal 155 amino acids and was characterized as a cysteine proteinase-like activity with C409 and H483 residues in the active site. Supporting their fundamental functional role, the amino acid residues at the core of the proteolytic activity are under significant negative selection.
We have detected several interdomains covariation groups. Coevolution between amino acid sites can result from their structural, functional, or physical interactions, or from their phylogenetic convergence. The majority of coevolving sites were identified in the C-terminal regions of particular domains. The stronger covariation was observed between Domains II and III, which have been previously described as being involved in self-interactions (Plisson et al. 2003). The movement function also involves Domain III plus half of Domain II (Fig. 1). Some of the coevolving sites are spatially close, supporting a functional and maybe structural complicity highly constrained by selection, as amino acids under diversification evolution have been also identified in these regions.
In this study, we have entirely focused on the interactions between amino acids belonging to the same or different functional domains of HC-Pro and the constraints that such interactions may exert in the evolution of the protein. Obviously, the evolutionary history of HC-Pro cannot be understood only in terms of these intramolecular constraints: interactions with other viral proteins and with host proteins must had modulated the protein evolution as well. At the one side, HC-Pro occupies a central position in the potyvirus’ network of protein interactions. Yeast two-hybrids and bimolecular fluorescence complementation experiments have shown that HC-Pro can physically interact with P1, P3, CI, VPg, and CP (reviewed in Lalić and Elena 2012). At the other side, HC-Pro has been shown to establish interactions in vivo with a number of important host factors, several of which are related to the process of RNA silencing and to the 20S proteasome (all these interactions have been reviewed in Elena and Rodrigo 2012). Integrating all these intra- and intermolecular interactions, although a complex task, must necessarily be the ultimate goal of evolutionary virologists.
In conclusion, our study grounds in evolutionary terms the division of potyvirus HC-Pro multifunctional protein into three different functional domains. While functional Domain I shows a coordinated pattern of positive selection events with Domains II and III, this pattern is not necessarily significant for overlapping Domains II and III, suggesting that this functional overlap generates adaptive trade-offs: optimization of functions mainly performed by Domain II (e.g., suppression of RNA silencing) comes with a cost in the performance of Domain III (e.g., proteinase) and vice versa. Moreover, our covariation analysis has found patterns of variable complexity in the number and connectivity of variables sites at each domain. While Domain I shows only a small cluster of covarying sites, Domain II has two clusters and Domain III shows the more complex and diverse pattern of covarying sites. Finally, only overlapping Domains II and III show significant interdomain covariation groups, once again supporting their functional (e.g., genome amplification and systemic movement) and evolutionary overlap.
References
Adams MJ, Antoniw JF, Beaudoin F (2005) Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol Plant Pathol 6:471–487
Atreya CD, Atryea P, Thornbury DW, Pirone TP (1992) Site-directed mutations in the potyvirus HC-Pro gene affect helper component activity, virus accumulation and symptoms expression in infected tobacco plants. Virology 191:106–111
Blanc S, López-Moya JJ, Wang R, García-Lampasona S, Thornbury DW, Pirone TP (1997) A specific interaction between coat protein and helper component correlates with aphid transmission of a potyvirus. Virology 231:141–147
Blanc S, Ammar ED, García-Lampasona S, Dolja VV, Llave C, Baker J, Pirone TP (1998) Mutations in the potyvirus helper component protein: effects on interactions with virions and aphid stylets. J Gen Virol 79:3119–3122
Cantó T, López-Moya JJ, Serra-Yodi MT, Díaz-Ruiz JR, López-Abella D (1995) Different helper component mutations associated with lack of aphid transmissibility in two isolates of potato virus. Phytopathology 85:1519–1524
Carrington JC, Freed DD, Sanders TC (1989) Autocatalytic processing of the potyvirus helper component proteinase in Escherichia coli and in vitro. J Virol 63:4459–4463
Chung BY, Miller WA, Atkins JF, Firth AE (2008) An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci USA 105:5897–5902
Cronin S, Verchot J, Haldeman-Cahill R, Schaad MC, Carrington JC (1995) Long distance movement factor: a transport function of the potyvirus helper component-proteinase. Plant Cell 7:549–559
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797
Elena SF, Rodrigo G (2012) Towards and integrated molecular model of plant-virus interactions. Curr Opin Virol 2:713–718
Fares MA (2004) SWAPSC: sliding-window analysis procedure to detect selective constraints. Bioinformatics 20:2867–2868
Fares MA, McNally D (2006) CAPS: coevolution analysis using protein sequences. Bioinformatics 22:2821–2822
Fares MA, Travers AA (2006) A novel method for detecting intramolecular coevolution: adding a further dimension to selective constrains analyses. Genetics 173:9–23
Fares MA, Elena SF, Ortiz J, Moya A, Barrio E (2002) A sliding window-based method to detect selective constraints in protein-coding genes and its application to RNA viruses. J Mol Evol 55:509–521
Gibbs A, Ohshima K (2010) Potyviruses and the digital revolution. Annu Rev Phytopathol 48:205–223
Guo D, Mertis A, Saarma M (1999) Self-association and mapping of interaction domains of helper component of Potato virus A potyvirus. J Gen Virol 80:1127–1131
Guo B, Lin J, Ye K (2011) Structure of the autocatalytic cysteine protease domain of potyvirus helper-component proteinase. J Biol Chem 286:21937–21943
Haikonen T, Rajamäki ML, Tian YP, Valkonen JPT (2013) Mutation of a short variable region in HC-Pro protein of Potato virus A affects interactions with microtubule-associated protein and induces necrotic responses in tobacco. Mol Plant Microbe Interact 26:721–733
Hall TA (1999) BIOEDIT: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Hughes AL (2009) Small effective population sizes and rare nonsynonymous variants in potyviruses. Virology 393:127–134
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202
Kasschau KD, Carrington JC (1995) Requirement for HC-Pro processing during genome amplification of Tobacco etch potyvirus. Virology 209:268–273
Kasschau KD, Carrington JC (2001) Long-distance movement and replication maintenance functions correlate with silencing suppression activity of potyviral HC-Pro. Virology 285:71–81
Kasschau KD, Cronin S, Carrington JC (1997) Genome amplification and long-distance movement functions associated with the central domain of Tobacco etch potyvirus helper component-proteinase. Virology 228:251–262
Kosakovsky Pond SL, Frost SDW (2005a) DATAMONKEY: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21:2531–2533
Kosakovsky Pond SL, Frost SDW (2005b) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SDW (2006) Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol 23:1891–1901
Lakatos L, Csorba T, Pantaleo V, Chapman EJ, Carrington JC, Liu YP, Dojla VV, Calvino LF, López-Moya JJ, Burgyan J (2006) Small RNA binding is a common strategy to suppress RNA silencing by several viral suppressors. EMBO J 25:2768–2780
Lalić J, Elena SF (2012) Magnitude and sign epistasis among deleterious mutations in a positive-sense plant RNA virus. Heredity 109:71–77
Leigh JW, Susko E, Baumgartner M, Roger AJ (2008) Testing congruence in phylogenomic analysis. Syst Biol 57:104–115
Li WH (1993) Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol 36:96–99
Llave C, Kasschau KD, Carrington JC (2000) Virus-encoded suppressor of posttranscriptional gene silencing targets a maintenance step in the silencing pathway. Proc Natl Acad Sci USA 97:13401–13406
Maia S, Haenni AL, Bernardi F (1996) Potyviral HC-Pro: a multifunctional protein. J Gen Virol 77:1335–1341
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
Moroni E, Morra G, Colombo G (2012) Molecular dynamics simulations of Hsp90 with an eye to inhibitor design. Pharmaceuticals 5:944–962
Peng YH, Kadoury D, Gaol-On A, Huet H, Wang Y, Raccah B (1998) Mutations in HC-Pro gene of Zucchini yellow mosaic potyvirus: effects on aphid transmission and binding to purified virions. J Gen Virol 79:897–904
Plisson C, Drucker M, Blanc S, German-Retana S, Le Gall O, Thomas D, Bron P (2003) Structural characterization of HC-Pro a plant virus multifunctional protein. J Biol Chem 278:23753–23761
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Revers F, Le Gall O, Candresse T, Maule J (1999) New advances in understanding the molecular biology of plant/potyvirus interaction. Mol Plant Microbe Interact 12:367–376
Riechmann JL, Lain S, García JA (1992) Highlights and prospects of potyvirus molecular biology. J Gen Virol 73:1–16
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738
Ruiz-Ferrer V, Boskovic J, Alfonso C, Rivas G, Llorca O, López-Abella D, López-Moya JJ (2005) Structural analysis of Tobacco etch potyvirus HC-pro oligomers involved in aphid transmission. J Virol 79:3758–3765
Shiboleth YM, Haronsky E, Leibman D, Arazi T, Wassenegger M, Whitham SA, Gaba V, Gal-On A (2007) The conserved FRNK box in HC-Pro, a plant viral suppressor of gene silencing, is required for small RNA binding and mediates symptom development. J Virol 81:13135–13148
Smoot M, Ono K, Ruschelnski J, Wang PL, Ideker T (2011) CYTOSCAPE 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432
Syller J (2006) The roles and mechanisms of helper component proteins encoded by potyviruses and caulimoviruses. Physiol Mol Plant Pathol 67:119–130
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Torres-Barceló C, Martín S, Daròs JA, Elena SF (2008) From hypo- to hypersuppression: effect of amino acid substitutions on the RNA-silencing suppressor activity of Tobacco etch potyvirus HC-Pro. Genetics 180:1039–1049
Torres-Barceló C, Daròs JA, Elena SF (2010a) Compensatory molecular evolution of HC-Pro, an RNA-silencing suppressor from a plant RNA virus. Mol Biol Evol 27:543–551
Torres-Barceló C, Daròs JA, Elena SF (2010b) HC-Pro hypo- and hypersuppressor mutants: differences in viral siRNA accumulation in vivo and siRNA binding activity in vitro. Arch Virol 155:251–254
Urcuqui-Inchima S, Walter J, Drugeon G, German-Retans S, Haeni AL, Candresse T, Bernardi F, Le Gall O (1999) Potyvirus HC-Pro self-interaction in the yeast two hybrid system and delineation of the interaction domain involved. Virology 258:95–99
Urcuqui-Inchima S, Maia IG, Arruda P, Haenni AL, Bernardi F (2000) Deletion mapping of the potyviral helper component-proteinase reveals two regions involved in RNA binding. Virology 268:104–111
Urcuqui-Inchima S, Haenni AL, Bernardi F (2001) Potyvirus proteins: a wealth of functions. Virus Res 74:157–175
Varrelmann M, Maiss E, Pilot R, Palkovics L (2007) Use of pentapeptide-insertion scanning mutagenesis for functional mapping of the Plum pox virus helper component proteinase suppressor of gene silencing. J Gen Virol 88:10051015
Ward CW, Shukla DD (1991) Taxonomy of potyviruses: current problems and some solutions. Intervirology 32:269–296
Wu S, Zhang Y (2007) LOMETS: a local meta-threading-server for protein structure prediction. Nucl Acids Res 35:3375–3382
Yang Z, Bielawski JP (2000) Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15:496–503
Yap YK, Duangjit J, Panyim S (2009) N-terminal of Papaya ringspot virus type-W (PRSV-W) helper component proteinase (HC-Pro) is essential for PRSV systemic infection in zucchini. Virus Genes 38:461–467
Zheng H, Yan F, Lu Y, Sun L, Lin L, Cai L, Hou M, Chen J (2010) Mapping the self-interaction domains of TuMV HC-pro and the subcellular localization of the protein. Virus Genes 42:110–116
Acknowledgments
This work was supported by grants BFU2012-30805 (SFE) and BFU2012-36346 (MAF) from the Spanish Dirección General de Investigación Científica y Técnica and by an EMBO Short-Term Fellowship and the Mentoring Program from the Foundation for Polish Science (BHJ).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hasiów-Jaroszewska, B., Fares, M.A. & Elena, S.F. Molecular Evolution of Viral Multifunctional Proteins: The Case of Potyvirus HC-Pro. J Mol Evol 78, 75–86 (2014). https://doi.org/10.1007/s00239-013-9601-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-013-9601-0