
Abstract
In this study, a novel analytical approach for the determination of 11 monohydroxylated polycyclic aromatic hydrocarbon metabolites (OH-PAHs) in urine was developed and validated. The rapid, simple and high-throughput sample preparation procedure based on ethyl acetate extraction and subsequent purification by dispersive solid-phase extraction (d-SPE) employing a Z-Sep sorbent is used for the first time. For the identification/quantification of target compounds, ultra-high-performance liquid chromatography (U-HPLC) interfaced with tandem mass spectrometry (MS/MS) was applied. The results of validation experiments performed on the Standard Reference Material (SRM) 3673 (organic contaminants in non-smokers’ urine) were in accordance with the certified values. The method recoveries ranged from 77 to 114 % with the relative standard deviation lower than 20 % and the quantification limits in the range of 0.010–0.025 ng mL−1 (except for benzo[a]pyren-3-ol with 0.9 ng mL−1). Within the pilot study, the new method was used for the analysis of OH-PAHs in 50 urine samples. The concentrations of ΣOH-PAHs were in the range of 0.87–63 ng mL−1 (1600–33,000 ng g−1 creatinine), with naphthalen-2-ol (2-OH-NAP) and phenanthren-1-ol (1-OH-PHEN) being the most abundant exposure biomarkers detected in all samples.

Flow-chart of the analytical method for the determination of major OH-PAHs in urine including the concentrations of detected metabolites in real samples (n = 50)
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAHs), produced by incomplete combustion of organic materials, are ubiquitous contaminants present in the environment. The toxicity of some PAHs has been extensively explored due to their carcinogenic, mutagenic and teratogenic potency [1]. The human organism may be exposed to PAHs through various pathways; inhalation of polluted air or cigarette smoke together with dietary intake represents the major ones [2, 3]. As soon as PAHs enter the human body, rapid biotransformation process starts with phase I metabolism in which PAHs are oxidised by the hepatic cytochrome P450 monooxygenases to form reactive epoxide intermediates, followed by reduction or hydrolysis to hydroxylated derivates (monohydroxylated PAH metabolites (OH-PAHs)). In phase II metabolism, the OH-PAHs are conjugated to glucuronic acid or sulphate to increase the water solubility of the metabolite [4]. Depending of the molecular weight, the OH-PAH conjugates (typically glucuronides and/or sulphates) are excreted either into urine (species with two to three benzene rings) or in faeces (greater than or equal to four benzene rings) [5]. These biological processes lead to the formation of multiple metabolites including epoxide, dihydrodiols and monohydroxylated and polyhydroxylated PAHs. The selection of a reliable biomarker(s) for human PAH exposure is a crucial point since a PAH pattern may fairly vary among the particular exposure sources. Moreover, each PAH is metabolised in the exposed organism to more than one metabolite constituting different positional isomers [6]. Considering various environmental and/or occupational studies [7–11] concerned with monitoring of PAH emissions associated with petrol or diesel combustion, pyren-1-ol (1-OH-PYR), the major monohydroxylated metabolite of pyrene, is one of the most popular exposure biomarkers. However, pyrene itself is not carcinogenic; thus, determination of its biotransformation product is not fully relevant to a health risk assessment process. Bearing in the mind that benzo[a]pyrene is the key representative of carcinogenic PAHs, the monitoring of its major metabolite benzo[a]pyren-3-ol (3-OH-BaP) is a more realistic option. Nevertheless, the measurement of urinary 3-OH-BaP is a rather difficult task. Due to its predominant elimination via biliary excretion in faeces, concentrations of urinary 3-OH-BaP are under common exposure conditions by 3 orders of magnitude lower compared to those of the pyrene metabolite, 1-OH-PYR [12]. With regard to multiple exposures to different PAHs under real-life conditions, measurements of other hydroxylated metabolites originated from naphthalene, phenanthrene, fluorene and chrysene have been realised by many authors in the recent decade [4, 6, 13–19].
The quantitative determination of multiple OH-PAHs and other PAH-related biomarkers in human urine is an analytical challenge due to a large range of their polarities and typically (ultra)-trace levels. Currently, typical sample preparation procedures for isolation of OH-PAHs from urine include enzymatic hydrolysis with β-glucuronidase/sulfatase and a subsequent extraction by applying liquid/liquid extraction (LLE) using pentane [4, 20], pentane/toluene mixture [17] or, alternatively, hexane [6] and, as an extraction/preconcentration alternative, also solid-phase extraction (SPE) [13, 14, 16, 18, 19, 21, 22] or solid-phase microextraction (SPME) [23, 24] were employed. Analytical platforms, which have proved to be sufficiently sensitive for the quantification of urinary OH-PAHs, include high-performance liquid chromatography with fluorescence detection (HPLC–FLD) [12, 18], gas chromatography (GC) coupled with (high-resolution) mass spectrometry ((HR)MS) [6, 13, 19, 20] or tandem mass spectrometry (MS/MS) [25], and liquid chromatography (LC) with MS/MS, either with [22, 26] or without prior derivatisation [13–17, 27]. When selecting the most relevant instrumental set-up, both advantages and disadvantages should have to be considered. For example, GC–MS (/MS) methods for OH-PAHs require derivatisation of the analytes but, on the other hand, thanks to their high separation power, differentiation of the isomeric metabolites is enabled. Moreover, applications of isotope dilution techniques for quantification offer a high-precision analysis. HPLC–FLD, representing a common equipment of many laboratories, benefits from the high sensitivity of fluorescence detection to analytes with PAH structures. Contrary to GC, LC separation suffers of lower separation efficiency which may lead to, together with limited detection, misidentification of analytes; also, the impossibility to compensate matrix effects by isotopically labelled surrogates may complicate accurate quantification [28, 29]. Owing to these facts, a LC–MS/MS technique seems to be, nowadays, a gold standard in this field because of its applicability for highly sensitive quantification of a wide range of substances including multiple PAH biomarkers in a complex biological matrix such as urine.
The main aims of this study were to (i) develop and validate a simple, rapid and high-throughput sample preparation procedure for the simultaneous isolation of most often monitored OH-PAHs (due to their toxicity or potential negative effect on human health) represented by metabolites of various PAH classes such as naphthalene, fluorene, phenanthrene, chrysene, pyrene and benzo[a]pyrene using ultra-HPLC (U-HPLC)–MS/MS and to (ii) apply this method for the examination of 50 urine samples collected from the Czech women within the project ‘Impact of air pollution to genome of newborns’ (No. 13-13458S).
Materials and methods
Standards
Certified standards of OH-PAHs represented by naphthalen-1-ol (1-OH-NAP, 1000 μg mL−1 of methanol, purity 99 %) and naphthalen-2-ol (2-OH-NAP, 1000 μg mL−1 of methanol) were obtained from Absolute Standards, Inc. (USA). Fluoren-2-ol (2-OH-FLUO), phenanthren-1-ol (1-OH-PHEN), phenanthren-2-ol (2-OH-PHEN) and phenanthren-4-ol (4-OH-PHEN) were supplied by Toronto Research Chemicals, Inc. (USA). Phenanthren-3-ol (3-OH-PHEN), phenanthren-9-ol (9-OH-PHEN), 1-OH-PYR and 3-OH-BaP were purchased from Neochema (Germany). Chrysen-6-ol (6-OH-CHR) was obtained from AccuStandard® (USA). Isotopically labelled analogues, specifically [2 H]7-naphthalen-1-ol (d 7-1-OH-NAP), [2 H]7-naphthalen-2-ol (d 7-2-OH-NAP), [2 H]9-fluoren-2-ol (d 9-2-OH-FLUO), [2 H]9-phenanthren-1-ol (d 9-1-OH-PHEN), [2 H]9-phenanthren-2-ol (d 9-2-OH-PHEN), [2 H]9-phenanthren-3-ol (d 9-3-OH-PHEN), [2 H]8-phenanthren-9-ol (d 8-9-OH-PHEN), [2 H]9-pyren-1-ol (d 9-1-OH-PYR) and [2 H]11-benzo[a]pyren-3-ol (d 11-3-OH-BaP), were supplied by Toronto Research Chemicals, Inc. Creatinine was delivered by Sigma-Aldrich (USA). The purity of all standards and their isotopically labelled analogues was at least 98 %.
Individual OH-PAHs delivered as solids were dissolved with respect to the manufacturers’ recommendations. Mixtures of OH-PAHs and their isotopically labelled analogues (d-OH-PAHs) were prepared in methanol at concentrations of 10, 100 and 1000 ng mL−1. Each calibration solution of OH-PAHs corresponding to the calibration curve at levels of 0.1, 0.25, 0.5, 1, 2.5, 5, 10, 25, 50 and 100 ng mL−1 in methanol contained an internal standard at 10 ng mL−1 except for d 11-3-OH-BaP, for which the concentration was 10 times higher, 100 ng mL−1, because of its lower instrumental sensitivity compared to other d-OH-PAHs. All solutions were stored at −20 °C in the freezer.
The Standard Reference Material® 3673 (organic contaminants in non-smokers’ urine) used for the method evaluation and validation experiments was supplied by the US National Institute of Standards and Technology (NIST, Gaithersburg, Maryland, USA).
Chemicals, reagents and other materials
Analytical grade ethyl acetate for pesticide residue analysis, SupraSolv® n-hexane for gas chromatography ECD and FID (purity ≥ 98 %), picric acid, enzyme β-glucuronidase (type HP-2, glucuronidase activity ≥ 100,000 units mL−1, sulfatase activity ≤ 7500 units mL−1) and sorbents, specifically Supel™ QuE Z-Sep, Supel™ QuE Z-Sep + and Supelclean™ ENVI-Carb™, were supplied by Sigma-Aldrich. Acetone and sodium hydroxide were purchased from Lach-Ner (Czech Republic). LC–MS CHROMASOLV® methanol (purity ≥ 99.9 %) was delivered by Merck (Germany). Sorbents C18 silica gel and primary–secondary amine (PSA), both Bondesil (40 μm), were obtained from Agilent Technologies (USA). Sep-Pak® Vac 3 cc (500 mg) SPE cartridges were supplied by Waters (USA). Unsterile polytetrafluoroethylene (PTFE, 5.0 μm, Ø 25 mm) filters were purchased from Rotilabo® (Germany). Ninety-six-well microtiter plates were obtained from Gama Group (Czech Republic). Polypropylene (PP) centrifuge tube filters (nylon, pore size 0.22 μm) were supplied by Sigma-Aldrich.
Sample collection
All 50 urine samples were collected during winter in 2013/2014 from Czech women living in the Karviná (situated in highly polluted North Moravian industrial agglomeration with a black coal mining and coke and steel production) within the project Impact of air pollution to genome of newborns (No. 13-13458S). The urine was stored in the freezer at −20 °C before analysis. Prior to the analysis, thawed samples were filtered through PTFE filters in order to remove solid impurities.
Method description
Determination of creatinine
The creatinine values were used to normalise the urine concentration/dilution in individual samples, thus ensuring improved data comparability. Samples with excessive physiological dilution or concentration outside the range of 0.3 mg mL−1 ≤ creatinine ≤ 3.0 mg mL−1 were excluded. A spectrophotometric method for the estimation of urinary creatinine was based on Jaffé’s reaction [30, 31], where a coloured complex of creatinine with alkaline picrate formed and then measured at 505 nm. The reaction mixture consisted of 75 μL creatinine calibration standards at 8, 16, 24, 32 and 40 μg mL−1, or a 75 μL 100 times diluted urine, together with 75 μL of saturated picric acid solution, 75 μL of 1 M sodium hydroxide and 75 μL of deionised water, was transferred directly into a well of a microtiter plate. Jaffé’s reaction was carried for 20 min at laboratory temperature, and then the absorbance was measured using the spectrophotometer Epoch (BioTek, USA). The obtained data were evaluated by Gen5™ Microplate Data Analysis software.
Enzymatic hydrolysis
An amount of 5 mL of urine was transferred into a 50-mL polypropylene centrifuge tube, and 20 μL of β-glucuronidase and 10 mL of acetate buffer (pH 5) were added. After the addition of an internal standard (d x -OH-PAHs) at 100 ng mL−1, the mixture was incubated overnight (37 °C, 15 h) to release OH-PAHs from conjugated forms.
SPE
The SPE cartridges (sorbent C18) were conditioned with 5 mL of methanol followed by 10 mL of acetate buffer (pH 5). After the loading of 15 mL urine hydrolysate with the addition of an internal standard into the cartridges, washing with 5 mL of deionised water and 5 mL of 25 % (v/v) aqueous methanol was performed. During these procedures, the flow rate was held lower than 1 mL min−1. The cartridges were dried using a vacuum pump and centrifuged for 2 min at 5000 rpm (Hettich, Germany). Finally, the target analytes were eluted with 10 mL of ethyl acetate, methanol or acetone. Collected eluates were allowed to evaporate (Büchi Rotavapor, Flawil, Switzerland) and the residual solvent was removed under the gentle stream of nitrogen, if needed. The residues were dissolved in 250 μL of methanol and transferred into the vial for the U-HPLC–MS/MS analysis.
LLE with clean-up using dispersive solid phase extraction
In total, 15 mL of ethyl acetate was added to the enzymatic hydrolysate and the tube was vigorously shaken for 1 min. The tube was then centrifuged for 5 min at 10,000 rpm to facilitate phase separation. An amount of 12 mL of the upper organic layer was transferred into the new tube containing 180 mg of Z-Sep, Z-Sep+, C18, PSA or ENVI-Carb sorbent and 1.8 g anhydrous MgSO4. The tube was shaken again for 1 min and centrifuged for 5 min at 10,000 rpm. Subsequently, 8 mL of a purified extract was allowed to evaporate using a rotary vacuum evaporator near to dryness. The residual solvent was removed under a gentle stream of nitrogen. The residues were dissolved in 250 μL of methanol. The reconstituted extract was filtered through a 0.22-μm nylon centrifuge tube filter and transferred into the vial for the U-HPLC–MS/MS analysis.
Quality assurance/quality control
The validation of the final method (LLE followed by clean-up based on dispersive solid-phase extraction (d-SPE)) was evaluated through the analysis of the NIST Standard Reference Material® (SRM) 3673 (organic contaminants in non-smokers’ urine) in six replicates. In the case of 6-OH-CHR and 3-OH-BaP, for which the concentrations are not certified, an artificially contaminated urine sample, previously tested for the content of target analytes, was analysed. The recoveries (REC, %) and repeatabilities (expressed as relative standard deviations, RSDs, %) for 6-OH-CHR and 3-OH-BaP were calculated from six analyses at a concentration level of 0.05 and 1 ng mL−1 (based on a regular occurrence of these metabolites in urine). In the case of 4-OH-PHEN and 6-OH-CHR, for which their deuterated analogues were not available, d 8-9-OH-PHEN and d 9-1-OH-PYR, respectively, were chosen. To control background contamination by target analytes, the procedural blank (i.e. in the same sample procedure, only instead of urine, the same volume of deionised water was used) was prepared together with each batch of 20 samples (per day). The determined concentration of OH-PAHs in each procedural blank was subtracted from the respective sample. The limits of quantification (LOQs) were determined as the lowest calibration standard at which analytes provided a signal-to-noise ratio (S/N) of >10. For the compensation of the unexpected influence of matrix or losses of targeted analytes, the entire method was validated using isotopically labelled surrogates.
As regards the miniaturised spectrophotometric method based on Jaffe’s reactions, which was implemented within our study, the concentration of creatinine was also determined in the SRM 3673 in six replicates.
Instrumental analysis
The U-HPLC analyses of OH-PAHs were performed using an Acquity Ultra-Performance LC system (Waters, USA) equipped with a 10-μL sample loop. Analytes were separated on a pentafluorophenyl (PFP; Kinetex, Phenomenex, USA) column (100 mm × 2.1 mm × 1.7 μm) maintained at 40 °C. Water (A) and methanol (B) were used as mobile phases at a flow rate of 300 μL min−1 and with a gradient, specifically 10–40 % B over 0.5 min then 40–100 % B over 11 min followed by an isocratic hold at 100 % B for 2 min. The total run time for each injection was 13 min. The flow rate began at 0.3 mL min−1, and the sample volume injected was 5 μL at 10 °C. The U-HPLC system was coupled to a triple–quadrupole mass spectrometer Xevo TQ-S (Waters, USA) with electrospray ionisation (ESI-) that was operated in the negative ion mode with a capillary voltage of −2000 V, and ionisation and desolvation temperatures were 150 and 400 °C, respectively. The instrument was operated in multiple reaction monitoring (MRM) mode. The retention times and quantitative/qualitative MRM transitions of target analytes are listed in Table S1 in the Electronic Supplementary Material (ESM).
Results and discussion
U-HPLC–MS/MS method development
In the first stage, the instrumental method for the determination of OH-PAHs was optimised. In order to achieve the lowest LOQs required for real-sample analysis, MS/MS and ionisation parameters (capillary voltage, ionisation temperature) were extensively tested. In the next phase, the chromatographic separation of isomeric compounds, mainly OH-PHEN compounds, was performed on the three types of columns.
To optimise mass spectrometric parameters, standard solutions of individual OH-PAHs at 5000 ng mL−1 in a mixture of methanol/water (50:50, v/v) were directly infused into ESI-. The MRM transition [M–H]− > [M–H–28]− was the most abundant for most of the analytes, with the exception of 2-OH-FLUO. A neutral loss of 28 Da has been observed by other investigators and has been attributed to a loss of CO from the parent ion. No further fragmentation of the [M–H–28]− ion occurred which indicated a high stability of this daughter ion under the experimental conditions. In the case of 2-OH-FLU, two fragment ions [M–H–28]− and [M–H–1]− were observed, the latter probably due to five membered rings present in its structure. The sensitivity of the MRM transition [M–H]− > [M–H–1]−, i.e. m/z 181 > 180, was 20 times higher compared to that of the reaction [M–H]− > [M–H–28]−, i.e. 181 > 154. Regarding with the capillary voltage, three different values, −2000, −3000 and −4000 V, were tested. The highest sensitivity was obtained when −2000 V was set. Compared to −3000 and −4000 V, the signals for all compounds were significantly higher, by 20 and 90 %, respectively. Also, the influence of mobile phase composition was recognised as an important factor affecting sensitivity. The best results under the above-mentioned mobile phase gradient were achieved when aqueous methanol was used (system 1). For example, the addition of ammonium acetate in water (system 2) caused the decrease in signal intensity by 50 % for most analytes compared to water and methanol (system 1). When acetonitrile was used instead of methanol (system 3), similar signal intensities were obtained for OH-NAP, 2-OH-FLUO and OH-PHEN compounds, contrary to later eluted compounds, 1-OH-PYR, 6-OH-CHR and 3-OH-BaP, when signal decreased by 50 % (Fig. 1).

The influence of mobile phase composition on the signal intensity of OH-PAHs (responses are normalised to the mobile phase composition of water (A) and methanol (B) of 100 %)
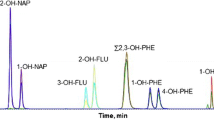
Because of identical MRM transitions of OH-PHEN and OH-NAP isomers, their individual chromatographic separation had to be optimised. Besides the different compositions of the mobile phase, gradient and total time of analysis, various types of columns were tested: Kinetex (i) PFP (100 × 2.1 mm; 1.7 μm), (ii) BEH (C18) (100 × 2.1 mm; 1.7 μm) and (iii) HSS T3 (C18) (100 × 2.1 mm; 1.8 μm). The most critical isomers were OH-PHEN compounds, when the total co-elution of pairs 2-OH-PHEN/3-OH-PHEN and 1-OH-PHEN/9-OH-PHEN using a column with C18 stationary phase under tested conditions was observed. Better selectivity for these aromatic isomers was achieved using PFP stationary phase, when individual OH-PHEN compounds were partially separated (Fig. 2). Due to the unique properties (high electronegativity, low polarizability and strong lipo- and hydrophobicity) of organofluorines, the fluorinated phases offered many possibilities that could not be accomplished by conventional C8 and C18 reverse phases. The PFP column offers dispersive, dipole–dipole, π–π, charge transfer and ion exchange interactions to enable the retention of different types of compounds including aromatics and polycyclic aromatic hydrocarbons [32]. From these reasons, Kinetex PFP (100 × 2.1 mm; 1.7 μm) column was used for the U-HPLC–MS/MS analysis.

Chromatographic separation of OH-PAHs on various columns (c = 10 ng mL−1): A Kinetex PFP (100 × 2.1 mm; 1.7 μm), B BEH (C18) (100 × 2.1 mm; 1.7 μm) and C HSS T3 (C18) (100 × 2.1 mm; 1.8 μm)
Extraction method development
Two different analytical approaches were tested for the isolation of OH-PAHs from urine. With regard to unsatisfactory results obtained by common SPE with C18 sorbent, mainly low method recovery and poor purification of extract, we decided to isolate the target compounds by LLE using ethyl acetate as a medium to which they are to be transferred. Due to a limited selectivity of this extraction step, purification was needed. To have the analytical procedure as simple as possible, d-SPE was chosen in this particular case. In paragraphs below, the development/modification steps are described in detail. The problem we had to overcome was the practical unavailability of a truly blank matrix. Given that the human body is exposed to PAHs practically throughout life (inhalation of polluted air or cigarette smoke, ingestion of contaminated food, dermal absorption), traces of OH-PAHs and their glucuronides/sulphates are unavoidably present in any urine sample. The only way to assess the performance characteristics of both methods during optimisation was to spike urine with a mixture of isotopically labelled analogues (d x -OH-PAHs) prior to the hydrolysis. For the most effective enzymatic deconjugation step, 10, 20 or 50 μL of β-glucuronidase was added to the naturally contaminated urine sample (pH 5, 37 °C, 15 h). The comparable amounts of OH-PAHs were released (20 and 50 μL) for both sample preparation procedures discussed below. From this reason, for the final validation experiments and examination of real samples, 20 μL of β-glucuronidase was used.
SPE
Considering the relatively non-polar nature of OH-PAHs compared to other substances occurring in urine, C18 silica-based SPE was selected for isolation/preconcentration of these compounds. Within the optimisation, various solvent mixtures were tested for both washing (starting with water and followed by 10, 25 or 50 % aqueous MeOH) and elution (methanol, acetone or ethyl acetate). Regarding with the washing step, the best set-up avoiding d-OH-PAH losses and, at the same time, enabling removing of some interfering substances was the combination of water and 25 % (v/v) aqueous MeOH. From tested elution solvents, the highest recoveries in the range of 50–70 % were achieved when ethyl acetate was used. On the other hand, we experienced a similar retention behaviour of yellow/amber urine pigments and other matrix components that eluted from the SPE cartridge together with analytes. These residues of matrix co-extracts had a negative impact on the analyte ionisation, which resulted in the signal suppression by up 90 %. For the compensation of these effects, matrix calibration can be used. However, for its preparation, it is necessary to have sufficient quantities of the blank matrix, which is complicated as described above. Other difficulties were associated with residues of water (100–300 μL) after evaporation of the collected eluate. Considering that the extract was reconstituted in 250 μL of methanol, the final volume was increased and resulted in a dilution of matrix followed by higher LOQs. Finally, after the enzymatic deconjugation, the consistency of hydrolysate compared to non-hydrolysed urine was changed. After the loading of the hydrolysed sample onto the column, the precipitate accumulated on top of the sorbent, which extended the time required for the elution. The clogging of cartridge, which resulted in the lost of samples, has been identified as problematic in a similar study [20]. From these reasons, the new analytical method had to be implemented. The major requirements were as follows: (i) LOQs below 0.05 ng mL−1, (ii) effective removing of matrix co-extracts and (iii) simple and fast extraction with regard to the consistency of the hydrolysate.
LLE with clean-up using d-SPE
For this purpose, a simple LLE extraction using ethyl acetate (for which the highest recoveries within the SPE method evaluation were observed) including d-SPE was implemented. Various sorbents, specifically C18, PSA, Z-Sep, Z-Sep+ and ENVI-Carb, were tested in the purification step. The best results were obtained when Z-Sep was used. Compared to the other tested sorbents, the response of analytes in the purified extract increased by 30–80 %. The removing of matrix interferences was significant especially for d 9-OH-PYR and d 11-3-OH-BaP. As can be expected, ENVI-Carb adsorbs planar compounds, which resulted in the elimination of most OH-PAHs from the extract. Similar results were achieved using C18, PSA and Z-Sep+; nevertheless, the signals in the purified extract decreased by approximately 50 % compared to Z-Sep. The effectivity of the clean-up step was documented by the assessment of the matrix effects. The comparison of matrix effects in both extracts obtained by SPE and LLE with and without d-SPE documented (Fig. 3) that the cleaning of the urine extract with d-SPE led to the reduction of these effects by approximately 50 %, with the exception of d 11-3-OH-BaP, when only 20 % reduction was observed.

Comparison of matrix effects (ME, %) for tested extraction techniques. ME (%) = [peak area of matrix-matched standards / peak area of solution standards − 1] × 100 (%). ME = 0 %, no matrix effects; ME < 0 %, ionisation suppression; ME > 0 %, ionisation enhancement [33]
Method validation
Recoveries and repeatabilities (expressed as RSD, %) of the final method (LLE with clean-up using d-SPE) for OH-NAP isomers, 2-OH-FLUO, OH-PHEN isomers and 1-OH-PYR were calculated from six replicate analyses of the SRM 3673 (Table 1). The results were in good agreement with the certified values except for 1-OH-NAP, the measured concentration of which was slightly lower, 162 ± 11 ng mL−1, compared to the certified value (211 ± 34 ng mL−1). A similar trend as regards the concentrations of 1-OH-NAP determined in the SRM 3673 was observed also in other studies, for instance 151 ± 11 ng mL−1 by Motorykin et al. [19] and 197 ± 3 ng mL−1 by Li et al. [25]. Recoveries of nine OH-PAHs were in the range of 77–113 %, and repeatabilities were less than 20 %. LOQs for OH-PAHs were in the range of 0.01–0.025 ng mL−1. For the 6-OH-CHR and 3-OH-BaP, which were not certified in the SRM 3673, the performance parameters were determined by the analysis of the artificially contaminated urine blank sample. The validation level was 0.05 ng mL−1 for 6-OH-CHR and 1 ng mL−1 for 3-OH-BaP (the concentration was higher because of its lower instrumental sensitivity). The recoveries for 6-OH-CHR and 3-OH-BaP were 95 % (RSD 13 %, LOQ 0.01 ng mL−1) and 97 % (RSD 16 %, LOQ 0.9 ng mL−1), respectively. As overviewed in Table 2, present LOQs (0.01–0.025 ng mL−1) are comparable with the other reported values within the similar multi-analyte determination of OH-PAHs using LC–MS/MS [13–17, 34, 35]. The only exception is for 3-OH-BaP, when higher LOQ 0.9 ng mL−1 was achieved as compared to Xu et al. [13] (0.005 ng mL−1) or Barbeau et al. [12], who use the LC–FLD detection for the single determination of this metabolite and achieved 0.05 ng L−1.
Finally, the determined concentration of creatinine (508 ± 4 mg mL−1, RSD 0.8 %, LOQ 0.1 ng mL−1) was in accordance with the reference value in the certificate (505 ± 2 mg mL−1). Regarding with the control of background contamination, procedural blanks were prepared. From all target OH-PAHs, traces of 2-OH-NAP, 2-OH-PHEN and 3-OH-PHEN were detected in the concentration range of 0.01–0.09 ng mL−1.
Method applicability
Following the successful method validation, 50 samples of urine (collected within the project Impact of air pollution to genome of newborns, No. 13-13458S) were analysed. The samples were obtained from Czech women living in the industrial city Karviná, which represents a locality with high PAH air pollution, especially during winter season due to emissions from the residential heating [36, 37]. The results, expressed both in nanograms per milliliter and nanograms per gram creatinine, are summarised in Table 3. The total concentration of OH-PAHs (ΣOH-PAHs) was in the range of 0.87–63 ng mL−1 (1600–33,000 ng g−1 creatinine). The most abundant metabolites were 2-OH-NAP and 1-OH-PHEN, which were detected in all examined urine samples. Their concentrations were in the range of 0.51–53 ng mL−1 (720–25,000 ng g−1 creatinine) and 0.07–3.2 ng mL−1 (90–2400 ng g−1 creatinine), respectively. The predominance of 2-OH-NAP in the urine of general population (children, non-smokers) was published within recent studies in the range of median concentrations 1.1–5.3 ng mL−1 [38–41] and 2600–7400 ng g−1 creatinine [35, 42, 43]. The other detected OH-PAHs, namely 1-OH-NAP, 2-OH-FLUO, 2-OH-PHEN, 3-OH-PHEN, 4-OH-PHEN, 9-OH-PHEN and 1-OH-PYR, were found in more than 72 % of samples. 6-OH-CHR and 3-OH-BaP were not detected in any tested urine, and their occurrence was reported only in highly exposed subjects [44], since they are mainly excreted in faeces [5]. The example of U-HPLC–MS/MS chromatogram of urine sample is documented in Fig. S1 (see ESM).
Conclusions
Within our study, the novel analytical approach for the multiple determination of OH-PAHs including the metabolites of carcinogenic PAHs in urine has been developed and validated. The sample preparation procedure based on extraction with ethyl acetate and further purification of a crude urine extract with d-SPE using the sorbent Z-Sep is demonstrated for the first time in the analysis of PAH metabolites in the biological matrices such as urine and substitutes the demanding and time-consuming SPE on columns (with the risk of clogging of cartridge resulting in the loss of the sample or analytes) and thus significantly streamlines the analysis of urine in human biomonitoring studies. Great performance characteristics were obtained during the validation experiments on the SRM 3673, and the method recoveries ranged from 77 to 114 % with the relative standard deviation lower than 20 % and the quantification limits in the range of 0.010–0.025 ng mL−1 (except for 3-OH-BaP with 0.9 ng mL−1). With regard to the optimisation of the U-HPLC–MS/MS method, the chromatographic separation of OH-PHEN isomers was achieved using the PFP stationary phase.
The final method was successfully used for the analysis of 50 urine samples obtained from Czech women who are residents of Karviná region, which represent the highly industrialised locality with relatively high PAH exposure. The concentrations of ΣOH-PAHs were in the range of 0.87–63 ng mL−1 (1600–33,000 ng g−1 creatinine), with 2-OH-NAP and 1-OH-PHEN being the most abundant contaminants. Their concentrations were in the range of 0.51–53 ng mL−1 (720–25,000 ng g−1 creatinine) and 0.07–3.2 ng mL−1 (90–2400 ng g−1 creatinine), respectively. These results were comparable to those reported in similar studies since 2014.
This is the very first study which reported the use of ethyl acetate for the isolation of OH-PAHs from urine, followed by the clean-up step of extraction by d-SPE with the sorbent Z-Sep. We recommend that it became a routine method to measure urinary OH-PAHs, which was documented by the investigation of OH-PAH occurrence in the urine of Czech general population.
References
Scientific opinion of the panel on contaminants in the food chain on a request from the European Commission on Polycyclic Aromatic Hydrocarbons in Food. EFSA J. 2008;724:1–114.
Ma Y, Harrad S. Spatiotemporal analysis and human exposure assessment on polycyclic aromatic hydrocarbons in indoor air, settled house dust, and diet: a review. Environ Int. 2015;84:7–16.
Moustafa GA, Xanthopoulou E, Riza E, Linos A. Skin disease after occupational dermal exposure to coal tar: a review of the scientific literature. Int J Dermatol. 2015;54:868–79.
Li Z, Romanoff LC, Trinidad DA, Hussain N, Jones RS, Porter EN, et al. Measurement of urinary monohydroxy polycyclic aromatic hydrocarbons using automated liquid-liquid extraction and gas chromatography/isotope dilution high-resolution mass spectrometry. Anal Chem. 2006;78:5744–51.
Ramesh A, Walker SA, Hood DB, Guillen MD, Schneider K, Weyand EH. Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int J Toxicol. 2004;23:301–33.
Campo L, Rossella F, Fustinoni S. Development of a gas chromatography/mass spectrometry method to quantify several urinary monohydroxy metabolites of polycyclic aromatic hydrocarbons in occupationally exposed subjects. J Chromatogr B. 2008;875:531–40.
Jongeneelen FJ. Benchmark guideline for urinary 1-hydroxypyrene as biomarker of occupational exposure to polycyclic aromatic hydrocarbons. Ann Occup Hyg. 2001;45:3–13.
Jacob J, Seidel A. Biomonitoring of polycyclic aromatic hydrocarbons in human urine. J Chromatogr B. 2002;778:31–47.
Zhang W, Xu D, Zhuang G, Ding C, Wang G, Chang J, et al. A pilot study on using urinary 1-hydroxypyrene biomarker for exposure to PAHs in Beijing. Environ Monit Assess. 2007;131:387–94.
Freire C, Abril A, Fernández MF, Ramos R, Estarlich M, Manrique A, et al. Urinary 1-hydroxypyrene and PAH exposure in 4-year-old Spanish children. Sci Total Environ. 2009;407:1562–9.
Yamano Y, Hara K, Ichiba M, Hanaoka T, Pan G, Nakadate T. Urinary 1-hydroxypyrene as a comprehensive carcinogenic biomarker of exposure to polycyclic aromatic hydrocarbons: a cross-sectional study of coke oven workers in China. Int Arch Occup Environ Health. 2014;87:705–13.
Barbeau D, Maître A, Marques M. Highly sensitive routine method for urinary 3-hydroxybenzo [a] pyrene quantitation using liquid chromatography-fluorescence detection and automated off-line solid phase extraction. Analyst. 2011;136:1183–91.
Xu X, Zhang J, Zhang L, Liu W, Weisel CP. Selective detection of monohydroxy metabolites of polycyclic aromatic hydrocarbons in urine using liquid chromatography/triple quadrupole tandem mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:2299–308.
Onyemauwa F, Rappaport SM, Sobus JR, Gajdošová D, Wu RA, Waidyanatha S. Using liquid chromatography–tandem mass spectrometry to quantify monohydroxylated metabolites of polycyclic aromatic hydrocarbons in urine. J Chromatogr B. 2009;877:1117–25.
Ramsauer B, Sterz K, Hagedorn HW, Engl J, Scherer G, McEwan M, et al. A liquid chromatography/tandem mass spectrometry (LC-MS/MS) method for the determination of phenolic polycyclic aromatic hydrocarbons (OH-PAH) in urine of non-smokers and smokers. Anal Bioanal Chem. 2011;399:877–89.
Fan R, Wang D, Ramage R, She J. Fast and simultaneous determination of urinary 8-hydroxy-2′-deoxyguanosine and ten monohydroxylated polycyclic aromatic hydrocarbons by liquid chromatography/tandem mass spectrometry. Chem Res Toxicol. 2012;25:491–9.
Fan R, Ramage R, Wang D, Zhou J, She J. Determination of ten monohydroxylated polycyclic aromatic hydrocarbons by liquid–liquid extraction and liquid chromatography/tandem mass spectrometry. Talanta. 2012;93:383–91.
Chetiyanukornkul T, Toriba A, Kameda T, Tang N, Hayakawa K. Simultaneous determination of urinary hydroxylated metabolites of naphthalene, fluorene, phenanthrene, fluoranthene and pyrene as multiple biomarkers of exposure to polycyclic aromatic hydrocarbons. Anal Bioanal Chem. 2006;386:712–8.
Motorykin O, Schrlau J, Jia Y, Harper B, Harris S, Harding A, et al. Determination of parent and hydroxy PAHs in personal PM 2.5 and urine samples collected during native American fish smoking activities. Sci Total Environ. 2015;505:694–703.
Romanoff LC, Li Z, Young KJ, Blakely NC, Patterson DG, Sandau CD. Automated solid-phase extraction method for measuring urinary polycyclic aromatic hydrocarbon metabolites in human biomonitoring using isotope-dilution gas chromatography high-resolution mass spectrometry. J Chromatogr B. 2006;835:47–54.
Li Y, Li X, Zhou Z. A novel facile method using polyetheretherketone as a solid phase extraction material for fast quantification of urinary monohydroxylated metabolites of polycyclic aromatic hydrocarbons. RSC Adv. 2014;4:39192–6.
Luo K, Gao Q, Hu J. Derivatization method for sensitive determination of 3-hydroxybenzo [a] pyrene in human urine by liquid chromatography–electrospray tandem mass spectrometry. J Chromatogr A. 2015;1379:51–5.
Smith CJ, Walcott CJ, Huang W, Maggio V, Grainger J, Patterson DG. Determination of selected monohydroxy metabolites of 2-, 3-and 4-ring polycyclic aromatic hydrocarbons in urine by solid-phase microextraction and isotope dilution gas chromatography–mass spectrometry. J Chromatogr B. 2002;778:157–64.
Luan T, Fang S, Zhong Y, Lin L, Chan SM, Lan C, et al. Determination of hydroxy metabolites of polycyclic aromatic hydrocarbons by fully automated solid-phase microextraction derivatization and gas chromatography–mass spectrometry. J Chromatogr A. 2007;1173:37–43.
Li Z, Romanoff LC, Trinidad DA, Pittman EN, Hilton D, Hubbard K, et al. Quantification of 21 metabolites of methylnaphthalenes and polycyclic aromatic hydrocarbons in human urine. Anal Bioanal Chem. 2014;406:3119–29.
Jacob P, Wilson M, Benowitz NL. Determination of phenolic metabolites of polycyclic aromatic hydrocarbons in human urine as their pentafluorobenzyl ether derivatives using liquid chromatography–tandem mass spectrometry. Anal Chem. 2007;79:587–98.
Li X, Zenobi R. Use of polyetheretherketone as a material for solid phase extraction of hydroxylated metabolites of polycyclic aromatic hydrocarbons in human urine. Anal Chem. 2013;85:3526–31.
Hagedorn HW, Scherer G, Engl J, Riedel K, Cheung F, Errington G, et al. Urinary excretion of phenolic polycyclic aromatic hydrocarbons (OH-PAH) in nonsmokers and in smokers of cigarettes with different ISO tar yields. J Anal Toxicol. 2009;33:301–9.
Zhang X, Hou H, Xiong W, Hu Q. Development of a method to detect three monohydroxylated polycyclic aromatic hydrocarbons in human urine by liquid chromatographic tandem mass spectrometry. J Anal Methods Chem. 2015;2015:1–8.
Butler AR. The Jaffè reaction: identification of the colored species. Clin Chim Acta. 1976;59:227–32.
Bonsnes RW, Taussky HH. On the colorimetric determination of creatinine by the Jaffe reaction. J Biol Chem. 1954;158(3):581–91.
Euerby MR, McKeown AP, Petersson P. Chromatographic classification and comparison of commercially available perfluorinated stationary phases for reversed-phase liquid chromatography using principal component analysis. J Sep Sci. 2003;26:295–306.
Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–30.
Guo Y, Senthilkumar K, Alomirah H, Moon HB, Minh TB, Mohd MA, et al. Concentrations and profiles of urinary polycyclic aromatic hydrocarbon metabolites (OH-PAHs) in several Asian countries. Environ Sci Technol. 2013;47:2932–8.
Wang Z, Zheng Y, Zhao B, Zhang Y, Liu Z, Xu J, et al. Human metabolic responses to chronic environmental polycyclic aromatic hydrocarbon exposure by a metabolomic approach. J Proteome Res. 2015;14:2583–93.
Svecova V, Topinka J, Solansky I, Rossner P, Sram RJ. Personal exposure to carcinogenic polycyclic aromatic hydrocarbons in the Czech Republic. J Expo Anal Environ Epidemiol. 2013;23:350–5.
Sykorova B, Kucbel M, Raclavska H, Drozdova J, Raclavsky K. Seasonal variations of polycyclic aromatic hydrocarbons (PAHs) in the air of Moravian-Silesian region, Czech Republic. In: Sung WP, Kao J, editors. Environment, energy and applied technology. London: Taylor & Francis Group; 2015. p. 367–72.
Thai PK, Li Z, Sjödin A, Fox A, Diep NB, Binh TT, et al. Biomonitoring of polycyclic aromatic hydrocarbons exposure in small groups of residents in Brisbane, Australia and Hanoi, Vietnam, and those travelling between the two cities. Chemosphere. 2015;139:358–64.
Jung KH, Lovinsky-Desir S, Perzanowski M, Liu X, Maher C, Gil E, et al. Repeatedly high polycyclic aromatic hydrocarbon exposure and cockroach sensitization among inner-city children. Environ Res. 2015;140:649–56.
Li J, Fan R, Lu S, Zhang D, Zhou Y, Lv Y. Exposure to polycyclic aromatic hydrocarbons could cause their oxidative DNA damage: a case study for college students in Guangzhou, China. Environ Sci Pollut Res. 2015;22:1770–7.
Kim HW, Kam S, Lee DH. Synergistic interaction between polycyclic aromatic hydrocarbons and environmental tobacco smoke on the risk of obesity in children and adolescents: the US National Health and Nutrition Examination Survey 2003–2008. Environ Res. 2014;135:354–60.
Li J, Lu S, Liu G, Zhou Y, Lv Y, She J, et al. Co-exposure to polycyclic aromatic hydrocarbons, benzene and toluene and their dose–effects on oxidative stress damage in kindergarten-aged children in Guangzhou, China. Sci Total Environ. 2015;524:74–80.
Fan R, Li J, Chen L, Xu Z, He D, Zhou Y, et al. Biomass fuels and coke plants are important sources of human exposure to polycyclic aromatic hydrocarbons, benzene and toluene. Environ Res. 2014;135:1–8.
Förster K, Preuss R, Roßbach B, Brüning T, Angerer J, Simon P. 3-Hydroxybenzo[a] pyrene in the urine of workers with occupational exposure to polycyclic aromatic hydrocarbons in different industries. J Occup Environ Med. 2008;65:224–9.
Acknowledgments
This study was funded by the Czech Science Foundation Project No. 13-13458S. The authors would like to thank the gynaecological and maternity ward at the Hospital and Polyclinic Karviná for organising the logistic and collection of samples. The ‘Operational Program Prague – Competitiveness’ (CZ.2.16/3.1.00/22197) and ‘National Program of Sustainability’ (NPU I (LO) MSMT - 34870/2013) are also gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study has been approved by the appropriate ethics committee and has been performed in accordance with the ethical standards.
Conflict of interest
The authors declare that they have no competing interests.
Informed consent
Informed consent was obtained from all participants for being included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1353 kb)
Rights and permissions
About this article

Cite this article
Lankova, D., Urbancova, K., Sram, R.J. et al. A novel strategy for the determination of polycyclic aromatic hydrocarbon monohydroxylated metabolites in urine using ultra-high-performance liquid chromatography with tandem mass spectrometry. Anal Bioanal Chem 408, 2515–2525 (2016). https://doi.org/10.1007/s00216-016-9350-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9350-1