
Abstract
Polycyclic aromatic hydrocarbons (PAH) are products of the incomplete combustion of organic materials and, therefore, occur ubiquitously in the environment and also in tobacco smoke. Since some PAH have been classified as carcinogens, it is important to have access to suitable analytical methods for biomarkers of exposure to this class of compounds. Past experience has shown that measuring a profile of PAH metabolites is more informative than metabolites of a single PAH. Assessment of environmental and smoking-related exposure levels requires analytical methods with high sensitivity and specificity. In addition, these methods should be fast enough to allow high throughput. With these pre-conditions in mind, we developed and validated a high-performance liquid chromatographic method with tandem mass spectrometric detection (LC-MS/MS) for the determination of phenolic metabolites of naphthalene, fluorene, phenanthrene and pyrene in urine of smokers and non-smokers. Sample work-up comprised enzymatic hydrolysis of urinary conjugates and solid-phase extraction on C18 cartridges. The method showed good specificity, sensitivity, and accuracy for the intended purpose and was also sufficiently rapid with a sample throughput of about 350 per week. Application to urine samples of 100 smokers and 50 non-smokers showed significant differences between both groups for all measured PAH metabolites, and strong correlations with markers of daily smoke exposure in smoker urine. Urinary levels were in good agreement with previously reported data using different methodologies. In conclusion, the developed LC-MS/MS method is suitable for the quantification of phenolic PAH metabolites of naphthalene, fluorene, phenanthrene, and pyrene in smoker and non-smoker urine.

Cumulative frequencies of urinary 4-OH-Phe (left, not significantly influenced by smoking) and 1-OH-Pyr (right, significantly elevated in smokers)
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polycyclic aromatic hydrocarbons (PAH) are an important class of environmental contaminants, which are formed during the incomplete combustion of organic materials [1]. Single PAH have been classified by the International Agency for Research on Cancer (IARC) as Class 1, “human carcinogens” (benzo[a]pyrene, BaP), Class 2A, “probably carcinogenic to humans” (dibenz[a,b]anthracene, dibenzo(a,l)pyrene, cyclopenta[c,d]pyrene), Class 2B, “possibly carcinogenic to humans” (14 PAH, including naphthalene, Nap) or Class 3, “not classifiable as to carcinogenicity to humans” (another 10 PAH) [2]. Sources of PAH exposure for non-occupationally exposed subjects are the environment (by inhalation and dermal contact), coal-tar-containing medications, diet and tobacco smoke [1, 3, 4].
Given the widespread distribution of PAH and their potential carcinogenicity, a reliable assessment of the internal dose by measuring suitable biomarkers of exposure is of importance. Biomonitoring of non-occupational PAH exposure is complicated by at least two adversities: (1) PAH occur almost exclusively as a complex mixture of several hundred compounds [5, 6]; (2) except for naphthalene, all potentially carcinogenic PAH (IARC classes 1, 2A, and 2B) are higher condensed compounds with more than four ring systems, the amounts of which in the complex mixture are far lower than those of the lower condensed PAH (less than or equal to four rings). The second point indicates that biomarkers for non-occupational PAH exposure are primarily derived from lower condensed compounds, namely Nap, fluorene (Flu), phenanthrene (Phe) and pyrene (Pyr) [7, 8]. In a few studies, biomarkers for BaP (the lead compound for PAH) have been measured as a surrogate for the smoking-related exposure to PAH [9–12]. However, the rather challenging measurement of urinary 3-hydroxybenzo[a]pyrene does not have major advantages as a surrogate biomarker for exposure to PAH over the more frequently used marker 1-hydroxypyrene (1-OH-Pyr). Another question concerns whether the exposure to a complex mixture of PAH is better reflected by a profile of biomarkers rather than by a single biomarker. The US Environmental Protection Agency has set up a list of 16 PAH as “priority pollutants”, which are most frequently used for environmental PAH studies; namely, naphthalene, acenaphthylene, acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene, pyrene, benzo[a]anthracene, chrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, dibenz[a,h]anthracene, benzo[g,h,i]perylene, and indeno[1,2,3-cd]pyrene [13]. The obvious approach for the biological monitoring of environmental PAH exposure would be to determine stable urinary metabolites of these 16 PAH. However, this procedure is prohibited by the fact that urinary metabolites have not been identified for all compounds and that some of the higher condensed PAH are probably excreted in only trace amounts. There is a clear trend in the last decade from single PAH biomarker determinations (most frequently phenolic metabolites of Pyr, Phe and Nap) to multi PAH biomarker methods which comprise phenolic metabolites of four or more PAH [14–27]. Analytes included in the multi-methods for PAH most frequently comprise monohydroxy metabolites of Nap, Flu, Phe, and Pyr. In some methods, phenolic metabolites of benzo[c]phenanthrene, benz[a]anthracene, chrysene, and BaP have also been included [22, 25–27]. However, data from a large US population study has shown that a majority of the urine samples analyzed had levels of these metabolites below the limit of detection (LOD) [27].
All analytical methods for phenolic PAH in urine comprise enzymatic hydrolysis with β-glucuronidase/sulfatase, and subsequent extraction applying liquid/liquid extraction [16, 21, 22, 24], solid-phase extraction [17, 18, 23, 26–28], solid-phase microextraction [14, 25] or column switching techniques [15, 23]. Analytical platforms, which have proved to be sufficiently sensitive for quantifying environmental and smoking-related urinary OH-PAH, include high-performance liquid chromatography with fluorescence detection (HPLC-FD) [15, 17, 20, 23], gas chromatography coupled with high-resolution mass spectrometry after trimethylsilylation [18, 22, 25, 27], and liquid chromatography with tandem mass spectrometry (LC-MS/MS), either with [19, 21] or without [26], [29] prior derivatization. We have also developed a GC-MS/MS method using derivatization with pentafluoropropionic acid anhydride, which was suitable for quantifying OH-PAH metabolites originating from exposure to PAH from the environment or tobacco smoke (publication in preparation).
The purpose of our study was to improve sample throughput, while simultaneously retaining the required method performance. To this end, we have developed an LC-MS/MS method without derivatization for the OH-PAH depicted in Fig. 1. The mass transition used for quantification comprised the loss of CO (−28 Da), as described by others [26], [29]. The method was validated according to US FDA guidelines [30] and applied to urine samples from 50 non-smokers and 100 smokers.

Chemical structures of OH-PAH determined in this investigation
Experimental
Chemicals
1-OH-Naphthalene (1-OH-Nap), 2-OH-naphthalene (2-OH-Nap) and 2-OH-fluorene (2-OH-Flu) were purchased from Sigma-Aldrich (Deisenhofen, Germany). 1-Hydroxypyrene (1-OH-Pyr) was purchased from Acros Organics (Geel, Belgium). 1-, 2-, 3-, 4-, and 9-OH-Phenanthrene (1-, 2-, 3-, 4-, and 9-OH-Phe) were generously supplied by Dr. A. Seidel (Grosshansdorf, Germany). 1- and 2-OH-Nap-d7, 1-OH-Pyr-d9 were purchased from CDN Isotopes, Quebec, Canada. 2-OH-Flu-d9 as well as a mixture of 1-, 2-, 3-, 4-, and 9-OH-Phe-d9 were synthesized according to a published method [21].
HPLC-grade methanol and all other analytical grade organic solvents were from LGC Standards (Wesel, Germany). Water was purified using a Seraldest purification system (Seraldest, Munich, Germany). Other chemicals and reagents used were at least of analytical grade. β-Glucuronidase/arylsulfatase (enzyme preparation from Helix pomatia, 4.5/14 U∙mL−1) was purchased from Roche (Mannheim, Germany).
Urine sample preparation
Extraction of OH-PAH from human urine followed a previously published method [17] with modifications. Urine was thawed at room temperature and briefly agitated. For enzymatic hydrolysis of glucuronides and sulfates, 5 mL urine was mixed with 10 mL acetate buffer (100 mM, pH 5.0). The pH of the mixture was adjusted to 5.0 ± 0.1 with 4 M HCl. After addition of 10 μl β-glucuronidase (0.05 U)/arylsulfatase (0.14 U), the sample was incubated overnight (37 °C, 16–20 h) with gentle agitation in the dark. The following amounts of internal standards (IS) were added to the hydrolysate: 250 ng of 1-/2-OH-Nap-d7, 50 ng of 2-OH-Flu-d9, 5 ng of 1-OH-Pyr-d9 and 5–25 ng of OH-Phe-d9. The sample was transferred to a C18 cartridge (Bond Elut-C18, 500 mg, 3 mL, Varian, Darmstadt, Germany) which was conditioned with 7.5 mL methanol and 10 mL water. The loaded cartridge was washed with 10 mL water and 10 mL methanol/water (1:4, v/v), vacuum-dried and centrifuged (3,000×g, 10 min 10 °C) to remove the excess solvent. Finally, the cartridge was thoroughly dried with a stream of nitrogen (1 bar, 20 min) and eluted with 7.5 mL n-hexane/methanol (100:0.7, v/v). The eluate was evaporated to dryness under reduced pressure at room temperature (Speed Vac concentrator, RC 10.22, Thermo, Dreieich, Germany). The residue was re-dissolved in 100 μl methanol.
LC-MS/MS analysis
Ten μl of the extract were injected into an Agilent HPLC system Series 1200 (Agilent Technologies, Waldbronn, Germany), consisting of a binary pump G1312B, a column oven G1316B, a degasser G1379B and a CTC PAL HTC autosampler (Axel Semrau, Sprockhoevel, Germany). The HPLC system was coupled to a Sciex API 5000 MS/MS system (ABI Sciex, Darmstadt, Germany). Chromatography was performed on a Kinetex C18 column, 10 × 100 mm, 2.6 μm (Phenomenex, Aschaffenburg, Germany) at 40 °C column temperature with a flow rate of 0.4 mL/min and a gradient consisting of A (0.1% ammonium acetate in water) and B (methanol) programmed as follows: 0.00 min, 95% A; 20.01 min, 20% A; 20.01 min, 95% A; 26 min, 95% A. Negative electrospray ionization at 650 °C was applied and the MS/MS was run in the multi-reaction-monitoring (MRM) mode. Used mass transitions and retention times for analytes and IS are shown in Table 1.
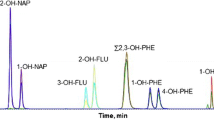
Note that 1- and 9-OH-Phe were not sufficiently separated chromatographically, and were therefore evaluated as a sum of both these metabolites. Although the IS 2-OH-Flu-d9 and its native analogue show the same fragmentation in the tuning mode, the IS did not show the mass transition of 190→162 (CO loss) which would correspond to the transition of 181→153 for the analyte 2-OH-Flu. Also the loss of CO + D (−30), observable for the native analogue (−29) as well, has been measured but did not give a detectable signal for the IS. Therefore, 2-OH-Flu-d9 was quantified in single ion monitoring mode.
One explanation is the very weak intensities of these mass transitions, which are selectively disturbed for the isotopically labeled compound in real samples. The background level for 190→162 and 190→160 sis 64-fold higher than for 181→152. Together with the weak intensity of the daughter ions (daughter/parent ion ≈ 0.01 compared to 0.1–0.4 for the other compounds) the different behavior of analyte and internal standard in real urine samples can be explained. The reason for choosing 181→152 instead of 181→153 is the lower intensity of the latter for this partly aromatic analyte. It is interesting to see that Onyemauwa et al. [29] have also not been using the mass transition of CO loss in the fragmentation pattern of 2-OH-Flu. This behavior is different to the other hydroxylated PAHs investigated.
Calibration
Calibration was performed by spiking a 1:1 mixture of synthetic urine [21] and non-smoker urine with the following levels of the analytes: 1-OH- and 2-OH-Nap: 0.10, 0.50, 1.00, 5.00, 10.0, 25.0 ng/mL; 2-OH-Flu: 0.05, 0.50, 1.00, 2.50, 5.00, 10.0, 15.0 ng/mL; 1-/9-OH-Phe: 0.02, 0.04, 0.10, 0.20, 0.40, 1.00, 2.00, 4.00 ng/mL; 2-OH, 3-OH-, 4-OH-Phe: 0.01, 0.02, 0.05, 0.10, 0.20, 0.50, 1.00, 2.00 ng/mL; 1-OH-Pyr: 0.010, 0.025, 0.050, 0.250, 0.500, 1.00, 2.00 ng/mL. Authentic human urine was necessary in order to achieve the required sensitivity due to the positive matrix effect with 1-OH-Pyr (analyte and IS, see Table 2). The samples were prepared as described above. All calibrators, including the unspiked samples (zero calibrators), were analyzed in duplicate. The analyte/IS ratio of the zero calibrator was subtracted from the ratio of each of the spiked calibrators. Calibration lines were constructed by plotting the area ratios (area of analyte/area of IS) against the nominal concentrations. Linear regression, using lines forced through the origin, were calculated.
Method validation
Method validation was performed according to FDA guidelines [30]. Specificity was measured using six different urine matrices, which were checked for interference with the analytes. As a criterion, the accuracy at a medium concentration level tested in these six matrices should be in the range of 85–115%. Intra-day precision was determined by evaluating three analyte concentration levels in authentic human urine samples, to which only 4-OH-Phe was spiked in appropriate concentrations. For intra-day precision, each sample was analyzed five times. For inter-day precision, samples were analyzed once on six different days within 2 weeks. Acceptance criteria for precision were ±15% and ±20% at levels up to three times the limit of quantification (LOQ). Accuracies at low, medium, and high concentrations were determined with human urine samples spiked at low, medium and high levels. Each level was analyzed five times. Acceptance criteria were rates of 85–115% (80–120% at levels up to 3× LOQ) as well as coefficient of variations (CV) of ±15% (±20% <3× LOQ). Recovery rates were determined by comparing the analyte concentrations at the three levels, measured when non-smoker urine extract was spiked after sample work-up and before the LC-MS/MS measurements (reference, 100%), and when the non-smoker urine was spiked at the beginning of the sample work-up procedure. LOD and LOQ were estimated with the signal/noise (=3× standard deviation) method using the integrated function of the Analyst software (Version 1.4.2, AB Sciex, Darmstadt, Germany). Signal/noise ratios of 3:1 and 9:1 were applied for estimating the LOD and LOQ, respectively. As a second criterion for the LOQ, lowest calibration level was used, which showed an accuracy of 80–120% with a CV of ±20% for a duplicate analysis. Matrix effects were determined by calculating the peak area ratios of the analytes at low and high concentrations when spiked to worked-up urine extracts (with the background areas subtracted) and the same amounts of analytes in solvent (methanol). Stability under post-sample preparation conditions was determined by storing the extracts 13–20 day at −20 °C and subsequently for 24 h at room temperature. Carry-over effects in the chromatographic system were tested by injecting urine extracts with high analyte concentrations five times, followed by blank (methanol) injection. This was repeated 3 times. The solvent runs were checked for analyte peaks.
Urine samples
One hundred and fifty (150) 24-h urine samples stored at −25 °C were taken from a previous clinical study performed in Germany (full publication in preparation). The study protocol and informed consent forms for this study were approved by the Ethics Committee (EC) of the Ärztekammer Hamburg, Germany and the clinical study was conducted in accordance with the World Medical Association Declaration of Helsinki and International Conference on Harmonisation (ICH) Guidelines for Good Clinical Practice [31, 32].
The urine samples were obtained from healthy smoking (N = 100) and non-smoking (N = 50) volunteers. The smokers were enrolled for 6 weeks into the study and all habitually smoked between 6 and 30 king sized cigarettes per day. In all cases their usual brand was of typical German blend style with a tar yield of 6 to 7 mg/cigarette (as determined according to the ISO (International Standard Organization) standard smoking regime). All smokers made an ambulatory visit to the clinic where they were supplied with 2 weeks supply of a control 6 mg ISO tar yield king size cigarette of similar blend style to their normal brand. They returned home for approximately 2 weeks, during which time they were required to smoke only the supplied cigarette, and were asked to smoke normally in terms of frequency and daily consumption. In addition, subjects kept a diary that recorded daily cigarette consumption, diet, exercise and general health status. During this non-residential part of the study, subjects were asked to refrain from grilled, smoked, fried or barbequed food and cruciferous vegetables. After 12 days, the smokers were admitted into the clinic for a residential 2 day period. The 24-h urine samples used for the purposes of this paper were obtained during the first 24 h in the clinic. While resident in the clinic, subjects received a standardized bulk diet that excluded grilled, smoked, fried or barbequed food and cruciferous vegetables.
The non-smoking group was enrolled for a total of 8 weeks (also recording diet, exercise and health details and adhering to the same dietary limitations as the smokers). Again, for the purposes of this paper, the urine samples used for this study were taken from those collected during the first 24 h period resident in the clinic.
Statistics
All statistical analyses were carried out with SPSS Version 13.0 (SPSS GmbH Software, Munich, Germany). Summary statistics were computed for each OH-PAH. A correlation matrix was produced to test relationships between the different OH-PAH and daily cigarette consumption.
Results
Performance of the analytical method
MRM chromatograms for OH-PAH and the corresponding IS of urine extracts from a non-smoker to a smoker are shown in Fig. 2. The isomeric pair of 1-OH-Phe/9-OH-Phe could not be separated and these two isomers were therefore evaluated as a sum.

MRM Chromatograms for OH-PAH and corresponding internal standards of urine samples from a non-smoker (a) and a smoker (b). Mass transitions and actual analyte concentrations are shown in the graphs
The method performance data are shown in Table 2. Intra- and inter-day precisions for all analytes were clearly below 10%. Accuracy rates were in the range of 91–114%. Recovery rates for the complete analytical procedure were >80% for all OH-PAH at medium and high concentrations, and between 40% and 70% at the lowest concentrations. LODs were approximately 0.03 and 0.01 ng/mL for 1- and 2-OH-Nap, respectively, 0.02 ng/mL for 2-OH-Flu, 0.01 ng/mL for the 1-/9-OH-Phe, 0.003, 0.002 and 0.001 ng/mL for 2-, 3- and 4-OH-Phe, and 0.007 ng/mL for 1-OH-Pyr.
Method selectivity, determined as accuracy at a medium concentration in six different urine matrices, was in the range of 88–115%.
Slightly positive matrix effects of 102–134% were observed for all analytes, except for 1-OH-Pyr, which showed a significant positive matrix effect of about 550%.
The analytes were stable under post-preparative conditions. No carry-over effects were observed in the LC-MS/MS system used (data not shown).
Urinary excretion of OH-PAH in non-smokers and smokers
Subject demography, cigarette consumption and a summary table of OH-PAH excretion levels of the study population are shown in Table 3. Non-smokers and smokers in this study featured equal gender distributions and similar 24-h urine volumes. The smokers in this study were significantly younger than the non-smokers. The average daily cigarette consumption was 17.5 cigarettes/day, with a range of 5–32 cigarettes/day. Cigarette consumption per day was available only for 99 out of 100 smokers.
Smokers excreted significantly higher amounts of all measured OH-PAH than non-smokers, except for 4-OH-Phe, which showed similar excretion levels in smokers and non-smokers. Cumulative frequencies of all measured OH-PAH in non-smokers and smokers are depicted in Fig. 3.

Cumulative frequency plots of the measured OH-PAH classified according to smoking habit
In smokers, there was significant correlation between each of the OH-PAH, and the daily cigarette consumption (Table 4). The correlation with cigarettes/day was lowest for 4-OH-Phe (r = 0.319), but still significant.
Discussion
PAHs are an important class of toxicants in workplaces, the environment and also in tobacco smoke. BaP, a lead compound belonging to the class of PAH, has been recommended for mandated lowering in cigarette smoke by a working group of the World Health Organization (WHO) [33]. Assessing the exposure to toxicants by measuring suitable biomarkers in body fluids is generally regarded to be more reliable than only measuring the external dose [34]. Furthermore, biomarkers of exposure are valuable tools for the evaluation of potentially reduced exposure products [35].
The described LC-MS/MS method for measuring urinary OH-PAH at environmental and smoking-related levels, is the third in a series of three methods developed in our lab for this purpose. The recently published HPLC-FD method [17] was sensitive and specific enough for the intended purpose of assessing the environmental and smoking-related exposure to PAH. However, major drawbacks of the HPLC-FD method included the limited specificity of the fluorescence detection, the limited separation power of the HPLC for OH-PAH isomers and the impossibility of using authentic (labeled) IS for controlling all steps of the analytical method. Despite these limitations, results generated with the HPLC-FD method were in good agreement with other published data on urinary OH-PAH in environmental and smoking-related exposure ranges [17] (compare also Table 5). We have also developed a GC-MS/MS method using derivatization with pentafluoropropionic acid anhydride (publication in preparation). Since two SPE purification steps, derivatization and a chromatographic run time of 40 min were required, sample throughput was somewhat limited (~120 samples/week). However, the GC-MS/MS method performance was excellent, with complete chromatographic separation of all OH-PAH isomers.
The LC-MS/MS method performed well (Table 2), meeting all of the requirements for high sensitivity, specificity, precision and accuracy. In particular, sample throughput was increased from approximately 120 to about 350 samples per week. A major drawback of the method is certainly the inability to chromatographically separate the isomers 1- and 9-OH-Phe. At present, ultra-high pressure liquid chromatography is being applied to help resolve the problem. An unexpected outcome from this new LC-MS/MS method development was the significant positive matrix effect observed for 1-OH-Pyr (Table 2). Further investigations of this phenomenon revealed no linear dependency on the amount of urinary matrix present. Rather, the equivalent of 0.25 mL urine (5% of the usual amount of matrix present) accounted for the bulk (80%) of this effect. Further investigations may be required to explore the benefits of this outcome in terms of method sensitivity.
Application of the LC-MS/MS method to urine samples of 50 non-smokers and 100 smokers showed that smokers excreted significantly higher amounts of OH-PAH (Table 3, Fig. 3). Levels of urinary 4-OH-Phe were similar in smokers and non-smokers, and also exhibited the lowest concentration of all measured OH-PAH. For smokers, there was a significant correlation between urinary OH-PAH metabolites and the daily cigarette consumption (Table 4). In general, these findings are in agreement with other reports in the literature [15, 17, 29, 36–53]. Reported levels of urinary OH-PAH in non-occupationally exposed individuals applying OH-PAH multi-methods are compiled in Table 5. The observed levels for non-smokers and smokers in this study are in good agreement to those reported in other studies. A detailed publication on the cigarette-type related biomarkers of exposure levels, including OH-PAH, is in preparation.
In conclusion, the described LC-MS/MS method for the determination of the phenolic metabolites of naphthalene, fluorene, phenanthrene, and pyrene is suitable for assessing a profile of environmental and smoking-related OH-PAH in human urine. A main advantage compared to other methods is its improved sample throughput. Results obtained for OH-PAH in urine of smokers and non-smokers are in good agreement with reported levels using other methods.
The observed dose–response relationship between daily cigarette consumption and urinary OH-PAH excretion in smokers, combined with the fact that the OH-PAH were also quantifiable in non-smoker urine samples, suggests that this method is suitable for the evaluation of potentially reduced tobacco exposure products.
References
International Agency for Research on Cancer (1983) IARC monographs on the evaluation of the carcinogenic risk of chemicals to humans. Polynuclear aromatic compounds, Part 1, chemical, environmental and experimental data, Volume 32. International Agency for Research on Cancer, Lyon
International Agency for Research on Cancer. Agents classified by the IARC Monographs Volumes 1–100. http://monographs.iarc.fr/ENG/Classification/ClassificationsAlphaOrder.pdf. Accessed 25 Aug 2010
Hattemer-Frey HA, Travis CC (1991) Benzo-a-pyrene: environmental partitioning and human exposure. Toxicol Ind Health 7:141–157
Phillips DH (1999) Polycyclic aromatic hydrocarbons in the diet. Mutat Res 443:139–147
Dipple A, Chen SC, Bigger AH (1990) Polycyclic aromatic hydrocarbon carcinogens. In: Parizia MW (ed) Mutagens and carcinogens in the diet. Wiley-Liss, LissNew York
Rodgman A, Perfetti TA (2006) The composition of cigarette smoke: a catalogue of the polycyclic aromatic hydrocarbons. Beitr TabForsch Int 22:13–69
Angerer J, Mannschreck C, Gündel J (1997) Biological monitoring and biochemical effect monitoring of exposure to polycyclic aromatic hydrocarbons. Int Arch Occup Environ Health 70:365–377
Jacob J, Seidel A (2002) Biomonitoring of polycyclic aromatic hydrocarbons in human urine. J Chrom B 778:31–47
Simpson CD, Wu MT, Christiani DC, Santella RM, Carmella SG, Hecht SS (2000) Determination of r-7, t-8, 9, c-10-tetrahydroxy-7, 8, 9, 10-tetrahydrobenzo[a]pyrene in human urine by gas chromatography/negative ion chemical ionization/mass spectrometry. Chem Res Toxicol 13:271–280
Lafontaine M, Champmartin C, Simon P, Delsaut P, Funck-Brentano C (2006) 3-Hydroxybenzo[a]pyrene in urine of smokers and non-smokers. Toxicol Lett 162:181–185
Leroyer A, Jeandel F, Maitre A, Howsam M, Deplanque D, Mazzuca M, Nisse C (2010) 1-Hydroxypyrene and 3-hydroxybenzo[a]pyrene as biomarkers of exposure to PAH in various environmental exposure situations. Sci Total Environ 408:1166–1173
Sarkar M, Liu J, Koval T, Wang J, Feng S, Serafin R, Jin Y, Xie Y, Newland K, Roethig HJ (2010) Evaluation of biomarkers of exposure in adult cigarette smokers using Marlboro Snus. Nic Tob Res 12:105–116
US Environmental Protection Agency (1984). Screening methods for PAH priority pollutants in waste water. http://nepis.epa.gov/. Accessed 06 May 2010
Gmeiner G, Krassnig C, Schmid E, Tausch H (1998) Fast screening method for the profile analysis of polycyclic aromatic hydrocarbon metabolites in urine using derivatisation—solid-phase microextraction. J Chrom B 705:132–138
Heudorf U, Angerer J (2001) Urinary monohydroxylated phenanthrenes and hydroxypyrene—the effects of smoking habits and changes induced by smoking on monooxygenase-mediated metabolism. Int Arch Occup Environ Health 74:177–183
Serdar B, Waidyanatha S, Zheng Y, Rappaport SM (2003) Simultaneous determination of urinary 1- and 2-naphthols, 3- and 9-phenanthrols, and 1-pyrenol in coke oven workers. Biomarkers 8:93–109
Hagedorn HW, Scherer G, Engl J, Riedel K, Cheung F, Errington G, Shepperd J, McEwan M (2009) Urinary excretion of phenolic polycyclic aromatic hydrocarbons (OH-PAH) in nonsmokers and in smokers of cigarettes with different ISO tar yields. J Anal Toxicol 33:301–309
Huang W, Caudill SP, Grainger J, Needham LL, Patterson DG Jr (2006) Levels of 1-hydroxypyrene and other monohydroxy polycyclic aromatic hydrocarbons in children: a study based on U.S. reference range values. Toxicol Lett 163:10–19
Benowitz NL, Jacob P III, Bernert JT, Wilson M, Wang L, Allen F, Dempsey D (2005) Carcinogen exposure during short-term switching from regular to “light” cigarettes. Cancer Epidemiol Biomark Prev 14:1376–1383
Chetiyanukornkul T, Toriba A, Kameda T, Tang N, Hayakawa K (2006) Simultaneous determination of urinary hydroxylated metabolites of naphthalene, fluorene, phenanthrene, fluoranthene and pyrene as multiple biomarkers of exposure to polycyclic aromatic hydrocarbons. Anal Bioanal Chem 386:712–718
Jacob P III, Wilson M, Benowitz NL (2007) Determination of phenolic metabolites of polycyclic aromatic hydrocarbons in human urine as their pentafluorobenzyl ether derivatives using liquid chromatography-tandem mass spectrometry. Anal Chem 79:587–598
Li Z, Romanoff LC, Trinidad DA, Hussain N, Jones RS, Porter EN, Patterson DG Jr, Sjodin A (2006) Measurement of urinary monohydroxy polycyclic aromatic hydrocarbons using automated liquid-liquid extraction and gas chromatography/isotope dilution high-resolution mass spectrometry. Anal Chem 78:5744–5751
Rossbach B, Preuss R, Letzel S, Drexler H, Angerer J (2007) Biological monitoring of occupational exposure to polycyclic aromatic hydrocarbons (PAH) by determination of monohydroxylated metabolites of phenanthrene and pyrene in urine. Int Arch Occup Environ Health 81:221–229
Rossella F, Campo L, Pavanello S, Kapka L, Siwinska E, Fustinoni S (2009) Urinary polycyclic aromatic hydrocarbons and monohydroxy metabolites as biomarkers of exposure in coke-oven workers. Occup Environ Med 66:509–516
Smith CJ, Walcott CJ, Huang W, Maggio V, Grainger J, Patterson DG Jr (2002) Determination of selected monohydroxy metabolites of 2-, 3- and 4-ring polycyclic aromatic hydrocarbons in urine by solid-phase microextraction and isotope dilution gas chromatography-mass spectrometry. J Chrom B 778:157–164
Xu X, Zhang J, Zhang L, Liu W, Weisel CP (2004) Selective detection of monohydroxy metabolites of polycyclic aromatic hydrocarbons in urine using liquid chromatography/triple quadrupole tandem mass spectrometry. Rapid Commun Mass Spectrom 18:2299–2308
Grainger J, Huang W, Patterson DG Jr, Turner WE, Pirkle J, Caudill SP, Wang RY, Needham LL, Sampson EJ (2006) Reference range levels of polycyclic aromatic hydrocarbons in the US population by measurement of urinary monohydroxy metabolites. Environ Res 100:394–423
Romanoff LC, Li Z, Young KJ, Blakely NC III, Patterson DG Jr, Sandau CD (2006) Automated solid-phase extraction method for measuring urinary polycyclic aromatic hydrocarbon metabolites in human biomonitoring using isotope-dilution gas chromatography high-resolution mass spectrometry. J Chrom B 835:47–54
Onyemauwa F, Rappaport SM, Sobus JR, Gajdosova D, Wu R, Waidyanatha S (2009) Using liquid chromatography-tandem mass spectrometry to quantify monohydroxylated metabolites of polycyclic aromatic hydrocarbons in urine. J Chrom B 877:1117–1125
Food and Drug Administration (FDA). Guidance for Industry—Bioanalytical Method Validation. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf Assessed 17 April 2009
Dixon JR Jr (1998) The International conference on harmonization good clinical practice guideline. Qual Assur 6:65–74
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (1996) ICH harmonised tripartite guideline. Guidline of good clinical practice E6(R1), http://www.ich.org/MediaServer.jser?@_ID=482&@_MODE=GLB Accessed 31 March 2009
Burns DM, Dybing E, Gray N, Hecht S, Anderson C, Sanner T, O'Connor R, Djordjevic M, Dresler C, Hainaut P, Jarvis M, Opperhuizen A, Straif K (2008) Mandated lowering of toxicants in cigarette smoke: a description of the world health organization TobReg proposal. Tob Control 17:132–141
Verberk MM (1995) Biomarkers of exposure versus parameters of external exposure; practical applications in estimating health risks. Toxicology 101:107–115
Institute of Medicine (2001) Clearing the smoke: assessing the science base for tobacco harm reduction, National Academy Press, http://www.nap.edu/catalog.php?record_id=10029, Washington, D.C
Chen B, Hu Y, Zheng L, Wang Q, Zhou Y, Jin T (2007) Urinary 1-hydroxypyrene concentrations in Chinese coke oven workers relative to job category, respirator usage, and cigarette smoking. Am J Ind Med 50:657–663
Preuss R, Drexler H, Böttcher M, Wilhelm M, Brüning T, Angerer J (2005) Current external and internal exposure to naphthalene of workers occupationally exposed to polycyclic aromatic hydrocarbons in different industries. Int Arch Occup Environ Health 78:355–362
Wang Y, Zhang W, Dong Y, Fan R, Sheng G, Fu J (2005) Quantification of several monohydroxylated metabolites of polycyclic aromatic hydrocarbons in urine by high-performance liquid chromatography with fluorescence detection. Anal Bioanal Chem 383:804–809
Hecht SS, Carmella SG, Le KA, Murphy SE, Li YS, Le C, Jensen J, Hatsukami DK (2004) Effects of reduced cigarette smoking on levels of 1-hydroxypyrene in urine. Cancer Epidemiol Biomark Prev 13:834–842
Hoepfner I, Dettbarn G, Scherer G, Grimmer G, Adlkofer F (1987) Hydroxy-phenanthrenes in the urine of non-smokers and smokers. Toxicol Lett 35:67–71
Hu Y, Zhou Z, Xue X, Li X, Fu J, Cohen B, Melikian AA, Desai M, Tang MS, Huang X, Roy N, Sun J, Nan P, Qu Q (2006) Sensitive biomarker of polycyclic aromatic hydrocarbons (PAHs): urinary 1-hydroxyprene glucuronide in relation to smoking and low ambient levels of exposure. Biomarkers 11:306–318
Ichiba M, Matsumoto A, Kondoh T, Horita M, Tomokuni K (2006) Decreasing urinary PAH metabolites and 7-methylguanine after smoking cessation. Int Arch Occup Environ Health 79:545–549
Jacob J, Grimmer G, Dettbarn G (1999) Profile of urinary phenanthrene metabolites in smokers and non-smokers. Biomarkers 4:319–327
Scherer G, Westphal K, Adlkofer F (1990) Urinary mutagenicity, hydroxyphenanthrene and thioether excretion after exposure to environmental tobacco smoke. In: Kasuga H (ed) Indoor air quality. Springer, Berlin
Sithisarankul P, Vineis P, Kang D, Rothman N, Caporaso N, Strickland P (1997) Association of 1-hydroxypyrene-glucuronide in human urine with cigarette smoking and broiled or roasted meat consumption. Biomarkers 2:217–221
Siwinska E, Mielzynska D, Bubak A, Smolik E (1999) The effect of coal stoves and environmental tobacco smoke on the level of urinary 1-hydroxypyrene. Mutat Res 445:147–153
Suwan-ampai P, Navas-Acien A, Strickland PT, Agnew J (2009) Involuntary tobacco smoke exposure and urinary levels of polycyclic aromatic hydrocarbons in the United States, 1999 to 2002. Cancer Epidemiol Biomark Prev 18:884–893
Suzuki K, Yoshinaga J (2007) Inhalation and dietary exposure to polycyclic aromatic hydrocarbons and urinary 1-hydroxypyrene in non-smoking university students. Int Arch Occup Environ Health 81:115–121
Tsai HT, Wu MT, Hauser R, Rodrigues E, Ho CK, Liu CL, Christiani DC (2003) Exposure to environmental tobacco smoke and urinary 1-hydroxypyrene levels in preschool children. Kaohsiung J Med Sci 19:97–104
Van Rooij JGM, Veeger MMS, Bodelier-Bade MM, Scheepers PTJ, Jongeneelen FJ (1994) Smoking and dietary intake of polycyclic aromatic hydrocarbons as sources of interindividual variability in the baseline excretion of 1-hydroxypyrene in urine. Int Arch Occup Environ Health 66:55–65
Scherer G, Frank S, Riedel K, Meger-Kossien I, Renner T (2000) Biomonitoring of exposure to polycyclic aromatic hydrocarbons of nonoccupationally exposed persons. Cancer Epidemiol Biomark Prev 9:373–380
Hecht SS (2002) Human urinary carcinogen metabolites: biomarkers for investigating tobacco and cancer. Carcinogenesis 23:907–922
Jongeneelen FJ (2001) Benchmark guideline for urinary 1-hydroxypyrene as biomarker of occupational exposure to polycyclic aromatic hydrocarbons. Ann Occup Hyg 45:3–13
Yang M, Koga M, Katoh T, Kawamoto T (1999) A study for the proper application of urinary naphthols, new biomarkers for airborne polycyclic aromatic hydrocarbons. Arch Environ Contam Toxicol 36:99–108
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramsauer, B., Sterz, K., Hagedorn, HW. et al. A liquid chromatography/tandem mass spectrometry (LC-MS/MS) method for the determination of phenolic polycyclic aromatic hydrocarbons (OH-PAH) in urine of non-smokers and smokers. Anal Bioanal Chem 399, 877–889 (2011). https://doi.org/10.1007/s00216-010-4355-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-4355-7