
Abstract
The 7-azaindoles derivatives, as a major category of compounds, show a widespread of biological activities. The compound 5-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1) was obtained by one-step substitution reaction. In the meantime, the structure of compound 1 was confirmed by 1H NMR, 13C NMR, FTIR and X-ray diffraction. The optimized molecular crystal structure was preliminarily determined by using density functional theory (DFT) and compared with the X-ray diffraction values. In addition, more physical and chemical properties of compound 1 have been studied by further studying the molecular electrostatic potential and frontier molecular orbital of compound 1.

Similar content being viewed by others
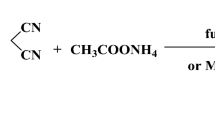
Avoid common mistakes on your manuscript.
INTRODUCTION
Indazole derivatives have been reported to have anticancer, antiviral, antibacterial, antiprotozoal, antipsychotic, anti-inflammatory, analgesic and radio sensitizing properties. Nitrogen-containing heterocyclic compounds are widely found in nature and in living organisms, and are of great interest in the fields of medicine, agriculture, functional materials and chemicals because of their high efficiency, low toxicity, environmental friendliness, and unique biological activity and structural diversity [1-4]. Novel molecularly targeted drugs usually have relatively clear targets, mainly acting on key molecules and their signal transduction pathways that regulate cell growth and proliferation, which differ greatly between normal and tumor cells, and can inhibit the growth of tumor cells while reducing the effect on normal cells, thus increasing the selectivity to tumors and reducing the toxicity to normal tissues. Among them, 7-azaindoles can inhibit the activity of various proteases in pharmacological effects, and have anticancer, antiviral, and other effects [5-8]. As widely studied intermediates of C3 and C5-substituted 7-azaindoles derivatives, 5-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (1) is available at the C3 position via Negishi reaction, Suzuki cross-coupling reaction, and at the C5 position via Buchwald-Hartwig coupling, Suzuki-Miyura coupling, which can be further modified to develop novel 7-azaindole derivatives. In addition, the synthesis method of the compound reported so far is to react under strong bases such as potassium hydroxide, sodium hydroxide and sodium hydroxide, while the method used in this paper is based on triethylamine and carried out under the condition of catalyst 4-dimethylaminopyridine (DMAP). The advantages of this method are mild conditions, simple post-treatment and high yield of the target compound, so it is suitable for industrial production. It is understood that the melting point of the measured substance can qualitatively identify the purity of the measured substance. The closer the melting point of the measured substance is to its standard melting point, the purer the substance will be. So we determined the melting point of compound 1. The melting range of compound 1 reported in the present literature is between 140-141 °C, while the melting point measured in this experiment is 138-140 °C, which is consistent with the melting range reported in the literature [9].

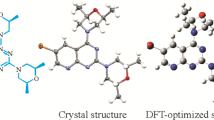
Crystal structure (a) and DFT optimized structure of compound 1 (b) and their overlap figure (c) (green: crystal structure; pink: DFT optimized structure) (see the electronic version).
In this paper, the target product 5-bromo-1-[(4-methylphenyl)sulfonyl]-1H-pyrrolo[2,3-b]pyridine was synthesized by a one-step substitution reaction (Fig. 1) using 5-bromo-7-azaindole as the starting material, based on the principles of collocation, local modification, bioelectronic equipartition and bioequivalence, while preserving the mode of action of the pharmacophore. The structure was confirmed by FTIR, hydrogen spectroscopy, carbon spectroscopy, NMR spectroscopy and mass spectroscopy. The dominant conformation was obtained by density functional theory (DFT) calculations using Gaussian09 software package in B3LYP/6-311+G(2d,p) mode [10, 11]. Crystallographic and conformational analysis of compound 1 and comparison of the structure of X-ray single-crystal diffraction with the DFT-optimized structure revealed that the structure of the title compound 1 is indeed identical to the single-crystal structure determined by experiment. In order to reveal the pharmaceutical aspects of 5-bromo-1-[(4-methylphenyl)sulfonyl]-1H-pyrrolo[2,3-b]pyridine, we also investigated the physicochemical characteristics, molecular electrostatic potential and frontier molecular orbitals of the title compound by DFT.
EXPERIMENTAL
General remarks
All the experimental materials used, including chemicals and reagents, were commercial suppliers and were used without further purification. The default TMS was an internal standard, and 13C and 1H NMR spectra (400 MHz) in DMSO-d6 solvent were recorded on a JEOL-ECX NMR spectrometer. Mass spectrometry studies were performed on the Agilent Technologies 1100 organic mass spectrometer. The UV study was performed on a UV spectrophotometer (Shanghai Yuanxi Instrument Co., Ltd.). The IR spectra of compound 1 were recorded in the 4000-400 cm–1 regions on the Bruker IFS-55V IR spectrometer (Bruker, Germany). X-ray diffraction data were recorded on a Bruker Apex II X-ray diffractometer and collected using graphite monochromatic Kα radiation (λ = 0.71073 Å). Compound 1 was synthesized by one step, and the reaction was monitored by thin layer chromatography.
Synthetic procedure
In a 500 mL single-mouth flask, 5-bromo-7-azaindole (20 g, 101.5 mmol) was dissolved in 200 mL tetrahydrofuran, and 4-dimethylaminopyridine (DMAP) (0.62 g, 5.1 mmol) and triethylamine (20.54 g, 202.6 mmol) were added sequentially. p-Toluenesulfonyl chloride was slowly added under stirring and the reaction was carried out at room temperature for 1 h. The reaction was monitored by TLC. After the reaction, the reaction solution was poured into 300 mL of water with slow stirring, a solid was precipitated, filtered, and the filter cake was washed with a little water to obtain a light yellow solid, the filter cake was dried to obtain 35.90 g of crude product, the crude product was pulped with petroleum ether to obtain 5-bromo-1-[(4-methylphenyl)sulfonyl]-1H-pyrrolo[2,3-b]pyridine as a white solid (34.30 g, yield: 96.2%). 1H NMR (400 MHz, CDCl3) 8.46(d, J = 4.0 Hz, 1H), 8.07 (d, J = 8.0 Hz, 2H), 7.98 (d, J = 4.0 Hz, 1H), 7.76 (d, J = 4.0 Hz, 1H), 7.31 (d, J = 12.0 Hz, 2H), 6.55 (d, J = 4.0 Hz, 1H), 2.39 (s, 3H); 13C NMR (101 MHz, CDCl3) δ, ppm 145.51, 145.45, 135.05, 131.72, 129.74, 128.09, 127.94, 124.43, 115.18, 104.48, 21.67.

Synthesis route of compound 1.
X-ray crystal structure determination
The crystals of compound 1 were successfully obtained by solvent evaporation method. 10 mg of compound 1 was weighed and dissolved in 5 mL of a mixture of acetonitrile and DMF, and the solution was filtered through an organonylon syringe filter (⊘ = 0.22 μm) and transferred to a 10 mL reagent bottle. The bottle was sealed with a film and three small holes of the same size were punched in the film with a clean needle; single crystals of 5-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine were obtained after standing at room temperature and slow evaporation of the solvent. Crystals that were transparent, free of cracks and had a clean and shiny surface were selected for data collection under microscopic observation, and the size of the selected crystals was 0.200×0.200×0.200 mm3. Multiple scans and absorption corrections were performed at 293 K using a Bruker APEX II X-diffractometer in scanning mode with rays (0.071073 Å). The collected diffraction data were corrected using the full matrix least squares method and then refined with the SHELXL-2017/1 program. The crystal data for compound 1 are shown in Table 1.
Quantum chemistry/DFT calculation
The DFT calculations, which for the purpose of verifying the dependability of the X-ray crystal data, were performed in the ground state (in vacuo) with Gaussian09 software package [12] by using the B3LYP method with the 6-311+G(2d,p) basis set [13]. Furthermore, based on the optimized structure [14], information such as geometrical, electronic and energy parameters, was extracted from the GuassView 5.0 program [12].
RESULTS AND DISCUSSION
Synthesis and characterization
The 13C NMR, 1H NMR spectroscopy and MS of title compound 1, as shown in Fig. S1-S3 of Supplementary Material, they all confirmed that the structure of the compound was correct.
Crystallographic analysis
Further study on the structural characteristics of the compound can better analyze its various physical and chemical properties. Therefore, X-ray diffraction is adopted to study its structure. A ORTEP diagram can clearly and intuitively display the structural characteristics of compound 1, as shown in Fig. 1. Among them, we stack the structure of the obtained compound with the theoretical calculation results, as shown in Fig. 1, the green part is the obtained crystal, and the pink part is the theoretical calculation. The crystal packing in the compound 1 is stabilized by C4–H4⋯O1 and weak C8–H8…π interactions, the perpendicular distance of different molecules is 2.765 Å, as shown in Fig. 2. In general, the crystal structure shows that the intermolecular packing is stabilized by π–π interactions, C–H…π interactions and C–H…O hydrogen bond. The crystallographic and refinement data are shown in Table 1. The experimental and theoretical values of all bond lengths, bond angles and torsion angles as well as the differences between the two values are within reasonable range, as shown in Table 2 and Table S1 of Supplementary Materials.

The crystal packing of title compound (a); π–π stacking of compound 1 and centroid–centroid distances between two molecules (b); the distance of the hydrogen bond of the title compound (c).
Conformational determination
The biological activity and physicochemical properties of a molecule is dramatically affected by its conformation [15, 16]. Therefore, what takes a salient role in further study the structure of a compound is the reliable conformational analysis. The initial conformation is very important for the study of the compound 1, and this initial conformation is searched by means of Spartan08 program [17] with an MMFF [18, 19] molecular mechanics force field. Whereafter, at the level of DFT/B3LYP/6-311+G** [20, 21], frequency calculation and geometric optimization of all possible conformations are realized using Gaussian09 software package. On the basis of the relative free energies, it was feasible for us to predicted the overall percentage of each conformation in the room-temperature equilibrium mixture. The Gibbs free energy (G), relative Gibbs free energy (ΔG = exp(–Gi/RT)) and Boltzmann distribution (Boltzmann weighting factor \(Pi=\frac{\exp (-Gi/RT)}{\sum\nolimits_{j}{\exp }(-Gj/RT)}\cdot 100\%\)) for different conformers of compound 1 are shown in Table 3.

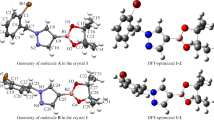
Stable conformers of compound 1.
There are two relatively stable conformers of the title compound 1 in Fig. 3, which are conformers 1-1 (56.1%), 1-2 (43.88%), and both of conformers are dramatically existed at room temperature. What the distinctions between the two conformers were principally classified into the rotational orientation of the S1–N1.
A comparison was drawn between the DFT-optimized structure and the crystal structure of the compound 1. The crystal conformation obtained using X-ray diffraction, as shown in Table 3 and Table S1, the experimental value of the crystal is nearly as same as the calculated value of the conformer 1-2. For example, bond angle C7–C2–C3, C7–C2–C1, C3–C2–C1, what the difference between the experimental values of crystal structure and the calculated values of conformer 1-2 are 0.6, –0.8, –0.03. Thus, as expected, the crystal conformation offered by X-ray diffraction is as consistent as the result of the conformer 1-2 which was calculated by DFT.
Not only the hydrogen bonds but the π–π stacking interactions were formed the molecular packing of the crystal structure of the compound 1, as clearly shown in Fig. 3. Hydrogen bonds, similar to the X–H⋯Y, can be formed between a hydrogen atom and two other atoms that are more electronegative but have smaller atomic radius. Hence, there is an inter-molecular hydrogen bonding: C4–H4…O1, which was found in the title compound 1 and played an extremely crucial part in stabilizing the molecule (Table 4). Except for the aforementioned interactions, what worth noting is that weak π–π stacking interactions have better ability to stabilize structures, including the distance between the ring formed by Cg1 and C1, between the ring formed by Cg1 and C1, between the ring formed by Cg1 and C2, as well as between the ring formed by Cg2 and C1, as a result, the perpendicular distance of two rings of two molecules respectively are dCg–Cg = 3.851 Å, dCg–Cg = 3.851 Å, dCg–Cg = 3.660 Å and dCg–Cg = 3.660 Å (Fig. 3). The detailed information of the title compound hydrogen bonds is listed in Table 4.
MEP
The B3LYP/6-311+G(2d,p) method was selected to investigate the molecular electrostatic potential of the conformer 1-2 (same as the crystal structure). Because it indicated the information about the intermolecular interaction region of the title compound 1, which was great help to further understand the compound 1. In the MEP map, as shown at the surface of the map, different colors suggest the different electrostatic potentials. And the underlying increases in the order of red < orange < yellow << green < blue. The color code of the maps in the range –5.0 e–2 (deepest red) to 5.0 e–2 (deepest blue) in title molecule surface, where red color denotes the electron rich area and blue region indicates electron deficient region. As shown in Fig. 4, The O1 and O2 atom in the 5-bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridine as well as the N1 and N2 atoms in the conformer 1-2 are surrounded by negative charges, which indicate some possible nucleophilic attack sites. Besides, the H atom of the C4 atom, C5 atom and the C6 are located on the positive charge regions.

Molecular electrostatic potential map of conformer 1-2.
FMOs
Frontier molecular orbitals investigation invariably plays an extremely important role in reactive prediction [22, 23]. As an electrons donor, on the one hand, the highest occupied molecular orbital (HOMO) is always related to potential electron delocalization directly. Indeed, the ability of charges transfer revealed from the energy of HOMO. On the other hand, the lowest unoccupied molecular orbital (LUMO) manifests an area where electrons can be accepted, and the energy of LUMO is regarded as electron affinity. What the energy of band gap point to the energy difference between this two important FMOs which implies the stability of molecular structure.
As a high-efficiency method to further investigate the chemical stability of conformer 1-2, the B3LYP/6-311+G(2d,p) was selected to calculate the energies of the highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), and their orbital energy gap were calculated. The pictorial illustration of the frontier molecular orbitals (FMOs) and their respective positive and negative regions represented by red and green colors are shown in Fig. 5. The values of LUMO and HOMO were –1.7374 eV and –6.7378 eV, respectively. The value of the energy separation between the HOMO and LUMO was 5.004 eV for conformer 1-2. The large HOMO–LUMO energy gap means high excitation energy of the excited state, good chemical stability and large hardness for the calculated conformer. In addition, the ionization energy and electron affinity can be expressed as: I = –EHOMO = 6.7378 eV, A = –ELUMO = 1.7374 eV. The hardness, which can be denoted as: η = (I – A)/2, indicates the resistance toward the deformation of the electron cloud of chemical systems under small perturbation encountered during chemical process [24-26]. Therefore, the hardness of title compound 1 is 2.5002.
Ultraviolet-visible spectral analysis
The experimental procedure of a 5 ppm concentration in the methanol was recorded. The experimental and theoretical UV-Vis spectra of compound 1 were relatively analyzed, as shown in Fig. 6, and the theoretical spectrum of compound 1 shifted by 34 nm to a higher wavelength agrees well with the experimental spectrum. For compound 1, the experimental spectra showed a strong absorption at 222 nm and a weak absorption at 241 nm and 291 nm. The strong band is obviously due to the conjugated double bond π → π* transition of the C=C double bond. The weak band is due to heteroatom N and n → π* transitions of the C–Br, C–S.

The highest occupied and lowest unoccupied molecular orbitals of conformer 1-2 obtained by the DFT/6-311+G(2d,p) method.

Theoretical and experimental UV spectra of compound 1 (a), the excited state energy (b).
The data in this paper are based on the analysis of DFT calculation of UV-Vis spectrum, we used the Gauss08 software package to perform the DFT calculation in the ground state. According to the optimized structure, the electronic, geometric and energy parameters were extracted from the GuassView 6.0 program. At the same time, we calculated the excited state of 40.
From TD-DFT calculations, the excited state energy and the corresponding absorption wavelength are shown together with the approximate curve in Fig. 6.
Vibration Analysis
It has been proved that the vibration of functional groups is bound to have different effects due to the different amplitudes of each functional group and the environment in which the molecule is located. Therefore, infrared spectroscopy is widely used to analyze and determine the structure of compounds. As shown in Fig. 7, the theoretical and experimental infrared spectra of compound 1 are clearly listed, and the measured, calculated.

Theoretical and experimental FTIR spectra of compound 1.
C–Br vibrations. Compound 1 has a bromine substituent with an absorption frequency of 690-515 cm–1 [27]. Therefore, 701.53 cm–1 in the IR spectrum can be considered as C–Br stretching vibration.
Ring vibrations. In general, the carbonyl vibrations of aromatic heterocycles such as pyridine ring, pyrrole ring, and benzene ring have about characteristic peaks around 1600 cm–1, 1580 cm–1, 1500 cm–1, and 1450 cm–1 [28]. Thus, the characteristic peaks at 1592.72 cm–1, 1513.88 cm–1, and 1439.48 cm–1 in the IR spectra can be attributed to the stretching vibrations of the aromatic rings.
For aromatic compounds containing benzene rings, absorption bands were observed between 1250 cm–1 and 950 cm–1 for C–H in-plane bending vibrations and between 900 cm–1 and 600 cm–1 for out-of-plane bending vibrations [28]. In the chromatogram of the title compound 1, the absorption band at the in-plane bending vibration band with the aromatic ring is clearly observed at 912.40 cm–1.
Ar–N vibrations. Amines with C–N bonds, such as aromatic or unsaturated amines, stretch at 1360-1250 cm–1. In secondary amines, when the aryl group is attached to the nitrogen atom, two peaks can be seen in the IR spectrum of compound 1 with C–N stretching vibrations at 1243.03 cm–1 and 1355.53 cm–1.
Sulfone (S=O=S) vibrations. The frequency of the absorption band where the sulfonyl group is located is 1340-1290 cm–1. In the IR spectrogram of compound 1, the position where the absorption band is 1335.53 cm–1 can be considered as the stretching vibration.
CONCLUSIONS
In this study, 5-bromo-1-[(4-methylphenyl)sulfonyl]-1H-pyrrolo[2,3-b]pyridine and was synthesized using a one-step reaction route. The structure of the compound was confirmed by MS and analyzed by 1H NMR spectra, IR spectra and 13C NMR spectra, in full agreement with the validation results. In addition, the conformation 1-2 obtained by DFT optimization are consistent with those determined by X-ray single crystal diffraction. The physical properties of compound 1 were further analyzed using the values of molecular electrostatic potential and frontier molecular orbitals.
REFERENCES
B. Liu, X. Yuan, B. Xu, H. Zhang, R. Li, X. Wang, Z. Ge, and R. Li. Synthesis of novel 7-azaindole derivatives containing pyridin-3-ylmethyl dithiocarbamate moiety as potent PKM2 activators and PKM2 nucleus translocation inhibitors. Eur. J. Med. Chem., 2019, 170, 1-15. https://doi.org/10.1016/j.ejmech.2019.03.003
A. Ponzi, E. Bernes, D. Toffoli, G. Fronzoni, C. Callegari, A. Ciavardini, M. Di Fraia, R. Richter, K. C. Prince, H. Sa′adeh, M. Devetta, D. Faccialà, C. Vozzi, L. Avaldi, P. Bolognesi, M. C. Castrovilli, D. Catone, M. Coreno, and O. Plekan. Carbon and nitrogen K-edge NEXAFS spectra of indole, 2,3-dihydro-7-azaindole, and 3-formylindole. J. Phys. Chem. A, 2021, 125(19), 4160-4172. https://doi.org/10.1021/acs.jpca.1c02570
M. A. Qhobosheane, R. M. Beteck, B. Baratte, T. Robert, S. Ruchaud, S. Bach, and L. J. Legoabe. Exploration of 7-azaindole-coumaranone hybrids and their analogues as protein kinase inhibitors. Chem. Biol. Interact., 2021, 343, 109478. https://doi.org/10.1016/j.cbi.2021.109478
U. K. Bandarage, M. P. Clark, E. Perola, H. Gao, M. D. Jacobs, A. Tsai, J. Gillespie, J. M. Kennedy, F. Maltais, M. W. Ledeboer, I. Davies, W. Gu, R. A. Byrn, K. Nti Addae, H. Bennett, J. R. Leeman, S. M. Jones, C. O′Brien, C. Memmott, Y. Bennani, and P. S. Charifson. Novel 2-substituted 7-azaindole and 7-azaindazole analogues as potential antiviral agents for the treatment of influenza. ACS Med. Chem. Lett., 2017, 8(2), 261-265. https://doi.org/10.1021/acsmedchemlett.6b00487
Z. Zhong, L. Shi, T. Fu, J. Huang, and Z. Pan. Discovery of novel 7-azaindole derivatives as selective covalent fibroblast growth factor receptor 4 inhibitors for the treatment of hepatocellular carcinoma. J. Med. Chem., 2022, 65(10), 7278-7295. https://doi.org/10.1021/acs.jmedchem.2c00255
C. Yang, M. Lu, Y. Chen, R. Xiang, T. Qiu, Y. Jia, Y. Yang, X. Liu, M. Deng, Y. Ling, and Y. Zhou. Development of anti-breast cancer PI3K inhibitors based on 7-azaindole derivatives through scaffold hopping: Design, synthesis and in vitro biological evaluation. Bioorg. Chem., 2021, 117, 105405. https://doi.org/10.1016/j.bioorg.2021.105405
M. A. Qhobosheane, L. J. Legoabe, B. Josselin, S. Bach, S. Ruchaud, J. P. Petzer, and R. M. Beteck. Synthesis and evaluation of 7-azaindole derivatives bearing benzocycloalkanone motifs as protein kinase inhibitors. Bioorg. Med. Chem., 2020, 28(11), 115468. https://doi.org/10.1016/j.bmc.2020.115468
R. García-Vázquez, E. P. Rebitski, L. Viejo, C. de los Ríos, M. Darder, and E. M. García-Frutos. Clay-based hybrids for controlled release of 7-azaindole derivatives as neuroprotective drugs in the treatment of Alzheimer′s disease. Appl. Clay Sci., 2020, 189, 105541. https://doi.org/10.1016/j.clay.2020.105541
Q. Zhou, A. F. Phoa, R. H. Abbassi, M. Hoque, T. A. Reekie, J. S. Font, R. M. Ryan, B. W. Stringer, B. W. Day, T. G. Johns, L. Munoz, and M. Kassiou. Structural optimization and pharmacological evaluation of inhibitors targeting dual-specificity tyrosine phosphorylation-regulated kinases (DYRK) and CDC-like kinases (CLK) in glioblastoma. J. Med. Chem., 2017, 60(5), 2052-2070. https://doi.org/10.1021/acs.jmedchem.6b01840
P. J. Stephens, F. J. Devlin, J. R. Cheeseman, and M. J. Frisch. Ab initio prediction of optical rotation: Comparison of density functional theory and Hartree–Fock methods for three 2,7,8-trioxabicyclo[3.2.1]octanes. Chirality, 2002, 14(4), 288-296. https://doi.org/10.1002/chir.10039
M. Kwit, N. D. Sharma, D. R. Boyd, and J. Gawronski. Determination of absolute configuration of conformationnally flexiblecis-dihydrodiol metabolites: Effect of diene substitution pattern on the circular dichroism spectra and optical rotations. Chirality, 2008, 20(5), 609-620. https://doi.org/10.1002/chir.20471
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox. Gaussian09, Revision C.01. Wallingford, CT, USA: Gaussian, Inc., 2009.
D. Ünlüer, Y. Ünver, E. Düğdü, Y. B. Alpaslan, Y. Köysal, M. S. Soylu, and K. Sancak. Novel 1,2,4-triazole derivatives: Structure, DFT study, X-ray analysis, and antimicrobial activity. Russ. J. Org. Chem., 2019, 55(2), 254-261. https://doi.org/10.1134/s1070428019020192
I. Romo, L. Abugoch, and C. Tapia. Soluble complexes between chenopodins and alginate/chitosan: Intermolecular interactions and structural-physicochemical properties. Carbohydr. Polym., 2020, 227, 115334. https://doi.org/10.1016/j.carbpol.2019.115334
A. Vera, M. A. Valenzuela, M. Yazdani-Pedram, C. Tapia, and L. Abugoch. Conformational and physicochemical properties of quinoa proteins affected by different conditions of high-intensity ultrasound treatments. Ultrason. Sonochem., 2019, 51, 186-196. https://doi.org/10.1016/j.ultsonch.2018.10.026
D. Marchesan, S. Coriani, C. Forzato, P. Nitti, G. Pitacco, and K. Ruud. Optical rotation calculation of a highly flexible molecule: the case of paraconic acid. J. Phys. Chem. A, 2005, 109(7), 1449-1453. https://doi.org/10.1021/jp047108b
V. V. Dotsenko, D. S. Buryi, D. Y. Lukina, and S. G. Krivokolysko. Recent advances in the chemistry of thieno[2,3-b]pyridines. 1. Methods of synthesis of thieno[2,3-b]pyridines. Russ. Chem. Bull., 2020, 69(10), 1829-1858. https://doi.org/10.1007/s11172-020-2969-2
B. Altava, M. Isabel Burguete, N. Carbó, S. V. Luis, V. Martí-Centelles, and C. Vicent. Bis(amino amides) derived from natural amino acids as chiral receptors for N-protected dicarboxylic amino acids. Tetrahedron Lett., 2013, 54(1), 72-79. https://doi.org/10.1016/j.tetlet.2012.10.099
N. Huang, C. Kalyanaraman, K. Bernacki, and M. P. Jacobson. Molecular mechanics methods for predicting protein–ligand binding. Phys. Chem. Chem. Phys., 2006, 8(44), 5166-5177. https://doi.org/10.1039/b608269f
W. Koch and M. C. Holthausen. Elementary Quantum Chemistry. In: A Chemist′s Guide to Density Functional Theory. John Wiley & Sons, 2001, Ch. 1.
P. Geerlings, F. De Proft, and W. Langenaeker. Conceptual density functional theory. Chem. Rev., 2003, 103(5), 1793-1874. https://doi.org/10.1021/cr990029p
K. Fukui, T. Yonezawa, and H. Shingu. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys., 1952, 20(4), 722-725. https://doi.org/10.1063/1.1700523
D. P. Holst, P. Friederich, A. Aspuru-Guzik, and T. P. Bender. Updated calibrated model for the prediction of molecular frontier orbital energies and its application to boron subphthalocyanines. J. Chem. Inf. Model., 2022, 62(4), 829-840. https://doi.org/10.1021/acs.jcim.1c01048
M. N. Elinson, Y. E. Ryzhkova, and F. V. Ryzhkov. Multicomponent design of chromeno[2,3-b]pyridine systems. Russ. Chem. Rev., 2021, 90(1), 94-115. https://doi.org/10.1070/rcr4972
G. Sievers, K. Österlund, and N. Ellfolk. Resonance Raman study on yeast cytochrome c peroxidase: Effect of coordination and axial ligands. Biochim. Biophys. Acta, Protein Struct., 1979, 581(1), 1-14. https://doi.org/10.1016/0005-2795(79)90215-0
X. Yang, F. Hu, Y. Wang, C. Yang, X. Zou, J. Liu, and Q. Zhang. Base-catalyzed cascade synthesis of 2,3-dihydrofuro[2,3-b]pyridines and 2,3-dihydro-1H-pyrrolo[2,3-b]pyridines from N-propargylic β-enaminones. Chem. Commun., 2017, 53(54), 7497-7500. https://doi.org/10.1039/c7cc03308g
L. Greff da Silveira, M. Jacobs, G. Prampolini, P. R. Livotto, and I. Cacelli. Development and Validation of Quantum Mechanically Derived Force-Fields: Thermodynamic, Structural, and Vibrational Properties of Aromatic Heterocycles. J. Chem. Theory Comput., 2018, 14(9), 4884-4900. https://doi.org/10.1021/acs.jctc.8b00218
S. E. Wiberley and R. D. Gonzalez. Infrared spectra of polynuclear aromatic compounds in the C–H stretching and out-of-plane bending regions. Appl. Spectrosc., 1961, 15(6), 174-177.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interests.
Additional information
Text © The Author(s), 2023, published in Zhurnal Strukturnoi Khimii, 2023, Vol. 64, No. 8, 113762.https://doi.org/10.26902/JSC_id113762
Supplementary material
Rights and permissions
About this article

Cite this article
Cheng, T., Jing, J., Yang, C.Y. et al. Synthesis, Crystal Structure and DFT Study of 5-Bromo-1-Tosyl-1H-Pyrrolo[2,3-b]Pyridine. J Struct Chem 64, 1369–1379 (2023). https://doi.org/10.1134/S0022476623080024
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476623080024