
Abstract
(2S,2′S,6R,6′R)-4,4′-(6-Bromopyrido[2,3-d]pyrimidine-2,4-diyl)bis(2,6-dimethylmorpholine) is a novel organic intermediate having pyrido[2,3-d]pyrimidine. It is synthesized by four steps and confirmed by 1H and 13C NMR and FTIR spectroscopy and MS. Meanwhile, the single crystal of the title compound is subjected to the crystallographic analysis and the conformation determination. Moreover, density functional theory (DFT) is used to calculate the optimized structures of the molecule which are compared with the X-ray measurement. The result of the molecular structure optimized by DFT is consistent with the crystal structure determined by single crystal X-ray diffraction. Finally, in order to further investigate some physical properties of the title compound by the B3LYP/6-311G(2d,p) method, the molecular electrostatic potential and frontier molecular orbitals are calculated. The calculated and experimental data show that the title compound has good chemical stability and nucleophilic reactivity. Hirshfeld surface analyses can explain the atom pair contacts of the crystal and the quantitative analysis of intermolecular interactions is performed.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Pyrido[2,3-d]pyrimidine derivatives are a class of compounds with potential biological and pharmacological activities. Compounds bearing a pyrido[2,3-d]pyrimidine scaffold possess a wide range of biological properties, such as antitumoral [1], antiviral [2], antiproliferative [3], antihypertensive [4], cardiotonic [5], antifungal [6], anti-inflammatory [7], and antihistaminic [8]. Some pyrido[2,3-d]pyrimidine derivatives have been confirmed to have biological activities. For instance, compound 2 [9] can inhibit breast cancer cells, compound 3 [10] was found to have an excellented antimicrobial activity; compound 4 [11] showed significant potent analgesic activities; and compound 5 [12] exhibited a superior antitumor activity (Fig. 1). Now, a number of pyrido[2,3-d]pyrimidine compounds with a high activity and a high selectivity, which can be developed as medicines or pesticides, are also constantly being discovered. The synthesis and research of pyrido[2,3-d]pyrimidine derivatives are in the ascendant in the chemical circles at home and abroad. This is a very meaningful subject with good application prospects. We believe that in the near future, pyrido[2,3-d]pyrimidine compounds will occupy a very important position in the field of organic synthesis and related research.

Structures of pyrido[2,3-d]pyrimidine derivatives.
At present, there have been many reports on the research of pyrido[2,3-d]pyrimidine derivatives, but crystal structure studies of pyrido[2,3-d]pyrimidine derivatives have never been reported. We studied the crystal structures of pyrido[2,3-d]pyrimidine derivatives, which are of great significance for the development of medicine. In this study we designed and synthesized (2S,2′S,6R,6′R)-4,4′-(6-bromopyrido[2,3-d]pyrimidine-2,4-diyl)bis(2,6-dimethylmorpholine) (1), a novel pyrido[2,3-d]pyrimidine compound, by substitution and cyclization reactions (Scheme 1). Its structure was confirmed by 1H and 13C NMR, FTIR spectroscopy and MS. In addition, the crystal structure of title compound 1 was confirmed by X-ray diffraction (XRD) and subjected to the conformational analysis. Moreover, density functional theory (DFT) was used to calculate the optimized structure of the molecule, which was compared with the X-ray measurement. The results showed that the molecular structure optimized by DFT is consistent with the crystal structure determined by single crystal XRD.

Synthetic route for 1.
EXPERIMENTAL
General remarks
All the chemicals and reagents used were purchased from commercial supplies. Using TMS as an internal standard, 1H and 13C NMR spectra (400 MHz) spectra were recorded in the DMSO-d6 solvent on a JEOL-ECX NMR spectrometer. MS studies were conducted on an Agilent 1100 organic mass spectrometer. The IR spectrum of compound 1 was recorded in the range of 4000-400 cm–1 on a Bruker IFS-55V IR spectrometer (Bruker, Germany). The XRD data were collected on a Bruker APEX II X-diffractometer with graphite monochromatized Kα radiation (λ = 0.71073 Å). The reactions mentioned here were monitored by TLC. The crude products synthesized were purified by recrystallization and column chromatography. TLC was performed on GF254 silica gel.
Synthetic procedure
Synthesis of 2-amino-5-bromonicotinic acid (7). 2-Aminonicotinic acid (100 g, 724 mmol) and glacial acetic acid (500 mL) were added to a 1000-mL three-necked flask in an ice-water bath at 0 °С. Then liquid bromine (37.1 mL, 724 mmol) was slowly added dropwise with stirring. After the completion of the dropwise addition, the reaction solution was heated to room temperature and refluxed for 5 h. The yellow solid precipitated in the reaction was filtered. The filter cake was washed with glacial acetic acid (100 mL). Then glacial acetic acid in the product was washed away with water (50 mL) to obtain 7 (151.1 g, yield 93.8%) as a white solid.
Synthesis of 6-bromopyrido[2,3-d]pyrimidine-2,4-diolee (8). Compound 7 (100 g, 461 mmol) and urea (276.7 g, 4610 mmol) were added to a 1000-mL three-necked flask. After stirring for 6 h at 160 °С, the reaction solution was cooled to 100 °С and quenched by adding water. At this temperature excess urea in the reaction solution was dissolved. A large amount of a white solid precipitated out when the reaction mixture was cooled to room temperature. Then, it was filtered, the filter cake was washed with saturated NaOH (150 mL), proper glacial acetic acid was added to the filtrate, and pH was adjusted to 6 to 7. After filtration, the filter cake was dried to obtain compound 8 (103.2 g, yield 93.8%) as a white solid.
Synthesis of 6-bromo-2,4-dichloropyrido[2,3-d]pyrimidine (9). Compound 8 (80 g, 330.5 mmol) and chlorobenzene (200 mL) were added to a 1000-mL three-necked flask. Subsequently, N,N-diisopropylethylamine (8.5 g, 66.1 mL) and POCl3 (308.1 mL, 3305 mmol) were slowly added to the flask. Then the reaction was stirred for 12 h at 110 °С under nitrogen. The reaction mixture was poured into ice water (150 mL) to afford a black solid. Then, it was filtered, the filter cake was washed with CH2Cl2, proper saturated NaHCO3 was added to the filtrate, and pH was adjusted to 8. The mixed solution was extracted with CH2Cl2 (100 mL×3). The extraction fluid was purified by silica gel column chromatography (CH2Cl2 as eluent) to obtain compound 9 (68.1 g, yield 73.3%) as a brown-red solid.
Synthesis of (2S,2′S,6R,6′R)-4,4′-(6-bromopyrido[2,3-d]pyrimidine-2,4-diyl)bis(2,6-dimethylmorpholine) (1). (2R,6S)-2,6-Dimethylmorpholine (12.4 mL, 106.7 mmol) was slowly added dropwise to a solution of compound 9 (15 g, 53.8 mmol) and triethylamine (16.3 g, 161.4 mmol) in CH2Cl2 (30 mL) in MeOH (200 mL). After stirring for 2 h at room temperature and quenching with water, the reaction solution was filtered. The filter cake was dried to obtain title compound 1 (21.4 g, yield 92.5%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H, 2-CH), 8.28 (s, 1H, 4-CH), 4.59 (d, J = 11.5 Hz, 2H, 9-CH, 11-CH), 4.09 (d, J = 12.8 Hz, 2H, 8″-CH, 12″-CH), 3.70 (s, 2H, 14″-CH, 18″-CH), 3.55 (s, 2H, 15-CH, 17-CH), 2.91 (t, J = 11.7 Hz, 2H, 8′-CH, 12′-CH), 2.58 (d, J = 12.0 Hz, 2H, 14′-CH, 18′-CH), 1.15 (dd, J = 12.8, 6.1 Hz, 12H, 10-CH, 13-CH, 16-CH,19-CH). 13C NMR (100 MHz, DMSO-d6) δ 164.26 (C6), 161.06 (C7), 159.45 (C5), 156.11 (C2), 136.52 (C3), 109.69 (C1), 106.97 (C4), 71.53 (C9, C11), 71.21 (C15, C17), 54.74 (C8, C12), 49.48 (C14, C18), 19.21(C10, C13), 19.07 (C16, C19). MS (ESI): m/z = 437.1389 [M+H]+.
X-ray crystal structure description
The crystals of title compound 1 were obtained by solvent evaporation. An appropriate amount of title compound 1 as a light yellow powder was dissolved in acetone/DMF and filtered. The filtrate was placed into the sample tube, and the tube mouth was covered with a plastic film with several small holes. After a period of time, the solvent was slowly evaporated at room temperature, and a single crystal of title compound 1 was obtained. The colorless transparent crystal with dimensions of 0.15×0.08×0.05 mm was selected and placed on the Bruker APEX II diffractometer for data collection. Using graphite monochromatized MoKα radiation (λ = 0.71073 Å), 15435 diffraction spots of 4.006° to 52.044° (index ranges: –11 ≤ h ≤ 11, –15 ≤ k ≤ 14, –22 ≤ l ≤ 21) were collected at 170.0 K by ω/2θ scanning; 3924 were independent (Rint = 0.0864, Rσ = 0.0875), out of which 2186 were observed with I > 2σ(I). The crystal was kept at 170.0 K during data collection. Using Olex2 [13], the structure was solved with the ShelXT [14] structure solution program using Intrinsic Phasing and refined with the ShelXL [15] refinement package with the least squares minimization. Final R = 0.0639 and wR = 0.1831 \((w=1/[{{\sigma }^{2}}(F_{\,0}^{2})+{{(0.0758P)}^{2}}+3.0949P],\ \ \text{where}\ \ P=(F_{\,\text{0}}^{2}+2F_{\,\text{c}}^{2})/3,\) (Δ/σ)max = 0.000, S = 1.032, (Δρ)max = 0.601 and (Δρ)min = –0.508 e/Å3.
The single crystal XRD data for compound 1 have been deposited with the Cambridge Crystallographic Data Center (CCDC 2060554). It can be freely obtained through the request at the website: www.ccdc.cam.ac.uk/data_request/cif.
DFT calculation
In order to verify the reliability of the XRD crystal data, we used the Gauss09 software package to perform the DFT calculation in the ground state (vacuo). Based on the optimized structures, geometrical, electronic, and energy parameters were excerpted from the GuassView 5.0 program [16].
RESULTS AND DISCUSSION
Synthesis and characterization
Title compound 1 was obtained in four steps: substitution, cyclization, substitution, and again substitution. Its structure was confirmed by 1H and 13C NMR, FTIR spectroscopy and MS, which is shown in Supporting Materials S1–S4 (Supplementary Materials).
Crystallographic analysis
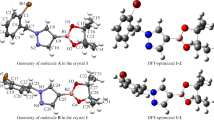
In order to further understand the structural characteristics of compound 1, it was analyzed by XRD. The measured values show that compound 1 has the monoclinic system with the P21/n space group (unit cell dimensions: a = 9.2481(7) Å, b = 12.4782(10) Å, c = 17.8247(14) Å). Experimental and DFT-optimized crystal structures of compound 1 are shown in Fig. 2. Crystal data and structure refinement for compound 1 are shown in Supporting Materials (Table S1, Supplementary Materials).
Bond lengths, bond and torsion angles in the crystal structure are within the normal range (Table S2, Supplementary Materials). From these data we can clearly see that the bond lengths, bond and torsion angles from the XRD data and calculated by DFT are different. This is because the DFT calculation is performed for a single molecule, and the X-ray crystal will be affected by the interaction between other molecules. The dihedral angle of the 2-substituted 2,6-dimethylmorpholine ring plane and the pyrido[2,3-d]pyrimidine ring plane is 8.57°, and the dihedral angle of the pyrido[2,3-d]pyrimidine ring plane and the 4-substituted 2,6-dimethylmorpholine ring plane is 53.81°, which indicate that 2,6-dimethylmorpholine and the pyrido[2,3-d]pyrimidine ring planes are not in the same plane.
The crystal molecular packing of compound 1 was formed by hydrogen bonds and π–π stacking interactions (Fig. 3). In the hydrogen bonded crystals, the bonding force mainly depends on the hydrogen atom and the two atoms with a large electronegativity and a small atomic radius to form X–H⋯Y. The hydrogen bond in the molecular structure of compound 1 includes two intermolecular C11–H11⋯N1 (2.60 Å), C17–H17⋯N1 (2.62 Å) hydrogen bonds and intramolecular C14–H14A⋯N4 (2.33 Å) hydrogen bonds (Table 1). It is worth noting that the crystal packing is further stabilized by two weak π–π stacking interactions (dCg–Cg = 3.523 Å) between N1–C2–C1–C3–C4–C5 and N2–C5–C4–C6–N4–C7. The sum of these interactions results in the formation of a three-dimensional framework (Fig. 3).

Experimental and DFT-optimized crystal structures of 1.

Crystal structure stacking (a) and hydrogen bonding (b) diagrams of 1.
Conformational determination
A reliable conformational analysis can enable us to further investigate the physical and chemical properties of compound 1. We first used the Spartan08 program [17] with a molecular mechanics force field (MM FF) [18, 19] to search for the initial conformation of compound 1, which was geometrically optimized and frequencies were calculated using DFT/B3LYP/6-311G** in the Gaussian09 package [16]. According to the relative free energy, we predicted the percentage of each conformation in a mixture equilibrated at room temperature. Table 2 shows the Gibbs free energy (G), the relative Gibbs free energy (ΔG = exp(–(Gi/RT)), and the Boltzmann weighting factor \(\left( Pi=\frac{\exp (-Gi/RT)}{\sum\nolimits_{j}{\exp }(-Gi/RT)}\cdot 100\ \% \right)\) of the two conformers of compound 1.
Four conformers of compound 1 (Fig. 4) are: 1-1 (33.83%), 1-2 (32.73%), 1-3 (24.70%), and 1-4 (8.74%). The difference between the three conformers is mainly caused by the orientation of the 4-substituted 2,6-dimethylmorpholine ring. The C6–N5 bond was used as a spin key to rotate the 2,6-dimethylmorpholine group by 360°, and the potential energy surface of the molecule was scanned (Fig. 5). It is interesting that conformers 1-2 (32.83%), 1-3(24.70%), and 1-4 (8.74%) belong to one local minimum.
The DFT-optimized structure was compared with the experimental crystal structure of compound 1. The crystal conformation obtained by XRD (Fig. 2) is consistent with that of conformer 1-3 calculated by DFT. For the C3–C4–C5, C3–C4–C6, C5–C4–C6 bond angles the experimental values are 117.8°, 126.3°, 115.6°, respectively, which are similar to the calculated values of conformer 1-3 of 118.3521°, 125.9202°, 115.5074°. From most of the geometric parameters (bond lengths, bond and torsion angles given in Table S2, Supplementary Materials), we can clearly see that the experimental value of the crystal is almost the same as the calculated value of conformer 1-3.

Relatively stable conformers of 1.

Potential energy surface scan curve of 1.
Molecular electrostatic potential (MEP)
In order to understand the interaction between the molecules and the reaction sites in the molecules of compound 1, MEP of conformer 1-3 was investigated using the B3LYP/6311G (2d,p) method. Different colors indicate different electrostatic potentials at molecular surfaces in the MEP diagram, and the electrostatic potential increases in the order red < orange < yellow < green < blue (see the electronic version). As shown in Fig. 6, the O1 and O2 atoms in the 2,6-dimethylmorpholine ring and the N1 and N2 atoms in the pyridine [2,3-d] pyrimidine ring of conformer 1-3 are surrounded by a negative charge, which may become nucleophilic attack sites. Moreover, the positive charge regions were localized on the hydrogen atoms of C12 and C18 atoms.
Frontier molecular orbitals (FMOs)
Using the B3LYP/6-311G(2d,p) method, we calculated the energies of the lowest unoccupied molecular orbital (ELUMO), the highest occupied molecular orbital (EHOMO), and their orbital energy gap (ΔE) to further investigate the structure stability of conformer 1-3. FMOs and their respective positive and negative regions in red and green colors are illustrated in Fig. 7. The ELUMO and EHOMO values are –2.0643 eV and –5.8534 eV, respectively, and their orbital energy gap is –3.7891 eV for conformer 1-3. The large HOMO–LUMO energy gap means a good chemical stability, high hardness and excitation energy of the excited state for the calculated conformer.

MEP diagram of conformer 1-3.

HOMO and LUMO of conformer 1-3.
Hirshfeld surface analyses
Hirshfeld surfaces analyses can clearly show the quantitative analysis of intermolecular interactions and contacts caused by supermolecular accumulation in crystals. The dnorm Hirshfeld curvatures for conformer 1-3 were displayed using a red-blue-white color scheme in Fig. 8a (see the electronic version), where red highlights shorter contacts, blue depicts areas with no interaction contacts, and white shows contacts around the Van der Waals separation. Large red spots indicate the existence of C–H⋯O and N⋯H hydrogen bonds on the surface. Breakdown of the Hirshfeld surfaces (fingerprint plot and dnorm property) into the contributions from H⋯H (58.3%), C⋯H (6.2%), O⋯H (5.9%) and N⋯H (10.6%) interactions, respectively, is shown in Fig. 8b–e.

Hirshfeld surface analyses mapped with dnorm of conformer 1-3 (a); 2D fingerprint plots of conformer 1-3 resolved into H⋯H contacts (b), C⋯H contacts (c), O⋯H contacts (d), and N⋯H contacts (e).
CONCLUSIONS
In this paper, (2S,2′S,6R,6′R)-4,4′-(6-bromopyrido[2,3-d]pyrimidine-2,4-diyl)bis(2,6-dimethylmorpholine) as a novel compound was synthesized in four steps. Its structure was confirmed by 1H and 13C NMR, FTIR spectroscopy, MS, and single crystal XRD. We conducted the conformational analysis and crystallographic studies on the title compound. Their data demonstrate an excellent correlation. The calculated data and experimental data on MEP and FMOs show that title compound 1 has good chemical stability and nucleophilic reactivity. Hirshfeld surface analyses can explain the atomic pair contacts of the crystal and the quantitative analysis of intermolecular interactions.
REFERENCES
A. Gangjee, O. Adair, and S. F. Queener. J. Med. Chem., 1999, 42, 2447-2455. https://doi.org/10.1021/jm990079m
V. Darias, S. S. Abdallah, M. L. Tello, L. D. Delgado, and S. Vega. Arch. Pharm., 1994, 327, 779-783. https://doi.org/10.1002/ardp.19943271205
C. Zhang, G. Sun, Q. Peng, S. Zhu, and D. Ni. RSC. Adv., 2016, 6, 73953–73958. https://doi.org/10.1039/C6RA17032C
B. Schmidt and B. Schieffer. J. Med. Chem., 2003, 46, 2261-2270. https://doi.org/10.1021/jm0204237
S. Furuya and T. Ohtaki. EP Patent 0608565A1, 1994.
M. M. Gineinah, M. N. A. Nasr, S. M. I. Badr and W. M. El-Husseiny. Med. Chem. Res., 2013, 22, 3943-3952. https://doi.org/10.1007/s00044-012-0396-0
S. P. Satasia, P. N. Kalaria, and D. K. Raval. Org. Biomol. Chem., 2014, 12, 1751-1758. https://doi.org/10.1039/C3OB42132E
J. M. Quintela, C. Peinador, L. Botana, M. Estévez, and R. Riguera. Bioorg. Med. Chem., 1997, 5, 1543-1553. https://doi.org/10.1016/S0968-0896(97)00108-9
R. Edupuganti, Q. Wang, C. D. J. Tavares, C. A. Chitjian, J. L. Bachman, P. Ren, E. V. Anslyn, and K. N. Dalby. Bioorg. Med. Chem., 2014, 22, 4910-4916. https://doi.org/10.1016/j.bmc.2014.06.050
B. Veeraswamy, D. Madhu, G. J. Dev, Y. Poornachandra, G. S. Kumar, C. G. Kumar, and B. Narsaiah. Bioorg. Med. Chem. Lett., 2018, 28, 1670-1675. https://doi.org/10.1016/j.bmcl.2018.03.022
S. R. Dasari, S. Tondepu, L. R. Vadali, and N. Seelam. Synth. Commun., 2020, 50, 2950-2961. https://doi.org/10.1080/00397911.2020.1787449
M. Fares, S. M. Abou-Seri, H. A. Abdel-Aziz, S. E.-S. Abbas, M. M. Youssef, and R. A. Eladwy, Eur. J. Med. Chem., 2014, 83, 155-166. https://doi.org/10.1016/j.ejmech.2014.06.027
O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, and J. A. K. Howard, H. Puschmann. J. Appl. Crystallogr., 2010, 42, 339-341.
G. M. Sheldrick. Acta Crystallogr., Sect. A, 2015, 71, 3-8. https://doi.org/10.1107/S2053273314026370
G. M. Sheldrick. Acta Crystallogr., Sect. C, 2015, 71, 3-8. https://doi.org/10.1107/S2053229614024218
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci,G. Petersson. Gaussian09, Revision A.01. Gaussian Inc.: Wallingford, CT, 2009.
B. J. Deppmeier, A. J. Driessen, T. S. Hehre, W. J. Hehre, J. A. Johnson, P. E. Klunzinger, J. M. Leonard, I. N. Pham, W. J. Pietro, and J. Yu. Spartan08. Wavefunction: Irvine, CA, 2009.
H. H. Brintzinger, M. H. Prosenc, F. Schaper, A. Weeber, and U. Wieser. J. Mol. Struct., 1999, 485/486, 409-419. https://doi.org/10.1016/S0022-2860(99)00184-2
N. Huang, C. Kalyanaraman, K. Bernacki, and M. P. Jacobson. Phys. Chem. Chem. Phys., 2006, 8, 5166-5177. https://doi.org/10.1039/b608269f
Funding
We have gratefully acknowledged the Guizhou Provincial Natural Science Foundation ([2020]1Y393) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Text © The Author(s), 2021, published in Zhurnal Strukturnoi Khimii, 2021, Vol. 62, No. 10, pp. 1606-1615.https://doi.org/10.26902/JSC_id80776
Supplementary material
Rights and permissions
About this article

Cite this article
Chen, Y.M., Chen, D.M., Wu, Q.M. et al. SYNTHESIS, CRYSTAL STRUCTURE AND DFT STUDY OF NOVEL (2S,2′S,6R,6′R)-4,4′-(6-BROMOPYRIDO[2,3-d]PYRIMIDINE-2,4-DIYL)BIS(2,6-DIMETHYLMORPHOLINE). J Struct Chem 62, 1501–1510 (2021). https://doi.org/10.1134/S0022476621100036
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476621100036