
Abstract
1-(2-Bromobenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole is an organic intermediate with both pyrazole heterocycle and borate functional group. In this paper, the title compound is obtained by the nucleophilic substitution reaction. The structure of the compound is confirmed by FTIR, 1H and 13C NMR spectroscopy, and MS. At the same time, the single crystal of the title compound is measured by X-ray diffraction and is subjected to the crystallographic and conformational analysis. The molecular structure is further calculated using Density Functional Theory (DFT) and compared with the X-ray diffraction value. The results of the conformational analysis indicate that the molecular structure optimized by DFT is consistent with the crystal structure determined by single crystal X-ray diffraction. In addition, the molecular electrostatic potential and frontier molecular orbitals of the title compound are further investigated by DFT, revealing molecular structure characteristics and molecular conformations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Nitrogen-containing heterocycle compounds are a very important class of compounds [1] with good biological activity and stability, which are widely distributed in nature and play an important role in the metabolism of organisms [2]. Pyrazole and its derivatives, as an extremely important class of nitrogen-containing heterocyclic compounds, have a broad antibacterial spectrum, strong efficacy, low toxicity, and unique mechanism of action. Due to its various biological activities, such as antitumor, antivirus, and antibacterial, it has become important raw materials and intermediates for the development of drugs [3]. For example, piroxib has a bactericidal effect [4]; celecoxib can selectively inhibit COX-2; clonazolic acid has an anti-inflammatory effect [5]; and so on. Arylboronic acid and arylborate ester compounds have the advantages such as good thermal stability, functional group compatibility and insensitivity to air and water. They are convenient for storage and have good reactivity, having become important intermediates in the fields of medicine, organic synthesis and fine chemicals [6]. In particular, they can participate in the Suzuki reaction to synthesize complex and active organic molecules which are widely used in the fields of natural product synthesis, pharmaceuticals, electronics industry, and advanced materials [7]. Title compound 1-(2-bromobenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1) possesses the characteristics of both pyrazole heterocycle and borate functional group, therefore, it is of great research significance to study its synthesis, the crystal structure and perform the conformational analysis.
Based on the above viewpoints, in this research we designed and synthesized title compound 1 by the nucleophilic substitution reaction (Scheme 1). Its structure was confirmed by FTIR, 1H and 13C NMR spectra, and MS. The single crystal X-ray diffraction of the title compound was performed, and the optimized structure of this molecule was calculated by the DFT/B3LYP method with the 6-311G(2d,p) basis set [8]. Then, the title compound was subjected to the conformational analysis, and the structure measured by X-ray diffraction was compared with the structure optimized by the DFT method. The results show that the molecular structure optimized by DFT is consistent with the crystal structure determined by single crystal X-ray diffraction. In addition, the molecular electrostatic potential and frontier molecular orbitals of the title compound were further studied by DFT.

Synthesis of title compound 1.
EXPERIMENTAL
General remarks. MS were taken in ESI mode on Agilent 1100 LC-MS (Agilent Technologies, Palo Alto, CA, USA). 1H and 13C NMR spectra were recorded on a Bruker ACF-400, 400 MHz spectrometer (Bruker Bioscience, Billerica, MA, USA) with TMS as the internal standard. The IR spectrum of the title compound was recorded in the range 4000-500 cm–1 using a KBr pellet technique with a 1.0 cm–1 resolution on a Bruker IFS-55V IR spectrometer (Bruker, Germany). The X-ray diffraction data on the title compound were recorded using a Bruker APEX II X-diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) at 170 K. For the title compound: data collection APEX2; cell refinement SAINT; program used to solve the structure SHELXS-2018/3; program used to refine the structure and draw molecular figures SHELXL-2018/3. All reagents and solvents for the synthesis and analysis were of analytical grade and used without further purification as purchased.
Synthetic procedure. 4-Pyrazoleboronic acid pinacol ester (2) (13.00 g, 67.00 mmol) was added to a 500-mL three-necked flask at room temperature, followed by the acetonitrile solvent (150 mL), 2-bromobromobenzyl (3) (15.22 g, 60.90 mmol), and potassium carbonate (4) (12.63 g, 91.40 mmol) (Scheme 1). The reaction mixture was heated to 90 °C and refluxed for 8 h. After the reaction was completed, the reaction mixture was poured into proper water and extracted with ethyl acetate (50 mL ×3). Then the organic phase was dehydrated with anhydrous sodium sulfate and concentrated at reduced pressure to obtain a yellow powder. The crude was washed by petroleum ether (10 mL ×2) to obtain 1 (13.6 g, yield 65%) as light yellow powder. 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H, –NCH=), 7.78 (s, 1H, –NCH=), 7.62 (d, J = 7.9 Hz, 1H, Ph–H), 7.32 (d, J = 6.9 Hz, 1H, Ph–H), 7.22 (t, J = 7.5 Hz, 1H, Ph–H), 7.06 (d, J = 7.5 Hz, 1H, Ph–H), 5.48 (s, 2H, –CH2–), 1.36 (s, 12H, –CH3).
X-ray crystal structure determination. The light yellow powder of title compound 1 was dissolved in hexane and the solvent was slowly evaporated under air at room temperature. A few days later, a single crystal of the title compound suitable for the X-ray crystallographic analysis was obtained. A colorless transparent crystal with dimensions 0.12×0.08×0.05 mm was selected to be mounted on the glass fiber in a random direction for data collection. The single crystal X-ray diffraction of the title compound was carried out on a Bruker APEX II diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å) at 170 K. A total of 27494 reflections were collected in the range from 2.336° to 27.476° (index ranges: –113 ≤ h ≤ 16, –17 ≤ k ≤ 28, –30 ≤ l ≤ 27) in a φ–ω scan mode; 7709 were independent with Rint = 0.0712, out of which 4478 observed reflections with I > 2σ(I) were used in the structure determination and refinements [9]. The structures were solved by direct methods using SHELXS-2018/3 [10] and refined by full-matrix least-squares procedure on F2 for all data using SHELXL-2018/3 [11]. The hydrogen atoms were determined by theoretical calculations. Final R = 0.0503 and wR = 0.1256 \((w=1/[{{\sigma }^{2}}(F_{\,0}^{2})+{{(0.0404P)}^{2}}+5.0967P],\) where \(P=(F_{\,\text{0}}^{2}+2F_{\,\text{c}}^{2})/3),\) (Δ/σ)max = 0.000, S = 1.009, (Δρ)max = 1.127 and (Δρ)min = –1.160 e/Å3.
The results of the single crystal X-ray diffraction analysis of compound 1 have been deposited with the Cambridge Crystallographic Data Centre (ССDC 1981713), and can be freely obtained by request via the web-site: www.ccdc.cam.ac.uk/data_request/cif.
Quantum chemistry/DFT calculation. The DFT calculations were performed in the ground state (in vacuo) with the Gaussian 09 software package [12] using the B3LYP method with the 6-311G(2d,p) basis set [8]. The geometrical, electronic, and energy parameters were extracted from the GuassView 5.0 program [13] based on the optimized structures [14].
RESULTS AND DISCUSSION
Synthesis and characterization. Title compound 1 was obtained expectedly by a one-step nucleophilic substitution reaction and its structure was confirmed by FTIR, 1H and 13C NMR spectroscopy, and MS (Fig. S1–S4, Supplementary Materials).
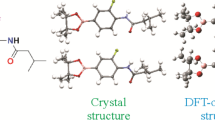
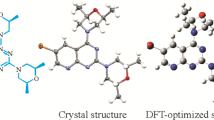
Conformational determination. In order to further investigate the structural characteristics of the synthesized compound, title compound 1 was subjected to the crystal structure X-ray diffraction analysis. The measured values reveal that 1 possess a monoclinic crystal system with the Pbca space group (unit cell dimensions: a = 13.0155(5) Å, b = 22.3880(8) Å, c = 23.4875(7) Å). There are two molecules in the asymmetric unit. The ORTEP diagram of the molecular structures and the atomic numbering schemes of 1 are shown in Fig. 1. The crystallographic and refinement data are shown in Supplementary Material, Table S1.

Crystal and DFT-optimized structures of compound 1.
The crystal structure of 1 was compared with the DFT optimized structures. Among all the conformers of 1-1, 1-2 were in accordance with molecules A and B, respectively. Some selected experimental and calculated geometry parameters for 1 are listed in Tables 1, 2.
Compound 1 crystallizes in the space group Pbca with Z = 16 and the conformer of the A crystal structure is shown in Fig. 1. In conformer A, the N1–C7 (1.457 Å) and N1–C8 (1.349 Å) bond lengths are shorter than a normal N–C single bond (1.470 Å). The O1–B1 bond length (1.374 Å) is close to the normal O–B bond (1.380 Å) and longer than O2–B1 (1.366 Å); N1–N2 (1.357 Å) is shorter than the normal N–N single bond (1.450 Å); N2–C10 (1.324 Å) is shorter than the normal N–N single bond (1.480 Å). The Br1–C1 bond length (1.929 Å) is close to the normal Br–C bond (1.920 Å). Bromobenzene and the pyrazole ring are in different planes (Fig. 2).

Dihedral angle of conformers A and B.
For conformer B, the spatial structure is similar to that of conformer A. The crystal structure of conformer B is shown in Fig. 1. The N3–C23 (1.448 Å) and N3–C24 (1.350 Å) bond lengths are shorter than the normal N–C single bond (1.470 Å). The O3–B2 (1.373 Å) and O4–B2 (1.372 Å) bond lengths are close to that of the normal O–B bond (1.380 Å); N3–N4 (1.358 Å) is shorter than the normal N–N single bond (1.450 Å); N4–C26 (1.324 Å) is shorter than the normal N–N single bond (1.480 Å). The Br2–C17 (1.927 Å) bond length is close to that of the normal Br–C bond (1.920 Å). The bromobenzene and pyrazole ring planes are orthogonal (Fig. 2).
The 1 molecules are linked by two intermolecular C7–H7A…Br(1) (2.726 Å), C24–H24…N2 (2.395 Å) hydrogen bonds and two intramolecular C21–H21…N3 (2.512 Å), C32–H21C…Br2 (2.929 Å) hydrogen bonds (Table 3). The hydrogen bonding plays an important role in the stability of the crystal structure. In addition to the afore-described interactions, it is worth noting that three π–π stacking interactions are found in three crystal structures. The distance between H7A and the benzene ring was found to be 2.894 Å. The distance between H23A and the benzene ring was found to be 2.845 Å. The distance between H 14A and the benzene ring was found to be 2.981 Å (Fig. 3d). Hence, the crystal structure is further stabilized by three π–π stacking interactions.
The molecular conformation seriously influences its physical and chemical properties. Therefore, a reliable conformational analysis plays a key role in the understanding of the structure. Initial conformational searching for title compound 1 was performed by the Spartan 08 program [15] with the MMFF [16, 17] molecular mechanics force field. Then, the geometry optimizations and frequency calculations of all the possible conformers were performed at the DFT/B3LYP/6-311G** level in the Gaussian 09 package [8, 13]. Based on the relative free energies, the percentage of each conformer can be predicted in the equilibrium mixture at room temperature (T = 295.15 K). The Gibbs free energy (G) for different conformers of compound 1 are shown in Table 4.

Crystal structure stacking diagram and hydrogen bonding diagram of compound 1: crystal packing of the title compound (a), the hydrogen bond length of the title compound (b), π–π stacking of compound 1 and cen-troid–centroid distances between two molecules (c), the π bond length of the title compound (d).
The three conformers of compound 1 are given in Fig. 4. The difference between the three conformers is mainly caused by the orientation of the bromobenzene group. The C6–C7 bond was used as a spin key to rotate the bromo benzene group by 360°, and the potential energy surface of the molecule was scanned (Fig. 4). Compound 1 was detected to have two relatively stable conformers 1-1 (58.02%), 1-2 (39.55%), and relatively unstable conformer 1-3 (2.43%).
Molecular electrostatic potential (MEP). MEP can affect the binding ability of the drug molecule with the target protein, and it also can affect the reaction performance of the compound, therefore the MEP study is essential. To gain information on the region in which conformers 1-1 and 1-2 (the major conformers) undergo intermolecular interactions, MEP was investigated by the B3LYP/6311G(2d,p) method [8]. In the MEP map, different electrostatic potentials on the surface are represented by different colors, and the potential increases in the order red < orange < yellow < green < blue (see electronic version). The color code of the maps ranges from –5.0 e–2 (deepest red) to 5.0 e–2 (deepest blue) on the title molecule surface. As shown in Fig. 5a, the positive charge regions are located on the H atom attached to the C3 atom. As shown in Fig. 5b, the positive charge regions are located on the H atom attached to the C(18) atom. The imine nitrogen atom (N2 in molecule A and N4 in molecule B) is surrounded by negative charges, hence, the imine nitrogen atom is the preferred site of the nucleophilic attack.

Relatively stable and relatively unstable conformers of compound 1.

MEP map of conformer A, MEP map of conformer B.
Frontier molecular orbitals (FMOs). The FMO investigation is frequently important in the reactive prediction [18, 19]. The highest occupied molecular orbital (HOMO), which is regarded as an electron donor, is directly related to potential electron delocalization. The HOMO energy reveals the ability of charge transfer. On the other hand, the lowest unoccupied molecular orbital (LUMO) describes an area where electrons can be accepted, and the LUMO energy is regarded as the electron affinity [20]. The band gap energy expresses the energy difference between these two important FMOs, which signifies the stability of the molecular structure.
The size of the HOMO–LUMO energy gap affects the stability and reaction performance of the compound structure, and the stability of the compound is related to the drug storage, transportation, and use. Therefore, the research of HOMO–LUMO is necessary. To investigate the chemical stability of conformers 1-1 and 1-2, the HOMO and LUMO energies, and their orbital energy gap were calculated by the B3LYP/6-311G(2d,p) method [8]. The pictorial illustration of the FMOs and their respective positive and negative regions represented by red and green colors are shown in Fig. 6 (see electronic version). The large HOMO–LUMO gap automatically implied high excitation energies of the excited states and good stability. The EHOMO and ELUMO values are –6.7384 eV and –1.0373 eV respectively, and the value of the energy separation between the HOMO and LUMO is –5.7011 eV for conformer 1-2 (Fig. 6a). Compared with conformer 1-2, the HOMO and LUMO energy separation of conformer 1-1 was –6.0014 eV (Fig. 6b). The difference between the two conformers is mainly caused by the orientation of the bromobenzene group. The hardness that can be denoted as η = (I – A)/2 indicates the resistance toward the deformation of the electron cloud of chemical systems under small perturbation encountered during the chemical process [21-23]. The hardness of compound 1-2 is 2.85055 eV. Compared with conformer 1-2, the hardness of compound 1-1 is 3.0007 eV. Therefore, conformer 1-1 is the most advantageous conformation for this compound.

HOMO and LUMO of conformer 1-2 (a). HOMO and LUMO of conformer 1-1 (b).
CONCLUSIONS
In this paper, the 1-(2-bromobenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole compound was synthesized by the one-step nucleophilic substitution reaction. Its structure was confirmed by spectroscopic techniques, MS, and single crystal X-ray diffraction. Crystallographic study and conformational analysis were performed for the title compound. The molecular structure optimized by DFT is consistent with the crystal structure determined by X-ray diffraction. The experimental and calculated data indicate that the title compound has a certain nucleophilic reactivity and good chemical stability, which has a potential medicinal value.
REFERENCES
R. Sreedevi, S. Saranya, K. R. Rohit, and G. Anilkumar. Adv. Synth. Catal., 2019, 361(10), 2236-2249.
J. F. Campos, M. Loubidi, M. C. Scherrmann, and S. Berteina-Raboin. Molecules, 2018, 23(3), 684.
K. Khalid, R. Smaail, R. Youssef, T. Jamal, M. Yahia, and A. A Faiz, A M. Hammed. Molecules, 2018, 23, 134.
S. Fustero, M. Sanchez-Rosello, P. Barrio, and A. Simónfuentes. Chem. Rev., 2011, 111(11), 6984-7034.
A. Ansari, A. Ali, and M. Asif. New J. Chem., 2017, 41(1), 16-41.
M. J. Masuda-Herrera, K. L. Dobo, M. O. Kenyon, J. D. Kenny, and J. P. Bercu. Environ. Mol. Mutagen., 2019, 60(9), 766-777.
N. J. Hiller, N. A. do Amaral e Silva, T. A. Tavares, R. X. Faria, M. N. Eberlin, and D. de Luna Martins. Eur. J. Org. Chem., 2020, 4841-4877.
J. S. Zhao, P. Jin, N. Xi, and D. D. Wei. Chin. J. Struct. Chem., 2017, 36, 937-942.
W. Y. Lin, F. Yang, A. N. Duan, W. W. You, and P. L. Zhao. Chin. J. Struct. Chem., 2018, 37, 1557-1562.
G. M. Sheldrick. SHELXS-2018/3. Program for Solution of Crystal Structures. University of Göttingen: Göttingen, Germany, 2018.
G. M. Sheldrick. SHELXL-2018/3. Program for Refinement of Crystal Structures. University of Göttingen: Göttingen, Germany, 2018.
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, and G. Petersson. Gaussian 09, Revision C.01. Gaussian: Wallingford, 2010.
A. Frish, A. B. Nielsen, and A. J. Holder. Gauss View User Manual. Gaussian: Pittsburg, PA, 2011.
Y. Y. Liu, K. Z. Lv, Y. Li, Q. L. Nan, and J. Y. Xu. Chin. J. Struct. Chem., 2019, 38, 171-186.
B. J. Deppmeier, A. J. Driessen, T. S. Hehre, W. J. Hehre, J. A. Johnson, P. E. Klunzinger, J. M. Leonard, I. N. Pham, W. J. Pietro, and J. Yu. Spartan08. Wavefunction: Irvine, CA, 2009.
H. H. Brintzinger, M. H. Prosenc, F. Schaper, A. Weeber, and U. Wieser. J. Mol. Struct., 1999, 485-486, 409-419.
N. Huang, C. Kalyanaraman, K. Bernacki, and M. P. Jacobson. Phys. Chem. Chem. Phys., 2006, 8, 5166-5177.
K. Fukui, T. Yonezawa, and H. Shingu. J. Chem. Phys., 2004, 20(10), 1653.
M. D. Rozeboom, I. M. Tegmo-Larsson, and K. Houk. J. Org. Chem., 1981, 46(11), 2338-2345.
C.-G. Zhan, J. A. Nichols, and D. A. Dixon. J. Phys. Chem. A, 2003, 107(20), 4184-4195.
R. G. Parr and R. G. Pearson. J. Am. Chem. Soc., 1984, 15(13), 7512-7516.
R. G. Pearson. Proc. Natl. Acad. Sci., 1986, 83(22), 8440-8441.
R. G. Parr and P. K. Chattaraj. J. Am. Chem. Soc., 1991, 113(5), 1854-1855.
Funding
This work was supported by Guizhou University of Traditional Chinese Medicine 2018 annual academic new seedling cultivation and innovation exploration special project cultivation project plan. (Qiankehe platform talent [2018]5766-14).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interests.
Additional information
Text © The Author(s), 2021, published in Zhurnal Strukturnoi Khimii, 2021, Vol. 62, No. 6, pp. 997-1006.https://doi.org/10.26902/JSC_id72898
Supplementary material
Rights and permissions
About this article

Cite this article
Yang, Z., Huang, P., Chen, J. et al. SYNTHESIS, CRYSTAL STRUCTURE, AND DFT STUDY OF 1-(2-BROMOBENZYL)-4-(4,4,5,5-TETRAMETHYL-1,3,2-DIOXABOROLAN-2-YL)-1H-PYRAZOLE. J Struct Chem 62, 928–937 (2021). https://doi.org/10.1134/S0022476621060123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476621060123