
Abstract—
We have synthesized tin dioxide with a large specific surface area (122 m2/g), prepared platinum materials supported on it, and characterized them. The results demonstrate that Pt/SnO2 + C composites can be used as catalysts for oxygen electroreduction. The stability of such a catalyst containing 50% carbon and 15 wt % Pt is better than that of commercially available Pt/C electrocatalyst containing 20% Pt.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
The engineering of environmentally safe chemical energy sources, such as fuel cells, is an important issue among modern research directions [1–4]. Efficient electrocatalysts for low-temperature fuel cells (FCs) include nanostructured composite materials containing platinum-based nanoparticles supported on nano- or microparticles of a carbon support [5–10]. A key issue pertaining to polymer membrane FCs is their limited lifetime, primarily because of the degradation of the membrane and catalytic layer [11–16]. Therefore, the ability to optimize the support for platinum nanoparticles is an important issue [17]. The use of some types of carbon supports more oxidation-resistant than pyrolytic carbon black, for example, carbon nanotubes, makes it possible to extend the life of membrane–electrode units [18, 19], thereby raising their efficiency. At the same time, the nature of the support material, capable of being oxidized, remains unchanged.
In recent years, a great deal of attention has been paid to degradation-resistant noncarbon catalyst supports. The use of various methods for the synthesis of inorganic materials [20, 21] makes it possible to considerably extend the range of systems suitable for practical application and stimulates a search for new approaches to modifying existing materials.
Tin dioxide is a promising support for Pt-containing electrocatalysts owing to a number of its advantages (stability, availability, simple synthesis, and others). The development of relatively simple methods for the synthesis of SnO2 supports and SnO2-based platinum electrocatalysts for polymer membrane Cs is a topical issue. In particular, in a study of the effects of solution pH and synthesis time (at a temperature of 190°C) on the size of SnO2 nanoparticles Jiang et al. [22] suggested a possibility of obtaining crystallites one to tens of nanometers in size, without however BET surface area measurements. Interesting results were reported by Kuriganova and Smirnova [23] and Kuriganova et al. [23] about the electrochemical synthesis of Pt/SnOx–C and Pt/MOx–C materials. Unfortunately, despite the possibility of controlling the crystallite size of SnO2 and the relative simplicity of the method, they reported a number of difficulties in controlling the chemical composition of SnO2 [23]. Besides, the characteristics of the supports were not described in sufficient detail [24]. Antoniassi et al. [25] studied an electrocatalyst of complex composition, consisting of Pt, SnO2, and carbon. They heralded the possibility of synthesizing a Pt/SnO2 + C composite with cubic platinum nanoparticles without using organic additives as stabilizers. Note that they paid most attention to ways of increasing the fraction of the (100) faces of Pt nanoparticles, without describing in detail SnO2 synthesis conditions.
Interesting results were reported by Zhang et al. [26], who obtained SnO2-supported platinum electrocatalysts offering enhanced stability in comparison with commercially available Pt/C materials. Results reported by Frolova and Dobrovolsky [27] and Frolova et al. [28] are also of obvious interest because they suggest the possibility of further improving functional characteristics of the Pt/(Sn,Sb)Ox materials obtained. Note that, in the above-mentioned studies [26–28], SnO2 was prepared from SnCl4 using rather complex sol–gel processing. Elezović et al. [29] studied Sb–SnO2 and Ru–SnO2 materials and related electrocatalysts, and Gutsche et al. [30] examined the effect of Ag+ impurities on the growth of platinum nanoparticles on the surface of SnO2 and found a direct relation: the fraction of faceted Pt nanoparticles was shown to increase with increasing Ag+ concentration.
Ruiz-Camacho et al. [31] studied Pt–Ag/SnO2–C composites as electrocatalysts for methanol oxidation, which is rather interesting from the viewpoint of enhancing the catalytic activity of Pt owing to the presence of SnO2. At the same time, it is clear that the addition of other metals to Pt/SnO2 increases the probability of contaminating the polymer components of the membrane–electrode unit with metal cations.
On the whole, the previously reported results on the behavior of Pt/SnO2 electrocatalysts demonstrate that SnO2 nanoparticles have a positive effect on their electrochemical performance and stability compared to Pt/C. At the same time, the problem of producing relatively simple, well-reproducible and scalable methods for the synthesis of supports and impurity-free Pt/SnO2 catalysts capable of ensuring stable functional characteristics remains to be resolved. Another important issue is the ability to enhance the mass activity of such catalysts. The composition of Pt/SnO2 composites with carbon remains to be optimized, and it is not yet clear whether or not carbon has a negative effect on the stability of electrocatalysts. Given that a porous catalytic layer should be produced, it is necessary to continue a search for optimal compositions of Pt/(SnO2 + C) composites capable of ensuring a combination of effective electron transport and contact of Pt nanoparticles with an ionomer and reactant molecules during the operation of the electrocatalyst.
The objectives of this work were to obtain highly dispersed SnO2 with a particle size in the nanometer range and, hence, a large surface area (at least 100 m2/g) and to use it for producing a stable Pt/SnO2 electrocatalyst for the electroreduction of oxygen. Given the low electronic conductivity of tin dioxide, it was important to study the electrochemical behavior of composite electrocatalysts based on mixtures of Pt/SnO2 with fine-particle carbon and assess their catalytic activity and stability in comparison with Pt/SnO2 and Pt/C catalysts.
EXPERIMENTAL
The support was synthesized using tin(IV) chloride dihydrate (SnCl2 · 2H2O), microcrystalline urea (NH2CONH2), saturated aqueous ammonia (NH4OH, 35 wt %), and a concentrated aqueous hydrogen peroxide solution (H2O2, 30 wt %, pure grade). To synthesize sample 1 (SnO2), to a 10% SnCl2 solution was added with constant stirring an excess of a NH3 solution (to adjust the solution to pH 12) and then a tenfold excess of a H2O2 solution. After that, stirring was continued for an hour at a temperature of 60°C. To prepare sample 2, an aqueous 10% SnCl2 solution was mixed with a tenfold excess of urea (under neutral conditions) in a round-bottomed refluxing flask, and the resultant suspension was then heated at 90°C in air on a water bath with constant stirring (15 h). The precipitate was repeatedly washed and separated by centrifugation (10 000 rpm, 5 min). In both cases, the product (SnO2) yield was 98–100%. Pt-containing materials were prepared via the chemical reduction of Pt(IV) in a SnO2 suspension. To prepare the suspension, we used samples 1 and 2 obtained in the preceding step, which were dispersed in a water + ethylene glycol mixture. Next, an appropriate volume of an aqueous solution of chloroplatinic acid (H2PtCl6 · 6H2O, Aurat, Russia) was added and the suspension was adjusted to pH 10 with aqueous ammonia (35 wt %). After that, a fourfold excess of a sodium borohydride (NaBH4) solution was added with vigorous stirring to the suspension, and the solid phase was then separated by centrifugation. The resultant samples were washed repeatedly (eight to ten times) with double-distilled water and ethanol. The product was dried to constant weight over P2O5. The Pt-containing materials thus prepared will hereafter be denoted as 1Pt and 2Pt. Pt/SnO2 + C composites were prepared by adding Vulcan XC72 carbon black to Pt/SnO2 in the process of preparing “electrochemical ink”—a catalyst suspension to be applied to an electrode [32].
X-ray diffraction patterns were collected on an ARL X’TRA automatic diffractometer with CuKα radiation (λav = 1.5418 Å). The average crystallite size Dav was evaluated as described elsewhere [33]. The specific surface area and pore size distribution of the samples were determined using an ASAP 2020 surface area and porosimetry system (Micromeritics, USA). In data processing, we used BET analysis [34].
The samples were examined by transmission electron microscopy (TEM) on a FEI Tecnai G2 F20 S-TWIN TMP analytical (scanning) transmission electron microscope equipped with an EDAX energy dispersive X-ray spectrometer system.
The electrochemically active surface area (S), activity, and stability of the electrocatalysts were assessed in a standard three-electrode cell using a Pine AFCBP1 bipotentiostat system (Pine Research Instrumentation, USA). The technique used to apply a catalyst layer to electrodes and the electrochemical measurement procedure were similar to those described previously [32].
RESULTS AND DISCUSSION
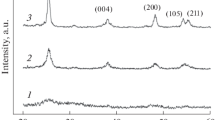
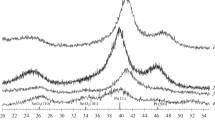
In preparing SnO2 from SnCl2, ammonia or urea was used as a precipitant. The oxidant used was H2O2 in the former case and atmospheric oxygen in the latter. The X-ray diffraction patterns of the materials obtained show reflections characteristic of the rutile structure, without reflections attributable to impurity phases (Fig. 1, Table 1). The observed diffraction line broadening is caused by the small crystallite size (Dav). We obtained two materials, similar in chemical composition (SnO2) but differing in average crystallite size (Table 1).
Pt nanoparticles were applied to the surface of SnO2 particles by a technique capable of ensuring reproducible nanoparticle size distributions and shapes.
Given the low electronic conductivity of SnO2, platinum electrocatalysts were produced by adding Vulcan XC72 carbon black to Pt/SnO2. The amount of the carbon black was such as to ensure electronic conductivity of the composite as a whole, as well as electron supply to and removal from the Pt nanoparticles. The electrocatalysts produced by adding 5 to 50 wt % carbon black to samples 1Pt and 2Pt will hereafter be denoted as 1PtC and 2PtC (Table 2).
The results of the S measurements in the Pt/SnO2 materials and the Pt/SnO2 + C composites differing in carbon content demonstrate that increasing the carbon content of the Pt/SnO2 + C electrocatalysts based on sample 2Pt to about 33% is accompanied by an increase in S (Table 2). At larger percentages of carbon in the composite, S stabilizes (Table 2). It seems likely that carbon content of 33% or above corresponds to “conversion” of all the Pt nanoparticles into an electrically active state.
Taking into account the above results, we also measured the S of the 1PtC50 electrocatalyst, containing 50% carbon (Table 2). Its S turned out to be smaller: 30 m2/g Pt. This can be the result of the poorer accessibility of catalytic centers because of the specific morphological features of the particles in sample 1 as a support. Note that the “working” surface area of the Pt in each carbon-containing composite considerably exceeded the S of the Pt in the carbon-free Pt/SnO2 materials (Table 2).
The parent supports (SnO2) have a large specific surface area. In particular, the surface area of sample 2 is 122 m2/g (BET measurements), the average total pore area in it is 35.5 m2/g Pt, and the average pore diameter is 4.1 nm. According to the TEM results, the composite 1PtC50 contains individual SnO2 nanoparticles 4 nm in size, agglomerates of such particles, and well-crystallized Pt nanoparticles about 5 nm in size (Fig. 3). Unfortunately, since the Pt and SnO2 nanoparticles differ little in contrast in TEM images, histograms of size distributions for the Pt and SnO2 nanoparticles are difficult to construct. X-ray element mapping results for the 1PtC50 electrocatalyst (Fig. 4) demonstrate that the Pt is evenly distributed over the surface of the SnO2 grains.
The smaller S of sample 1Pt50, in which the Pt crystallite size is smaller than that in 2Pt50 (Table 2), is attributable to both the presence of pores similar in size to the Pt nanoparticles (so that some of the nanoparticles reside in pores) and the higher degree of agglomeration of the platinum nanoparticles in 1Pt than in 2Pt.
Figure 5 compares cyclic voltammograms of sample 1PtC50 and a commercially available electrocatalyst. The shape of the voltammograms for oxygen reduction reaction (ORR) on the 2PtC50 electrocatalyst is typical of supported platinum electrocatalysts (Fig. 6). The half-wave potential in ORR at a rotation rate of 1600 rpm was 0.88 eV, and the number of electrons involved in the reduction of an O2 molecule, evaluated using the Koutecky–Levich equation, was determined to be 3.9. This confirms that oxygen electroreduction on the composite electrocatalyst follows a four-electron mechanism. At a potential of 0.85 V, the activity of the electrocatalyst is 1.73 A/m2 Pt, which is somewhat lower than that of the commercially available Pt/C analogue.
The stability of the 2PtC50 electrocatalyst (Fig. 7) was evaluated in comparison with a commercially available Pt/C electrocatalyst (JM20). The Pt/C electrocatalyst was found to undergo catastrophic degradation: its S dropped from 93 to 11 m2/g Pt in 500 cycles. At the same time, the S of the 2PtC50 composite electrocatalyst decreased by just about 10% relative to its original level.
CONCLUSIONS
The optimization of conditions for the synthesis of nanostructured SnO2 from a SnCl2 solution has made it possible to obtain a material in the form of nanoparticles with an average crystallite size near 3–4 nm and a large specific surface area (122 m2/g SnO2). The structure of the SnO2 nanocrystallites has been confirmed by direct TEM examination, electron diffraction data, and X-ray diffraction characterization results. The crystallite size agrees well with the nanoparticle size determined by TEM.
Pt/SnO2 electrocatalysts containing 30 wt % Pt and having a crystallite diameter of 5 and 7 nm have been prepared by chemical reduction. The addition of a Vulcan-XC72 fine-particle carbon material leads to an increase in the electronic conductivity of the composite. The largest S of the platinum, up to 47 m2/g Pt in the case of the 2PtC50 electrocatalyst, is observed at a carbon content of the composite above 33%.
The mass activity of the Pt/SnO2 + C electrocatalyst for ORR is slightly inferior to that of a commercially available Pt/C electrocatalyst containing considerably smaller Pt nanoparticles. The stability of the Pt/SnO2 + C electrocatalyst under harsh stress test conditions is significantly better than that of Pt/C. This confirms that Pt/SnO2 + C composites have considerable potential for use as electrocatalysts for polymer membrane hydrogen–air FCs. Reducing the size of Pt nanoparticles and optimizing the carbon content of such composites can improve the functional characteristics of the electrocatalysts.

Typical X-ray powder diffraction pattern of the synthesized tin dioxide samples.

X-ray powder diffraction patterns of the synthesized Pt/SnO2 materials (samples 1 and 2).

TEM images of parts of sample 1PtC50 (the arrows mark platinum nanoparticles).

X-ray element maps of a portion of the surface of sample 1PtC50.

Cyclic voltammograms of sample 2PtC50 and the commercially available Pt/C electrocatalyst JM20 (RHE = reversible hydrogen electrode).

Voltammograms for ORR.

Accelerated stress test results for electrocatalyst stability (500 cycles) at potentials in the range 0.6–1.4 V (ECASA = electrochemically active surface area).
REFERENCES
Kuzov, A.V., Tarasevic, M.R., and Bogdanovskaya, V.A., Catalysts of ethanol anodic oxidation for ethanol–air fuel cell with a proton-conducting polymer electrolyte, Russ. J. Electrochem., 2010, vol. 46, no. 4, pp. 422–430.
Sharaf, O.Z. and Orhan, M.F., An overview of fuel cell technology: fundamentals and applications, Renew. Sustain. Energy Rev., 2014, vol. 32, pp. 810–853.
Yaroslavtsev, A.B., Dobrovol’skii, Yu.A., Shaglaeva, N.S., Frolova, L.A., Gerasimova, E.V., and Sanginov, E.A., Nanostructured materials for low-temperature fuel cells, Usp. Khim., 2012, vol. 81, pp. 191–220.
Antolini, E., Structural parameters of supported fuel cell catalysts: the effect of particle size, inter-particle distance and metal loading on catalytic activity and fuel cell performance, Appl. Catal., B, 2016, vol. 181, pp. 298–313.
Antolini, E., Carbon supports for low-temperature fuel cell catalysts, Appl. Catal., B, 2009, vol. 88, nos. 1–2, pp. 1–24.
Stamenkovic, V.R., Fowler, B., Mun, B.S., Wang, G.F., Ross, P.N., Lucas, C.A., and Markovic, N.M., Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability, Science, 2007, vol. 315, no. 5811, pp. 493–497.
Stamenkovic, V.R., Mun, B.S., Arenz, M., Mayrhofer, K.J.J., Lucas, C.A., Wang, G.F., Ross, P.N., and Markovic, N.M., Trends in electrocatalysis on extended and nanoscale Pt–bimetallic alloy surfaces, Nat. Mater., 2007, vol. 6, no. 3, pp. 241–247.
Stamenkovic, V., Mun, B.S., Mayrhofer, K.J.J., Ross, P.N., Markovic, N.M., Rossmeisl, J., Greeley, J., and Norskov, J.K., Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure, Angew. Chem., Int. Ed., 2006, vol. 45, no. 18, pp. 2897–2901.
Oezaslan, M., Hasche, F., and Strasser, P., Pt-based core–shell catalyst architectures for oxygen fuel cell electrodes, J. Phys. Chem. Lett., 2013, vol. 4, no. 19, pp. 3273–3291.
Chen, A. and Holt-Hindle, P., Platinum-based nanostructured materials: synthesis, properties, and applications, Chem. Rev., 2010, vol. 110, no. 6, pp. 3767–3804.
Ferreir, P.J., La O, G.J., Shao-Horn, Y., Morgan, D., Makharia, R., Kocha, S., and Gasteiger, H.A., Instability of Pt/C electrocatalysts in proton exchange membrane fuel cells – a mechanistic investigation, J. Electrochem. Soc., 2005, vol. 152, no. 11, pp. A2256–A2271.
Borup, R., Meyers, J., Pivovar, B., Kim, Y.S., Mukundan, R., Garland, N., Myers, D., Wilson, M., Garzon, F., Wood, D., Zelenay, P., More, K., Stroh, K., Zawodzinski, T., Boncella, J., McGrath, J.E., Inaba, M., Miyatake, K., Hori, M., Ota, K., Ogumi, Z., Miyata, S., Nishikata, A., Siroma, Z., Uchimoto, Y., Yasuda, K., Kimijima, K.I., and Iwashita, N., Scientific aspects of polymer electrolyte fuel cell durability and degradation, Chem. Rev., 2007, vol. 107, no. 10, pp. 3904–3951.
Shao, Y.Y., Yin, G.P., and Gao, Y.Z., Understanding and approaches for the durability issues of Pt-based catalysts for PEM fuel cell, J. Power Sources, 2007, vol. 171, no. 2, pp. 558–566.
Hodnik, N., Dehm, G., and Mayrhofer, K.J.J., Importance and challenges of electrochemical in situ liquid cell electron microscopy for energy conversion research, Acc. Chem. Res., 2016, vol. 49, no. 9, pp. 2015–2022.
Kuzov, A.V., Tarasevich, M.R., Bogdanovskaya, V.A., Modestov, A.D., Tripachev, O.V., and Korchagin, O.V., Degradation processes in hydrogen–air fuel cell as a function of the operating conditions and composition of membrane–electrode assemblies, Russ. J. Electrochem., 2016, vol. 52, no. 7, pp. 705–715.
Venkatesan, S.V., Dutta, M., and Kjeang, E., Mesoscopic degradation effects of voltage cycled cathode catalyst layers in polymer electrolyte fuel cells, Electrochem. Commun., 2016, vol. 72, pp. 15–18.
Sharma, S. and Pollet, B.G., Support Materials for PEMFC and DMFC Electrocatalysts – A Review, J. Power Sources, 2012, vol. 208, pp. 96–119.
Bogdanovskaya, V.A., Kol’tsova, E.M., Tarasevich, M.R., Radina, M.V., Zhutaeva, G.V., Kuzov, A.V., and Gavrilova, N.N., Highly active and stable catalysts based on nanotubes and modified platinum for fuel cells, Russ. J. Electrochem., 2016, vol. 52, no. 8, pp. 723–734.
Wang, L., Chen, J., Rudolph, V., and Zhu, Z., Nanotubules-supported Ru nanoparticles for preferential CO oxidation in H2-rich stream, Adv. Powder Technol., 2012, vol. 23, no. 4, pp. 465–471.
Balakhonov, S.V., Vatsadze, S.Z., and Churagulov, B.R., Effect of supercritical drying parameters on the electrochemical properties of vanadium oxide-based aerogels, Inorg. Mater., 2017, vol. 53, no. 2, pp. 181–184.
Ogi, T., Nandiyanto, A.B.D., and Okuyama, K., Nanostructuring strategies in functional fine-particle synthesis towards resource and energy saving applications, Adv. Powder Technol., 2014, vol. 25, no. 1, pp. 3–17.
Jiang, L., Sun, G., Zhou, Z., Sun, S., Wang, Q., Yan, S., Li, H., Tian, J., Guo, J., Zhou, B., and Xin, Q., Size-controllable synthesis of monodispersed SnO2 nanoparticles and application in electrocatalysts, J. Phys. Chem. B, 2005, vol. 109, no. 18, pp. 8774–8778.
Kuriganova, A.B. and Smirnova, N.V., Pt/SnOx–C composite material for electrocatalysis, Mendeleev Commun., 2014, vol. 24, no. 6, pp. 351–352.
Kuriganova, A.B., Leontyeva, D.V., Ivanov, S., Bund, A., and Smirnova, N.V., Electrochemical dispersion technique for preparation of hybrid MOx–C supports and Pt/MOx–C electrocatalysts for low-temperature fuel cells, J. Appl. Electrochem., 2016, vol. 46, no. 12, pp. 1245–1260.
Antoniassi, R.M., Silva, J.C.M., Oliveira, N.A., and Spinacé, E.V., Synthesis of Pt + SnO2/C electrocatalysts containing Pt nanoparticles with preferential (100) orientation for direct ethanol fuel cell, Appl. Catal., B, 2017, vol. 218, pp. 91–100.
Zhang, P., Huang, S.-Y., and Popov, B.N., Mesoporous tin oxide as an oxidation-resistant catalyst support for proton exchange membrane fuel cells, J. Electrochem. Soc., 2010, vol. 157, no. 8, pp. B1163–B1172.
Frolova, L.A. and Dobrovolsky, Yu.A., Platinum electrocatalysts based on oxide supports for hydrogen and methanol fuel cells, Russ. Chem. Bull. Int. Ed., 2011, vol. 60, no. 6, pp. 1101–1111.
Frolova, L., Lyskov, N., and Dobrovolsky, Yu., Nanostructured Pt/SnO2–SbOx–RuO2 electrocatalysts for direct alcohol fuel cells, Solid State Ionics, 2012, vol. 225, pp. 92–98.
Elezović, N.R., Babić, B.M., Radmilović, V.R., and Krstajić, N.V., Synthesis and characterization of Pt catalysts on SnO2 based supports for oxygen reduction reaction, J. Electrochem. Soc., 2013, vol. 160, no. 10, pp. F1151–F1158.
Gutsche, C., Knipper, M., Plaggenborg, T., Parisi, J., and Kolny-Olesiak, J., Synthesis of facetted Pt nanoparticles on SnO2 as an oxygen reduction catalyst, CrystEngComm, 2017, vol. 19, no. 26, pp. 3666–3673.
Ruiz-Camacho, B., Medina-Ramírez, A., Fuentes-Ramírez, R., and Gómez, C.M., Simple synthesis of Pt–Ag/SnO2–C for use as a catalyst of methanol oxidation in alkaline media, J. Solid State Electrochem., 2017, vol. 21, no. 8, pp. 2449–2456.
Alekseenko, A., Ashihina, E., Shpanko, S., Volochaev, V., Safronenko, O., and Guterman, V., Application of CO atmosphere in the liquid phase synthesis as a universal way to control the microstructure and electrochemical performance of Pt/C electrocatalysts, Appl. Catal., B, 2018, vol. 226, pp. 608–615.
Kirakosyan, S.A., Alekseenko, A.A., Guterman, V.E., Volochaev, V.A., and Tabachkova, N.Y., Effect of CO atmosphere on morphology and electrochemically active surface area in the synthesis of Pt/C and PtAg/C electrocatalysts, Nanotechnol. Russ., 2016, vol. 11, no. 5, pp. 287–296.
Brunauer, S., Emmett, P.H., and Teller, E., Adsorption of gases in multimolecular layers, J. Am. Chem. Soc., 1938, vol. 60, no. 2, pp. 309–319.
ACKNOWLEDGMENTS
We are grateful to A.Yu. Nikulin, T.A. Lastovina and Shared Research Facilities Center, OOO SMA, Skolkovo for their assistance in the experimental work.
Funding
This research was supported by the Russian Federation Ministry of Science and Higher Education, project no. 13.3005.2017/4.6 (design stage).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Volochaev, V.A., Novomlinskii, I.N., Davydovich, Y.V. et al. Preparation of Nanostructured Tin(IV) Oxide and Supported Platinum Electrocatalysts Based on It. Inorg Mater 55, 1125–1131 (2019). https://doi.org/10.1134/S0020168519110165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0020168519110165