
Abstract
Catalysts containing bimetallic PtCu-nanoparticles deposited onto carbonaceous and composite SnO2/C supports are prepared by liquid-phase borohydride synthesis. The composition and structure of the synthesized materials, their catalytic activity in the reactions of oxygen electroreduction and methanol electrooxidation, as well as corrosion and morphological stability are investigated. The platinum doping with copper atoms is found to increase the materials’ catalytic activity and stability in comparison with Pt/C, regardless of the type of support used. In addition, the multicomponent PtCu/(SnO2/C) catalyst exhibits the highest tolerance to intermediate products of methanol electrooxidation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Direct methanol fuel cells attract ever growing attention as alternative energy source. Small release of pollutants [1], simplicity of the fuel storage and transportation [2], minor size and large current density [3] make them perspective current sources for applications in portable electronic devices [4–6]. Few factors prevent extensive commercialization of the methanol fuel cells: expensive electrocatalysts, which is mainly due to the usage of noble metals therein, and short in-service life duration (the membrane degradation, the Pt surface poisoning with intermediate products of methanol oxidation, the methanol crossover toward cathodic compartment and its caused changes in the cathode potential) [7, 8]. The methanol oxidation reaction includes stages of its adsorption, dehydrogenation, and further intermediate formation (such as HCOOH, HCOH, COad, etc.) [9]. The CO strong adsorption lowers the Pt-catalyst activity significantly because of the active centers’ blocking. The using of electrochemically active and stable anodic catalyst is crucial for high rate of the methanol oxidation reaction. Nowadays, most extensively studied are the PtRu/C-electrocatalysts, which gained widespread use. The PtRu/C-materials’ higher activity, as compared with Pt/C, in the organics’ oxidation reactions is used to be linked to the catalysis bifunctional mechanism: the OH groups facilitating oxidation of the CO molecules adsorbed at the platinum neighbor clusters do adsorb at the ruthenium cluster much easier than at platinum [10–12]. Further study of the mechanism of methanol oxidation at the PtRu-catalysts in more details showed that, along with the catalysis bifunctional mechanism, the effect of the metal electronic interaction also can contribute positively [13, 14]. Recently, numerous researchers were engaged in the preparation of novel platinum-metallic catalysts by means of the varying of the solid solution composition and the nanoparticles’ architecture. In so doing, they solved problems of the improving of both anodic and cathodic catalysts for the oxygen electroreduction reaction, handicapped by the methanol crossover interference. Meanwhile, the PtCu [15, 16], PtRu [17], PtSn [18] solid solutions were studied and materials synthesized that contained core–shell-structured nanoparticles whose shell consisted of the catalytically-active metals (Pt, Pd); the core, of nonprecious metal that promoted the platinum activity [19–22].
Another approach to the preparation of catalysts for the methanol oxidation and oxygen electroreduction reactions involves some oxide materials, e.g., CeO2 [23, 24], TiO2 [24], SnO2 [25] used as supports for the platinum nanoparticles. Here, the increase in the catalytic activity has been got because of positive effects of the support electronic interaction with the platinum nanoparticles [26, 27]. It was reported (see, e.g., works [28–31]) that the Pt/TiО2-catalysts are more active in the oxygen reduction reaction than the Pt/C ones because they do not suffer from the support corrosion and the following Pt nanoparticles’ falling-off. The further widespread of oxide supports in the electrocatalyst preparation is restricted by their fair electron conductivity. Therefore, to increase the conductivity, the oxides are often used as composite with highly disperse carbon black. Of special interest are nanostructured composites in which the oxide component is present as nanoparticles deposited onto the surface of carbonaceous-support microparticles and contacting the platinum nanoparticles [31–33]. It was shown [27, 34–37] that Pt-catalysts at carbonaceous supports containing tin dioxide are much more active in the methanol and ethanol oxidation reactions than similar metal-oxide-free catalysts. It was reported [27] that Pt-nanoparticles contact with SnO2-particles and carbon simultaneously, thus forming unique triple-contact nanostructures Pt/SnO2/C.
We have already mentioned that the methanol crossover through polymer membrane into cathodic compartment results in the Pt-surface poisoning with the methanol oxidation intermediate products [9] and drastic decrease of the catalyst electrochemical activity in the oxygen reduction reaction. On this reason, special demands are placed on the cathodic catalysts in the methanol fuel cells: they must combine activity in the oxygen reduction and methanol oxidation reactions with high tolerance to the products of the methanol incomplete oxidation and stability against degradation at high anodic potentials corresponding to the oxygen electrode exploiting. Results of our previous studies and literature data are indicative of the fact that the substituting of PtCu for the Pt nanoparticles [38, 39, 52], as well as the Pt nanoparticles’ applying to the MOx/C composite supports [23–25, 40] increased the catalyst activity in the oxygen reduction and methanol oxidation reactions. The question arises as to whether it is possible engaging in a single catalyst the positive effect of two factors, namely, the platinum alloying with a d-metal and the deposition of SnO2 nanoparticles’ contacting the metal nanoparticles onto the carbon surface? Will the modified material be superior to Pt/C and, possibly, PtRu/C as an anodic or cathodic catalyst?
In this work, we aimed at the preparing of a multicomponent PtCu/(SnO2/C)-catalyst containing bimetallic nanoparticles applied to the surface of nanostructured SnO2/C-support and the comparing of its performance in the oxygen reduction and methanol oxidation reactions with that of PtCu/C- and Pt/(SnO2/C)-electrocatalysts with a close platinum content; under some conditions, with the commercial PtRu/C-electrocatalyst. To lower external influence connected with methodical specifics of the porous catalytic layers formation at electrodes, we included the commercial Pt/C-catalyst into the list of studied materials.
EXPERIMENTAL
We deposited Pt-nanoparticles onto a SnO2/C-composite support containing tin nanoparticles at the surface of particles of the Vulcan XC72 carbonaceous support obtained by the procedure described elsewhere [40, 41]. To this purpose, 20 mL of ethylene glycol and 25.6 mL of 0.01 М H2PtCl6 aqueous solution were added to 0.2 g SnO2/C (30 wt % SnO2) powder. The obtained suspension was homogenized using ultrasound, then successively added with 1 mL 37% НСОН and 1 М NaOH solution in a water–ethylene glycol mixture (1:1) under constant agitation up to reaching рН 11. The suspension was exposed to 90°С for 2 h. After spontaneous cooling for 30 min the obtained Pt/(SnO2/C)-catalyst was isolated by filtering. The bimetallic PtCu-nanoparticles were deposited onto the carbonaceous and composite supports at a preselected Pt : Cu = 1 : 1 ratio in a single stage by co-reduction of platinum and copper precursors by excess of fresh-prepared 1 M NaBH4 solution in a water–ethylene glycol suspension at pH 9–10. The calculated metal mass fraction in the obtained materials is 20 wt % Pt and 9 wt % Cu.
The Pt:Cu metal ratio in the bimetallic catalysts was determined by X-ray fluorescent analysis using a RFS-001 spectrometer with full external reflection of X-ray radiation (Research Institute of Physics, the Southern Federal University, Russia). The metal mass fraction in the obtained materials was determined by thermogravimetry, by the oxidizing of the catalysts at 800°С. In the calculations, we took into consideration the actual component ratio determined by the X-ray fluorescent analysis, on the assumption of that the solid residue can contain Pt, CuO, and SnO2.
The catalyst phase composition was determined by the method of X-ray powder diffraction, using an ARL X’TRA (CuKα) diffractometer and performing the measurements over the 2θ angle diapason from 15° to 55°, in increments of 0.02° and with the registration rate of 2°/min. The crystallite mean size was determined using the Scherrer formula [42]: D = Kλ/(FWHM cosθ), where λ is the monochromatic radiation wavelength (Å); FWHM is the full peak width at its half maximum (radian); D is the crystallite mean diameter (nm); θ is the reflection angle (radian); K = 0.89 is the Scherrer constant.
The electrochemical measurements were carried out at a rotating disc electrode in a three-electrode cell, using a VersaStat 3 potentiostat (AMETEK Scientific Instruments, USA). To this purpose, a catalytic layer was formed at a disc electrode end-face. The quantity of substance of 0.006 g was added with 900 mL of isopropanol and 100 mL of 0.5% Nafion® polymer aqueous emulsion. The suspension was dispersed by its sonication for 15 min. Then, under steady agitation an aliquot of 6 mL was taken using a pipette and applied to the electrode end-face. The droplet was dried at the room temperature under the electrode rotation velocity of 700 rpm.
The catalytic layer was standardized by 100 cycles of potential, scanning at a rate of 200 mV/s in Ar-saturated 0.1 М HClO4 solution. Then, the electrochemically active surface area was calculated from the charge expended for the atomic hydrogen adsorption/desorption. To this purpose, two cyclic voltammograms were recorded in the same potential diapason, yet, at the potential scanning rate of 20 mV/s. The electrochemically active surface area was also measured by the monolayer of the chemisorbed CO oxidation. To this purpose, the electrolyte solution was bubbled-through with CO for 20 min, keeping the potential constant at 0.1 V. Then, the solution was bubbled-through with argon for 40 min, and two cyclic voltammograms were recorded, by which the calculation was done. The catalyst activity in the ORR was measured in 0.1 М HClO4 solution saturated with oxygen for 1 h, using rotating disc electrode. Linear-sweep voltammograms were recorded at the electrode rotation velocities of 400, 900, 1600, and 2500 rpm and potential scanning rate of 20 mV/s. To evaluate the catalysts’ activity in the methanol electrooxidation reaction, cyclic voltammogram was recorded in 0.1 М HClO4 + 0.5 М CH3OH solution. The methanol-oxidation-intermediate tolerance was evaluated by taking chronoamperogram in the same electrolyte at the potentials of 0.60 and 0.87 V.
The catalyst durability tests in MEAs is the best method of the stability evaluation [43, 44]. However, the tests are rather labor-consuming. Numerous publications suggest the carrying out of preliminary rapid evaluation of catalysts’ stability with the using of different regimes of stress-testing in electrochemical cell [45–50]. In this work, to evaluate the synthesized catalysts’ stability, we carried out 1000 voltammetric cycles over the 0.6–1.4 V potential range in 0.1 М HClO4 solution in argon atmosphere. Every 200 cycles, we measured electrochemically active surface area, as described above. We have successfully tried out such a stress-testing regime earlier [51]. The degree of catalysts degradation was calculated by the following formula:
where n is the cycle number; ESA100 and ESAn is the catalyst electrochemically active surface area after the carrying out of 100 test-cycles and n test-cycles, respectively. After the completing of the stress-testing, CH3OH solution was added into the electrochemical cell up to the achieving of the concentration 0.5 М. Afterwards, the catalyst residual activity in the methanol oxidation reaction and the methanol-oxidation-intermediate tolerance were evaluated.
The synthesized catalysts’ structural characteristics and electrochemical behavior were compared with those of the commercial Pt/C-catalyst HiSPEC3000 (Johnson Matthey), containing 20 wt % Pt. In what follows, it is designated JM20. In some cases, when the electrochemical behavior of the synthesized materials was studied in methanol solutions at relatively low potentials, we used the commercial PtRu/C-electrocatalyst (Pt : Ru = 1 : 1, 40 wt % Pt, 20 wt % Ru, Alfa Aesar) as a reference. The reason therefor is that the using of the PtRu/C-catalyst is expedient only at the methanol fuel cell anode, that is, at relatively low potentials [9–11, 13].
All potential values in the paper are given against reversible hydrogen electrode (RHE). Saturated silver/silver chloride electrode was used as reference electrode; platinum wire, as auxiliary electrode.
RESULTS AND DISCUSSION
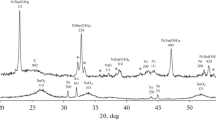
Powder diffraction patterns of PtCu/C-, PtCu/(SnO2/C)-, Pt/(SnO2/C)-, and Pt/C-materials show reflections corresponding to the platinum, carbon (2θ ~ 25°), and tin dioxide phases (Fig. 1). This confirms the metal component effective deposition onto both carbonaceous and composite supports. The maximum broadening is caused by the nanoparticles’ small size. At that, for the platinum–copper materials, the metal phase reflection maximums are shifted toward larger angles as compared with the platinum phase. The calculated lattice parameter for the bimetallic nanoparticles is less than for the polycrystalline platinum. This result can be explained by the presence of the copper atoms therein. The crystallite mean size calculated by the Scherrer formula is 1.8 nm for the platinum nanoparticles in Pt/(SnO2/C) and about 2.6 nm for PtCu-nanoparticles (Table 1). The platinum crystallite mean size in the commercial Pt/C-catalyst is 2.4 nm. Note that X-ray diffraction patterns of bimetallic catalysts has no reflections corresponding to the copper oxide phases, which does not exclude the possibility of their presence at smaller amount in the X-ray amorphous state.

X-ray diffraction patters of the studied catalysts: (1) PtCu/C, (2) Pt/C, (3) PtCu/(SnO2/C), (4) Pt/(SnO2/C).
According to the gravimetry data, all synthesized materials contain from 19 to 20% Pt. Both platinum–copper catalysts have the atomic ratio Pt : Cu = 1 : 0.7 (Table 1). This metals’ ratio somewhat differs from that of the precursors used in the synthesis, which can be a result of the incomplete reduction or minor loss of copper in the course of the synthesis, as noted elsewhere [52].
During the catalyst standardization, the metal nanoparticles’ surface is subjected to cleaning and developing. It is known that cyclic voltammograms of the platinum–copper materials can contain current peaks of the copper anodic dissolution from the copper phase or solid solution in the potential ranges 0.25–0.45 and 0.7–0.8 V, respectively [53, 54]. For the PtCu/C- and PtCu/(SnO2/C)-catalysts, no such maximums are observed. However, according to the X-ray fluorescent analysis, the copper amount in the post-standardized catalysts decreased (Table 1), which is due to selective dissolution of the alloying-component atoms from the nanoparticles and, possibly, copper oxide chemical dissolution. In order to prevent the effect of the Cu2+ ions transferred to the solution onto the studied electrode behavior, we replaced the electrolyte and saturated it with argon for 20 min after the completing of the catalyst standardization.
The catalyst electrochemically active surface area was calculated by the charge expended for the atomic hydrogen adsorption/desorption during the cyclic voltammogram recording (Fig. 2а, Table 2). The obtained values correlate well with the electrochemically active surface area calculated by the monolayer of the chemisorbed CO oxidation (Fig. 2b, Table 2). The electrochemically active surface area of the PtRu/C-catalyst was also calculated by the CO monolayer oxidation; it came to 68 m2/g (PtRu)Footnote 1.

(а) Cyclic voltammograms of standardized electrocatalysts taken in argon atmosphere; (b) the same, after the bubbling of СО. Electrolyte: 0.1 М HClO4 solution in argon atmosphere.
Our results showed that the electrochemically active surface area decreased in the series of catalysts Pt/C > Pt/(SnO2/C) \( \gg \) PtCu/C ≥ PtCu/(SnO2/C) from 78 down to 32–37 m2/g (Pt). The largest electrochemically active surface area is observed for the Pt/C- and Pt/(SnO2/C)-catalysts, which on the whole is coherent with the small crystallite size in these materials (Table 1), although the observed small difference in the crystallite size should not result in so drastic difference in the electrochemically active surface area. The significant decrease of the electrochemically active surface area when passing from platinum catalysts to the platinum–copper ones, PtCu/C and PtCu/(SnO2/C), is due to the larger agglomeration degree in the PtCu-nanoparticles [55]. Note that at the SnO2-containing catalysts the СО oxidation onset potential is lower and the СО oxidation current maximum in cyclic voltammogram is also shifted toward lower potentials (Fig. 2b). At that, the PtCu/(SnO2/C)-catalyst voltammogram contains even two СО oxidation current maximums (at 0.4 and 0.7 V, Fig. 2b). The presence of the two СО oxidation current peaks can be caused by various factors: nonuniformity in the metal nanoparticles’ composition or size-distribution, their agglomeration, special features in the behavior of the PtCu-nanoparticles contacting SnO2-nanoparticles, or special features of СО adsorption at different crystal faces of the nanoparticles [56‒60]. Taking into consideration some similarity in the performance of the two SnO2-nanoparticle-containing catalysts with respect to the СО oxidation (Fig. 2b), we may ascribe the observed effects right to the presence of tin dioxide.
To evaluate the catalyst activity in the oxygen reduction reaction, we used linear sweep voltammetry (Fig. 3а). The kinetic current at the potential of 0.90 V, normalized by the platinum mass, or the catalyst electrochemically active surface area—these quantities were used as descriptors of the catalytic activity (Table 2). The kinetic current and the number of electrons involved in the reaction were calculated by the Koutecký–Levich equation (Fig. 3b). We see that when the kinetic current has been normalized by the platinum mass, it is the Pt/(SnO2/C)-catalyst that demonstrated the lowest activity in the oxygen reduction reaction; when the kinetic current has been normalized by the electrochemically active surface area, the Pt/(SnO2/C)- and Pt/C-catalysts showed nearly the same activity. Despite the lesser values of the electrochemically active surface area, the platinum–copper catalysts showed higher activity in the oxygen reduction reaction, expressed both per unit platinum mass or per unit electrochemically active surface area, as compared with Pt/(SnO2/C)- and the commercial Pt/C-catalysts (Table 2). The special feature of all studied electrodes is that the oxygen reduction reaction passed by the 4-electron path (Table 2).

(а) Voltammograms of oxygen electroreduction at different catalysts at the disc rotation velocity 1600 rpm; (b) dependences in the coordinates 1/Isp–1/ω0.5 at E = 0.90 V plotted by the voltammograms. Electrolyte: 0.1 М HClO4 solution saturated with O2 under atmospheric pressure.
The catalyst performance in the methanol oxidation reaction was studied by the methods of cyclic voltammetry (Fig. 4а) and chronoamperometry (Figs. 4b, 4c). For the methanol oxidation reaction, the shape of cyclic voltammograms of the studied catalysts is characterized by two well pronounced oxidation current peaks in the direct and reverse runs of the potential sweeping. In terms of voltammetry, the catalyst activity in the methanol oxidation reaction is used to be characterized by the specific maximal current density in the direct run of the potential sweeping when taking cyclic voltammograms [60, 61], the methanol oxidation onset potential [61], or the charge \({{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}}\) (Table 3) expended in the methanol oxidation during the direct run of the potential sweeping when recording cyclic voltammograms [38]. Judging by the maximal current and the \({{Q}_{{{\text{C}}{{{\text{H}}}_{{\text{3}}}}{\text{OH}}}}},\) charge, the catalyst specific activity in the methanol oxidation reaction increased in the series Pt/C < Pt/(SnO2/C) \( \ll \) PtCu/C ≤ PtCu/(SnO2/C) (Fig. 4).

(а) Cyclic voltammograms (direct run) of methanol electrooxidation. Potential scanning rate 20 mV/s; (b), (c) chronoamperograms of methanol electrooxidation at the potentials 0.87 and 0.60 V, respectively. Electrolyte: 0.1 М HClO4 solution. The argon atmosphere.
The comparative evaluation of the catalyst activity in the methanol oxidation reaction and the methanol-oxidation-intermediate tolerance in terms of chronoamperometry is more complicated (Figs. 4b, 4c). At the potential of 0.87 V (approaching the potential of methanol fuel cell cathode), the largest specific current in the initial moment was detected with the copper-containing catalysts; however, the methanol-oxidation current decayed faster at these electrodes than at Pt/C and Pt/(SnO2/C) (Fig. 4b). The PtCu/C- and PtCu/(SnO2/C)-catalysts’ higher sensitivity to the methanol oxidation intermediate products can be explained, in particular, by the intermediates’ larger concentration in the near-electrode layer. At the same time, the current decay at the PtCu/(SnO2/C)-catalyst is slower than at PtCu/C. The slower current decay characteristic for the Pt/C- and Pt/(SnO2/C)-electrodes was observed at the higher methanol oxidation rate at Pt/(SnO2/C) as compared with Pt/C. Thus, the presence of SnO2 nanoparticles at the Pt/(SnO2/C)- and PtCu/(SnO2/C)-catalyst surfaces increased their tolerance to the methanol oxidation intermediate products at high potentials.
Chronoamperograms of metanol oxidation at the potential 0.60 V (Fig. 4c) were recorded also for the commercial PtRu/C-catalyst because it is the platinum–ruthenium catalysts that are known as highly active and methanol-oxidation-tolerant at the methanol fuel cell anodes. It turned out that under these conditions the largest anodic current was observed for the PtRu/C- and PtCu/(SnO2/C)-catalysts. The Pt/(SnO2/C)-catalyst demonstrated fair methanol oxidation rate and high current stability in time. Much lower currents were obtained with the Pt/C- and PtCu/C-catalysts.
Appearingly, the positive effect of the alloying metal (copper) manifesting itself at high potentials and the oxide component (SnO2) on the methanol electrooxidation rate is based on different mechanisms. At high potentials, in the initial moment when the number of СО molecules (the methanol-electrooxidation most stable intermediate) adsorbed at the catalyst active surface is not too large, it is the copper additive that affects most strongly the CH3OH oxidation rate. As time goes by, the number of СО adsorbed molecules increased which results in the platinum active surface blocking and the electrooxidation retardation. The presence of the SnO2 nanoparticles contacting those of Pt or PtCu favors the catalysis bifunctional mechanism realization in the СО oxidation over wide potential range, which increased the catalyst high tolerance. This mechanism of the SnO2 nanoparticles’ effect is also confirmed by the shift of the СО oxidation onset potential toward lower values in the corresponding voltammograms (Fig. 2b). Under the conditions of methanol oxidation at the potential of 0.60 V, the multicomponent PtCu/(SnO2/C)-catalyst practically is not inferior to the commercial PtRu/C-catalyst (Fig. 4b, Table 3).
On the whole, the platinum–copper catalysts demonstrated high activity in the oxygen reduction and methanol oxidation reactions at high potentials; those containing tin dioxide nanoparticles, the higher activity in the methanol oxidation reaction at lower potentials and methanol-oxidation-intermediate tolerance over wide potential range. Appearingly, based on the results of cyclic voltammetry we can formulate a concept of integral activity of the catalysts in the methanol oxidation reaction by comparing maximal currents and the charge expended for the methanol oxidation over wide potential range. The conclusion on the catalysts’ activity in potentiostatic conditions depends on the potential value (or narrow potential range) in which they are compared.
When stress-testing is carried out in methanol-free solution, for Pt/C and Pt/(SnO2/C) we observed rapid decrease in their electrochemically active surface area as compared with PtCu/C- and PtCu/(SnO2/C)-catalysts: for the platinum catalysts the degradation degree is about 75%; for the bimetallic ones, about 25–30% (Fig. 5). Thus, under the conditions of hard-mode stress-testing the studied catalysts rank in their stability as Pt/C ≤ Pt/(SnO2/C) \( \ll \) PtCu/(SnO2/C) ≤ PtCu/C. It is to be noted that during the first 100 cycles we observed an increase in the electrochemically active surface area of bimetallic catalysts; during further cycling, its decrease. Our data evidenced the higher corrosion resistance of the bimetallic catalysts, which is in good agreement with our earlier results and the data of other researchers [50–52]. It is not unthinkable that the PtCu-catalyst higher stability is caused by not only their composition but also larger size of nanoparticles, which results in a smaller electrochemically active surface area of the materials. The existence of the corresponding correlations was demonstrated in work [44]. According to the results of cyclic voltammetry data, after the completing of the stress-testing the PtCu/C- and PtCu/(SnO2/C) catalysts still retain much higher integral catalytic activity in the methanol oxidation reaction than the platinum catalysts (Table 4).

Dependence of the catalyst degradation degree on the cycle number during the electrode voltammetric stress-testing over the 0.6–1.4 V potential range.
CONCLUSIONS
Platinum-containing electrocatalysts in which Pt- or PtCu-nanoparticles are deposited onto carbonaceous supports or nanostructured composite support SnO2/C were prepared by liquid-phase borohydride synthesis. The composite support is a carbonaceous material on whose surface SnO2-nanoparticles were deposited. The Pt mass fraction in the catalysts is about 20%, the metal nanoparticle (crystallite) size increased in the row Pt/(SnO2/C) < Pt/C ≈ PtCu/(SnO2/C) < PtCu/C from 1.8 to 2.8 nm. The electrochemically active surface area of the catalysts decreased significantly when passing from platinum 70–78 m2/g (Pt) catalysts to platinum–copper ones 32–39 m2/g (Pt)).
By using cyclic voltammetry, it was found that the bimetallic catalysts PtCu/C and PtCu/(SnO2/C) are more active in the oxygen reduction reaction and more corrosion-stable in the voltammetric stress-testing (1000-fold potential cycling over the 0.6–1.4 V range) than their platinum analogs, Pt/C and Pt/(SnO2/C).
On the basis of the comparing of the maximal currents and charges expended in the methanol oxidation we can also concluded on the higher activity of the bimetallic catalysts in the methanol oxidation reaction. At the same time, the catalyst performance in potentiostatic conditions depends to a great extent on the potential at which the measurement is performed; this points to an important role of the tin oxide nanoparticles anchored at the carbon particles’ surface. At the potential of 0.87 V the SnO2/C-composite support-based platinum–copper and platinum catalysts demonstrated larger tolerance than the tin-dioxide-free analogs. The methanol oxidation at the potential of 0.60 V occurs more intensely at the SnO2/C-containing catalysts; here the PtCu/(SnO2/C)-material demonstrated the specific activity and tolerance which are practically not inferior to the commercial PtRu/C-catalyst.
The copper positive effect on the platinum activity can be caused by the decrease in the solid-solution crystal lattice parameter and the lowering in the energy of the platinum atom d-orbitals, which facilitated the oxygen molecule dissociative adsorption at the nanoparticles’ surface. The presence at the carbonaceous support surface of the SnO2 nanoparticles partly contacting metal nanoparticles increased the tolerance of both platinum–copper and platinum catalysts to the methanol oxidation intermediate products, presumably to СО.
To our view, the search for effective multicomponent platinum-containing catalysts for the oxygen electroreduction and methanol electrooxidation should be continued. In so doing, of importance is the increase of the materials' electrochemically active surface area, which will elevate their mass-activity significantly.
Notes
In the PtRu/C-catalyst, СО is chemisorbed both at platinum and ruthenium.
REFERENCES
Wee, J.-H., A feasibility study on direct methanol fuel cells for laptop computers based on a cost comparison with lithium-ion batteries, J. Power Sources, 2007, vol. 173, p. 424.
Brouzgou, A., Podias, A., and Tsiakaras P., PEMFCs and AEMFCs directly fed with ethanol: a current status comparative review, J. Appl. Electrochem., 2013, vol. 43, p. 119.
Burhan, H., Cellat, K., Yilmaz, G., and Sen, F., Chapter 3 Direct methanol fuel cells (DMFCs), in: Direct Liquid Fuel Cells: Fundamentals, Advances Future, 2021. p. 71.
Tarasevich, M.R. and Kuzov, A.V., Direct alcohol fuel cells, Al’ternat. Energetika Ekologiya (in Russian), 2010, no. 7(87), p. 86.
Meital, G., Menkin, S., and Peled, E., High power direct methanol fuel cell for mobility and portable applications, Int. J. Hydrogen Energy, 2019, vol. 44, p. 3138.
Pinto, A.M.F.R., Oliveira, V.S., and Falcao, D.S.C., Direct Alcohol Fuel Cells for Portable Applications: Fundamentals, Engineering and Advances 1st Edition, 2018. p. 287.
Kaur, B., Srivastava, R., and Satpati, B., Highly efficient CeO2 decorated nano-ZSM-5 catalyst for electrochemical oxidation of methanol, ACS Catal., 2016, vol. 6, p. 2654.
Liu, Y., Li, D., Stamenkovic, V.R., Soled, S., Henao, J.D., and Sun, S., Synthesis of Pt3Sn alloy nanoparticles and their catalysis for electro-oxidation of CO and methanol, ACS Catal., 2011, vol. 1, p. 1719.
Hamnett, A., Mechanism and electrocatalysis in the direct methanol fuel cell, Cat. Today, 1997, vol. 38, p. 445.
Watanabe, M. and Motoo, S., Electrocatalysis by adatoms. Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms, J. Electroanal. Chem., 1975, vol. 60, p. 267.
Gasteiger, H.A., Markovic, N., Ross, P.N., and Cairns, E.J., Methanol electrooxidation on well-characterized Pt–Ru alloys, J. Phys. Chem., 1993, vol. 97, p. 12029.
Markovic, N., Gasteiger, H.A., Ross, P.N., Jiang, X., Villegas, I., and Weaver, M.J., Electro-oxidation mechanisms of methanol and formic acid on Pt–Ru alloy surfaces, Electrochim. Acta, 1995, vol. 40, p. 91.
Tong, Y.Y., Kim, H.S., Babu, P.K., Waszczuk, P., Wieckowski, A., and Oldfield, E., An NMR investigation of CO tolerance in a Pt/Ru fuel cell catalyst, J. Am. Chem. Soc., 2002, vol. 124, p. 468.
Pinheiro, A.L.N., Zei, M.S., and Ertl, G., Electro-oxidation of carbon-monoxide and methanol on bare and Pt-modified Ru (1010) electrodes, PhysChemChemPhys., 2005, vol. 7, p. 1300.
Yang, H., Dai, L., Xu, D., Fang, J., and Zou, S., Electrooxidation of methanol and formic acid on PtCu nanoparticles, Electrochim. Acta, 2010, vol. 55, p. 8000.
Li, X., Zhou, Y., Du Y., Xu, J., Wang, W., Chen, Z., and Cao, J., PtCu nanoframes as ultra-high performance electrocatalysts for methanol oxidation, Int. J. Hydrogen Energy, 2019, vol. 44, p. 18050.
Santasalo-Aarnio, A., Borghei, M., Anoshkin, I.V., Nasibulin, A.G., Kauppinen, E.I., Ruiz, V., and Kallio, T., Durability of different carbon nanomaterial supports with PtRu catalyst in a direct methanol fuel cell, Int. J. Hydrogen Energy, 2012, vol. 37, p. 3415.
Çögenli, M. S. and Yurtcan, A. B., Catalytic activity, stability and impedance behavior of PtRu/C, PtPd/C and PtSn/C bimetallic catalysts toward methanol and formic acid oxidation. Int. J. Hydrogen Energy, 2018, vol. 43, p. 10698.
Stamenković, V., Schmidt, T.J., Ross, P.N., and Marković, N.M., Surface segregation effects in electrocatalysis: kinetics of oxygen reduction reaction on polycrystalline Pt3Ni alloy surfaces, J. Electroanal. Chem., 2003, vols. 554–555, p. 191.
Guofeng, W., van Hove, M.A., Ross, P.N., and Baskes, M.I., Quantitative prediction of surface segregation in bimetallic Pt–M alloy nanoparticles (M = Ni, Re, Mo), Prog. Surf. Sci., 2005, vol. 79, p. 28.
Noel, K. and Xin, W., Pt-shell-Au-core/C electrocatalyst with a controlled shell thickness and improved Pt utilization for fuel cell reactions, Electrochem. Commun., 2008, vol. 10, p. 12.
Hua, L.M. and Shan, D.J., Kinetics of oxygen reduction reaction on Co rich core–Pt rich shell/C electrocatalysts, J. Power Sources, 2009, vol. 188, p. 353.
Chen, L-N., Hou, K.-P., Liu, Y.-S., Qi, Z., Zheng, Q., Lu, Y.-H., Chen, J.-Y., Chen, J.-L., Pao, C.-W., Wang, S.-B., Li, Y.-B., Xie, S.-H., Liu, F-D., Prendergast, D., Klebanoff, L.E., Stavila, V., Allendorf, M.D., Guo, J., Zheng, L-S., Su, J., and Somorjai, G.A., Efficient Hydrogen Production from Methanol Using A Single-Site Pt1/CeO2 Catalyst, J. Amer. Chem. Soc., 2019, vol. 141, p. 17995.
Papavasiliou, J., Paxinou, A., Słowik, G., Neophytides, S., and Avgouropoulos, G., Steam Reforming of Methanol over Nanostructured Pt/TiO2 and Pt/CeO2 Catalysts for Fuel Cell Applications, Catalysts, 2018, vol. 8, p. 544.
Kuriganova, A., Chernysheva, D., Faddeev, N., Leontyev, I., Smirnova, N., and Dobrovolskii, Y., PAC Synthesis and Comparison of Catalysts for Direct Ethanol Fuel Cells, Processes, 2020, vol. 8, p. 712.
Zhang, K., Feng, C., He, B., Dong, H., Dai, W., Lu, H., and Zhang, X., An advanced electrocatalyst of Pt decorated SnO2/C nanofibers for oxygen reduction reaction, J. Electroanal. Chem., 2016, vol. 781, p. 198.
Zhang, N., Zhang, S., Du, C., Wang, Z., Shao, Y., Kong, F., Lin, Y., and Yin, G., Pt/Tin Oxide/Carbon Nanocomposites as Promising Oxygen Reduction Electrocatalyst with Improved Stability and Activity, Electrochim. Acta, 2014, vol. 117, p. 413.
Huang, S.-Y., Ganesan, P., and Popov, B.N., Titania supported platinum catalyst with high electrocatalytic activity and stability for polymer electrolyte membrane fuel cell, Appl. Catal. B: Environmental, 2011, vol. 102, p. 74.
Akalework, N.G., Pan, C.-J., Su, W.-N., Rick, J., Tsai, M.-C., Lee, J.- F., Lin, J.-M., Tsai, L.-D., and Hwang, B.-J., Ultrathin TiO2-coated MWCNTs with excellent conductivity and SMSI nature as Pt catalyst support for oxygen reduction reaction in PEMFCs, J. Mater. Chem., 2012, vol. 22, p. 20977.
Esfahani, R.A.M., Videla, A.H.M., Vankova, S., and Specchia, S., Stable and methanol tolerant Pt/TiOx–C electrocatalysts for the oxygen reduction reaction, Int. J. Hydrogen Energy, 2015, vol. 40, p. 14529.
Ando, F., Tanabe, T., Gunji, T., Tsuda, T., Kaneko, S., Takeda, T., Ohsaka, T., and Matsumoto, F., Improvement of ORR Activity and Durability of Pt Electrocatalyst Nanoparticles Anchored on TiO2/Cup-Stacked Carbon Nanotube in Acidic Aqueous Media, Electrochim. Acta, 2017, vol. 232, p. 404.
Kuriganova, A.B., Leontyev, I.N., Alexandrin, A.S., Maslova, O.A., Rakhmatullin, A.I., and Smirnova, N.V., Electrochemically synthesized Pt/TiO2–C catalysts for direct methanol fuel cell applications, Mendeleev Commun., 2017, vol. 27, p. 67.
Wang, J., Xu, M., Zhao, J., Fang, H., Huang, Q., Xiao, W., Li, T., and Wang, D., Anchoring ultrafine Pt electrocatalysts on TiO2–C via photochemical strategy to enhance the stability and efficiency for oxygen reduction reaction, Appl. Catal. B: Environmental, 2018, vol. 237, p. 228.
De Oliveira, M.B., Profeti, L.P.R., and Olivi, P., Electrooxidation of methanol on PtMyOx (M = Sn, Mo, Os or W) electrodes, Electrochem. Commun., 2005, vol. 7, p. 703.
Rousseau, S., Coutanceau, C., Lamy, C., and Léger, J.-M., Direct ethanol fuel cell (DEFC): Electrical performances and reaction products distribution under operating conditions with different platinum-based anodes, J. Power Sources, 2006, vol. 158, p. 18.
Cui, X., Cui, F., He, Q., Guo, L., Ruan, M., and Shi, J., Graphitized mesoporous carbon supported Pt–SnO2 nanoparticles as a catalyst for methanol oxidation, Fuel, 2010, vol. 89, p. 372.
Wang, X., Hu, X., Huang, J., Zhang, W., Ji, W., Hui, Y., and Yao, X., Electrospinning synthesis of porous carbon fiber supported Pt-SnO2 anode catalyst for direct ethanol fuel cell, Solid State Sci., 2019, vol. 94, p. 64.
Menshchikov, V.S., Alekseenko, A.A., Guterman, V.E., Nechitailov, A., Glebova, N.B., Tomasov, A.A., Spiridonova, O.A., Belenov, S.V., Zelenina, N.K., and Safronenko, O.I., Effective Platinum–Copper Catalysts for Methanol Oxidation and Oxygen Reduction in Proton-Exchange Membrane Fuel Cell, Nanomaterials, 2020, vol. 10, p. 742.
Guterman, V.E., Lastovina, T.A., Belenov, S.V., Tabachkova, N.Yu., Vlasenko, V.G., Khodos, I.I., and Balakshina, E.N., PtM/C (M = Ni, Cu, or Ag) electrocatalysts: effects of alloying components on morphology and electrochemically active surface areas, J. Solid State Electrochem., 2013, vol. 18, p. 1307.
Novomlinskiy, I.N., Guterman, V.E., Danilenko, M.V., and Volochaev, V.A., Platinum Electrocatalysts Deposited onto Composite Carbon Black–Metal Oxide Support, Russ. J. Electrochem., 2019, vol. 55, p. 690.
Guterman, V.E., Novomlinskij, I.N., Skibina, L.M., and Mauer, D.K., Method for obtaining nanostructural material of tin oxide on basis of carbon, Pat. 2656914 (Russia), 2017.
Langford, J.I. and Wilson, A.J.C., Scherrer after Sixty Years: A Survey and Some New Results in the Determination of Crystallite Size, J. Appl. Crystallography, 1978, vol. 11, p. 102.
Borup, R., Meyers, J., Pivovar, B., Kim, Yu.S., Mukundan, R., Garland, N., Myers, D., Wilson, M., Garzon, F., and Wood, D., More scientific aspects of polymer electrolyte fuel cell durability and degradation, Chem. Rev., 2007, vol. 107, p. 3904.
Pavlov, V.I., Gerasimova, E.V., Zolotukhina, E.V., Dobrovolsky, Y.A., Don, G.M., and Yaroslavtsev, A.B., Degradation of Pt/C electrocatalysts having different morphology in low-temperature PEM fuel cells, Nanotech. Russia, 2016, vol. 11, p. 743.
Zhang, Y., Chen, S., Wang, Y., Ding, W., Wu, R., Li, L., Qi, X., and Wei, Z., Study of the degradation mechanisms of carbon-supported platinum fuel cells catalyst via different accelerated stress test, J. Power Sources, 2015, vol. 273, p. 62.
Ohma, A., Shinohara, K., Iiyama, A., Yoshida, T., and Daimaru, A., Fuel cells by FCCJ membrane, catalyst, MEA WG membrane and catalyst performance targets for automotive, ECS Trans., 2011, vol. 41, p. 775.
Capelo, A., Esteves, M.A., Sa, A.I., Silva, R.A., Cangueiro, L., Almeida, A., Vilar, R., and Rangel, C.M., Stability and durability under potential cycling of Pt/C catalyst with new surface-functionalized carbon support, Int. J. Hydrogen Energy, 2016, vol. 41, p. 12962.
Hasche, F., Oezaslan, M., and Strasser, P., Activity, stability, and degradation mechanisms of dealloyed PtCu3 and PtCo3 nanoparticle fuel cell catalysts, ChemCatChem., 2011, vol. 3, p. 1805.
Park, Yu.-Ch., Kakinuma, K., Uchida, M., Uchida, H., and Watanabe, M., Deleterious effects of interim cyclic voltammetry on Pt/carbon black catalyst degradation during start-up/shutdown cycling evaluation, Electrochim. Acta, 2014, vol. 123, p. 84.
Riese, A., Banham, D., Ye, S., and Sun, X., Accelerated stress testing by rotating disk electrode for carbon corrosion in fuel cell catalyst supports, J. Electrochem. Soc., 2015, vol. 162, p. F783.
Moguchikh, E.A., Alekseenko, A.A., Guterman, V.E., Novikovsky, N.M., Tabachkova, N.Yu., and Menshchikov, V.S., Effect of the composition and structure of Pt(Cu)/C electrocatalysts on their stability under different stress test conditions, Russ. J. Electrochem., 2018, vol. 54, p. 979.
Alekseenko, A.A., Moguchikh, E.A., Safronenko, O.I., and Guterman, V.E., Durability of de-alloyed PtCu/C electrocatalysts, Int. J. Hydrogen Energy, 2018, vol. 43, p. 22885.
Oezaslan, M. and Strasser, P., Activity of dealloyed PtCo and PtCu nanoparticle electrocatalyst for oxygen reduction reaction in polymer electrolyte membrane fuel cell, J. Power Sources, 2011, vol. 196, p. 5240.
Oezaslan, M., Hasche, F., and Strasser, P., PtCu3, PtCu and Pt3Cu Alloy Nanoparticle Electrocatalysts for Oxygen Reduction Reaction in Alkaline and Acidic Media, J. Electrochem. Soc., 2012, vol. 159, p. 444.
Guterman, V.E., Belenov, S.V., Alekseenko, A.A., Rui, Lin, Tabachkova, N.Yu., and Safronenko, O.I., Activity and Stability of Pt/C and Pt–Cu/C Electrocatalysts, Electrocatalysis, 2018, vol. 9, p. 550.
Van der Vliet, Dr. D.F., Wang, D.F., Li, C., Paulikas, D., Greeley, A.P., Rankin, J., R.B., Strmcnik, D., Tripkovic, D., Markovic, N.M., and Stamenkovic, V.R., Unique Electrochemical Adsorption Properties of Pt-Skin Surfaces, Angew. Chem. Int. Ed., 2012, vol. 51, p. 3139.
Rudi, S., Cui, C., Gan, L., and Strasser, P., Comparative Study of the Electrocatalytically Active Surface Areas (ECSAs) of Pt Alloy Nanoparticles Evaluated by Hupd and CO-stripping voltammetry, Electrocatalysis, 2014, vol. 5, p. 408.
Ghavidel, Z.M.R., Monteverde Videla, A.H.A., Specchia, S., and Easton, E.B., The relationship between the structure and ethanol oxidation activity of Pt–Cu/C alloy catalysts, Electrochim. Acta, 2017, vol. 230, p. 58.
Maillard, F., Schreier, S., Hanzlik, M., Savinova, E.R., Weinkauf, S., and Stimming, U., Influence of particle agglomeration on the catalytic activity of carbon-supported Pt nanoparticles in CO monolayer oxidation, PhysChemChemPhys., 2005, vol. 7, p. 385.
Liu, C., Zhang, L., Sun, L., Wang, W., and Chen, Z., Enhanced electrocatalytic activity of PtCu bimetallic nanoparticles on CeO2/carbon nanotubes for methanol electro-oxidation, Int. J. Hydrogen Energy, 2020, vol. 45, p. 8558.
Wang, X., Wang, W., Qi, Z., Zhao, C., Ji, H., and Zhang, Z., Fabrication, microstructure and electrocatalytic property of novel nanoporous palladium composites, J. Alloys Compounds, 2010, vol. 508, p. 463.
Funding
The reported study was funded by the Russian Foundation of Basic Research according to the research project no. 19-33-90140.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by Yu. Pleskov
Rights and permissions
About this article

Cite this article
Menshchikov, V.S., Belenov, S.V., Novomlinsky, I.N. et al. Multi-Component Platinum-Containing Electrocatalysts in the Reactions of Oxygen Reduction and Methanol Oxidation. Russ J Electrochem 57, 587–597 (2021). https://doi.org/10.1134/S1023193521060070
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1023193521060070