
Abstract
Hybrid MOx–C (M = Sn, Ni) supports and Pt/MO x –C catalysts for direct alcohol fuel cells were prepared via the electrochemical oxidation and dispersion of a metal under the influence of a pulsed alternating current. We estimated conductivity and specific surface area of as-prepared SnO2, NiO and hybrid supports, as well as estimated their morphology and the morphology of Pt-based catalysts using X-ray diffraction, transmission and scanning electron microscopy analyses. Pt/MO x –C (M = Sn, Ni) catalytic systems with oxide contents of 30 and 60 % in hybrid supports and a Pt content of 25 % were used for the electrochemical oxidation of CO, methanol and ethanol in acidic and alkaline solutions. The presence of oxides in the hybrid supports for catalysts reduces the onset potential for the electrooxidation of CO and alcohols in an acidic solution. At Pt/SnO2–C, the onset potential of ethanol electrooxidation in an acidic solution decreases by 170 mV and increases the rate of the oxidation process by more than sevenfold at the low Tafel potential region. In alkaline solutions, the presence of metal oxides in the catalysts is not so effective, which is probably due to the high coverage of the platinum surface with oxygen-containing species. The Pt catalyst on the SnO2–C hybrid support exhibited superior electrochemical stability in an acidic solution and Pt/NiO–C—in an alkaline solution. The difference in the optimum oxide content (30 % for SnO2, 60 % for NiO) of the catalysts can be attributed to different morphologies of the oxides.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Low-temperature fuel cells (LTFCs) are promising future devices to provide power for vehicles, portable devices and emergency electric power sources because they result in less damage to the environment than current powering methods. Hydrogen and alcohols (methanol or ethanol) are usually used as a fuel for proton or anion exchange membrane fuel cells (PEMFCs or AEMFCs) and direct alcohol fuel cells (DAFCs), respectively [1–3]. For a more active extension of the scope of LTFCs, one needs to reduce their cost and increase their safety, durability, and life cycle. The main problem regarding the use of cheap hydrogen produced by hydrocarbon reforming is the poisoning of the catalyst layer by CO. The presence of over 10 ppm CO in hydrogen significantly reduces the operating current of the LTFC.
DAFCs have some advantages over hydrogen as a fuel: the use of alcohols does not involve the use of assistive systems, such as humidifiers and a gas pressure delivery system, and the storage of alcohols is more compact and safer compared with hydrogen [2]. Concerning their application in fuel cells, methanol and ethanol have both advantages and disadvantages when compared with each other. Methanol is oxidized much easier because it has only one carbon atom. On the other hand, methanol is toxic and highly soluble in water, making it very dangerous in an emergency. Ethanol is a more attractive fuel and can be easily produced in a large quantity by the fermentation of sugar-containing raw materials and biomass [4].
Nanodispersed Pt particles supported on carbon black are very effective catalysts for LTFCs. However, adsorbed CO or chemisorbed intermediate products of alcohol oxidation may block the active sites of the platinum catalyst and decrease its activity, which reduces the FC’s efficiency [5]. CO and alcohol adsorption as well as oxidation at Pt electrodes were studied by many researchers [3, 6–8]. All types of adsorbed particles are oxidized to CO2 via the Langmuir–Hinshelwood mechanism (Eq. 1) [9], and for their oxidation, the presence of adsorbed oxygen-containing species (OHads or Oads) that appear on platinum as a result of water molecule discharge is necessary. In particular, the low-surface concentration of the oxygen-containing species in the reaction zone causes a high overpotential of alcohol’s anodic oxidation process and a low tolerance to the CO poisoning of platinum [8, 10]
A number of metals, such as Ru, Sn, Ni, Co, and Fe, which adsorb oxygen at a higher cathodic potential than platinum, have been introduced in platinum-based catalysts. In this case, a bifunctional mechanism of electrocatalysis applies (Eq. 2) [11]:
As is generally known, ruthenium has the best oxygen absorbency and lability [7, 11], but many researchers are turning their attention to cheaper metals, such as tin and nickel. Those metals have been introduced in the catalytic system as ad-atoms [12, 13], as alloys with platinum [14–16] and metal oxides [17, 18]. An effective enhancement of the catalytic properties of single-platinum crystals by the deposition of irreversibly adsorbed ad-atoms was found for the oxidation of methanol [19] and ethanol [20].
In all of these cases, significant catalytic effects when using these electrodes have been obtained. However, it should be noted that the ad-atom approach is a model system that is necessary for developing an understanding of the regularity of the processes on electrodes and the nature of the catalyst, but it cannot be used in real fuel cells. Catalysts containing alloyed particles are more stable, but non-noble metal leaching will definitely reduce the catalytic effect with time [21]. Additionally, the bi-phase PtSn + SnO x compared with the purely alloyed PtSn catalysts were more active for ethanol electrooxidation at lower overpotentials, which is of practical interest for applications such as the catalytic system in direct oxidation fuel cells [22]
Another reason for the reduction of the fuel cell’s efficiency during operation is the degradation of the catalyst layer, which can be explained by catalyst particle agglomeration [23] as well as the oxidation of the carbon support [24, 25]. On this basis, the development of CO-tolerant and resilient to oxidative degradation catalytic materials is an important direction for the resolution of the problem described above.
Traditionally, carbon black is used as the support for Pt nanoparticles (Pt NPs) because it has a good combination of electronic conductivity, specific surface area and porosity. However, carbon corrosion during fuel cell operation is one of the main reasons for the decrease of the catalytic activity of Pt-based catalysts [23, 24]. To solve this problem, other support materials, such as metal oxides (MO x ), were investigated [18, 26–36]. Some of the results dedicated to this issue are summarized in Table 1:
It should be noted that all of those oxides (SnO2, NiO, TiO2, WO3, CeO2 and ZrO2) have shown good chemical and electrochemical stabilities and, in some cases, a good catalytic effect.
However, general problems in the syntheses of all of the given materials are that the syntheses are (i) multistage and complex (ii) time-consuming, as well as requiring (iii) the use of corrosive reagents and organic solvents and (iv) calcination at high temperatures.
Moreover, the values of the conductivity and specific surface area of the metal oxides or hybrid supports are not present in most of those studies. However, the metal oxides are semiconductors, and the complete replacement of the carbon support with MO x is problematic because of their lower electrical conductivity. Thus, hybrid MO x /C materials should have the optimal mixture of conductivity, catalytic properties and durability.
MO x and MO x /C composite supports for Pt NPs can be produced by different methods, including hydrothermal synthesis [37], Bönnemann method [38], spontaneous deposition [39], thermal decomposition of polymeric precursors [40] and precipitation [34, 41].
SnO2 and NiO are the most inexpensive and available metal oxides, whose properties and conductivities strongly depend on their preparation conditions. Moreover, it was found that these oxides promote Pt-based electrocatalysts during the electrooxidation of small organic molecules [42].
Nickel and tin oxides with a high electronic conductivity and appropriate structural characteristics can be synthesized by the electrochemical dispersion method [43–46]. This method is based on electrochemical oxidation and dispersion of metal electrodes under the action of an alternating pulse current. The technique is characterized by the simplicity of its procedure, high rate of the process, the lack of a need to use capping agents, its calcination procedure and the ability to produce nanosized platinum-based catalysts with various types of support—carbon black with different microstructures [47] as well as metal oxide support [48].
The main goal of the current study is to prepare hybrid MO x –C supports and Pt/MO x –C electrocatalysts an electrochemical dispersion approach and to study their activities and stabilities towards CO, methanol and ethanol oxidation in acidic and alkaline electrolytes. The fundamental investigation of the electrocatalytic properties of these materials is important for their further implementation in LTFCs, assisting the development of fuel cells.
2 Materials and methods
2.1 Electrochemical preparation of hybrid MO x –C supports and Pt/MO x –C catalysts (M = Sn, Ni)
For the preparation of hybrid MO x –C supports and Pt/MO x –C catalysts (M = Sn, Ni), we used different approaches: the preparation of hybrid MO x –C supports by the mechanical mixing of metal oxide powders with carbon black (Fig. 1(i)) and a one-pot electrochemical preparation of the hybrid MO x –C supports (Fig. 1(ii, iii)). Pt nanoparticles were prepared by the same electrochemical dispersion technique and deposited onto the hybrid MO x –C supports.

Scheme of Pt/MO x –C (M = Sn, Ni) catalysts synthesis via electrochemical oxidation and dispersion method. (a) Electrochemical oxidation and dispersion of Sn or Ni electrodes under action of alternating pulse current, current density 1 A cm−2 and current frequency 50 Hz; electrolyte 2 M NaCl for Sn, 2 M NaOH for Ni; mechanical mixing. (b) Filtrating, washing with distillate water, drying at 80 °C. (c) Mechanical mixing of MO x and Vulcan XC-72 in 2 M NaOH. (d) Electrochemical dispersion of Pt electrodes under action of alternating pulse current, current density 1 A cm−2 and current frequency 50 Hz; electrolyte 2 M NaOH + MO x –C, mechanical mixing. (e) Electrochemical oxidation and dispersion of Sn electrodes under action of alternating pulse current, current density 1 A cm−2 and current frequency 50 Hz; electrolyte 2 M NaCl + Vulcan XC-72; mechanical mixing. (f) Electrochemical oxidation and dispersion of Ni electrodes under action of alternating pulse current, current density 1 A cm−2 and current frequency 50 Hz; electrolyte 2 M NaOH + Vulcan XC-72; mechanical mixing
2.1.1 Metal oxide (MO x ) powder preparation
Two tin or two nickel foil electrodes (thickness 0.25 mm) were immersed in an aqueous solution of 2 M NaCl or 2 M NaOH, respectively [43, 45]. The electrodes were connected to a pulse alternating current (ac) source operating at 50 Hz. The current density was 1 A cm −2geom . Under pulse ac operation, the metal electrodes were oxidized and dispersed into the electrolyte (Fig. 1(i)). The as-prepared metal oxide powder formed after the electrolysis the products were collected by filtration, washed with double distilled water, and dried at 80 °C to constant weight. This treatment is referred to as purification.
2.1.2 Hybrid MO x –C support preparation
The hybrid MO x –C support preparation was carried out in two ways. The first way was the mechanical mixing of a desired amount of as-prepared metal oxide with a 30 g L−1 suspension of the carbon black powder Vulcan XC-72 (Cabot. Corp.) in a 2 M NaOH aqueous solution. The suspension was mixed with a magnetic stirrer for 1 h (Fig. 1(i)). The second way involved the one-pot preparation of the hybrid MO x –C supports (M = Sn, Ni) via electrochemical oxidation and the dispersion of the tin or nickel electrodes in a suspension of Vulcan XC-72 in 2 M NaCl or 2 M NaOH, respectively (Fig. 1(ii, iii)). It should be noted that the NiO–C hybrid supports in contradistinction to the SnO2–C hybrid supports do not need to be purified before the Pt dispersion process because the dispersions of the platinum electrodes and nickel electrodes are carried out in the same aqueous solution. The SnO2–C hybrid supports need to be purified (second stage) because 2 M NaCl was used as the electrolyte, and Cl− ions can subsequently have a negative effect on the platinum dispersion process.
2.1.3 Pt/MO x –C catalyst preparation
Two platinum foil electrodes were immersed in the suspension of the hybrid MO x –C support in a 2 M NaOH aqueous solution and were electrochemically dispersed under a pulsed ac operation at 50 Hz, and the current density was 1 A cm −2geom , as was reported in [46]. Finally, the freshly prepared Pt–MO x /C catalysts (M = Sn, Ni) were purified as described above. The metal oxide loading in the catalysts was 30 and 60 %, with a platinum loading of 25 %. For comparison, Pt/C catalysts were prepared the same way as was reported earlier [46].
2.2 Physical characterization of catalysts
The XRD studies and crystallite size determination were performed with an ARL X’TRA powder diffractometer, Thermo Scientific, (Cu K α target, λ = 1.5418 Å). XRD data were collected in the 2θ range of 20°–100° using step scan mode with a step width of 0.01° and a step time of 2.00 s. A sufficient intensity could be obtained with this step width and was found to be suitable for obtaining a satisfactory profile shape. The average grain size was determined after applying a correction for the instrumental and strain broadening using the well-known Scherrer equation (Eq. 3):
where λ is the wavelength, D is the volume averaged grain size, K = 0.89 is the Scherrer constant, FWHM is the full width at half maximum of the diffraction peak, and θ is the Bragg angle of the [h k l] reflection.
The morphology of the Pt/MO x –C catalysts was investigated using a ZEISS Supra 25 unit operated at 20 kV. A practical size distribution of samples was obtained with transmission electron microscopy (TEM) using a JEM-2100 (JOEL).
Low-temperature N2 adsorption and desorption measurements were performed using a Micromeritics ASAP 2020 analyser at 77 K. All of the investigated samples were degassed at 473 K for 24 h in a vacuum prior to the adsorption experiment. The specific surface area was determined in the relative pressure range 0.05 ≤ P/P 0 ≤ 0.20 according to the Brunauer–Emmett–Teller (BET) method for NiO, SnO2 and hybrid MO x –C materials.
The electrical conductivity (σ) of the metal oxide powders and hybrid MO x –C supports was measured at room temperature using a four-electrode arrangement. The powdered material was compressed (500 kPa) in a glass tube (0.4 cm diameter) between two brass electrodes. The value obtained for the electric conductivity was determined in the range between 0 and 1.5 A (scan rate 100 mA s−1).
2.3 Electrochemical measurements
The electrochemical measurements were performed at room temperature using a potentiostat/galvanostat P-30 J (Elins) in a three-electrode cell. Platinum wire was used as a counter electrode. The potentials were measured versus a saturated Ag/AgCl reference electrode. The potentials cited in this work were related to the reversible hydrogen electrode (RHE).
The catalytic ink was prepared with 14 mg of catalyst, 1 mL of isopropanol and 24 µL of a 10 wt% Nafion® solution (Du Pont). The mixture was mixed with a magnetic stirrer (30 min), and 60 µL of the catalytic ink was dropped onto a glassy carbon electrode (~0.2 mgPt) and dried at 80 °C in air for 40–50 min.
Cyclic voltammograms (CVs) were conducted in a nitrogen-saturated 0.5 M H2SO4 or 1 M NaOH electrolyte for a potential range of 50 to 1300 mV versus RHE. Before CV testing, the working electrode was cycled for 20 cycles to stabilize the system. The activity of the synthesized catalysts for the methanol oxidation reaction (MOR) and ethanol oxidation reaction (EOR) was measured by potential cycling between 50 to 1300 mV versus RHE in a 0.5 M H2SO4 + 0.5 M alcohol or 1 M NaOH + 0.5 M alcohol electrolyte.
The CO-stripping voltammograms were used for the determination of the electrochemically active surface area (ESA) of the Pt/C electrocatalysts. CO was adsorbed at the surface of the working electrode by bubbling CO through 0.5 M H2SO4 for 15 min, while the electrode was kept at a constant potential of 300 mV versus RHE. After CO bubbling, the electrolyte was purged with N2 (30 min) to remove the dissolved CO, and the CV curves were measured. The charge required for the desorption of a COads monolayer was 420 µC cm−2 of the true surface, i.e. only linear COads adsorption was assumed.
The ESA was evaluated by calculating the charge accumulated during CO adsorption after subtracting the double-layer charging current (Eq. 4),
where Q CO (µC) is the charge passed during the CO oxidation, g Pt is the mass of Pt on the working electrode and Q m = 420 (µC cm−2) [49]—the charge required for the desorption of a COads monolayer.
3 Results and discussion
3.1 Physical characterization of oxides MO x , hybrid supports MO x –C and Pt/MO x –C catalysts (M = Sn, Ni)
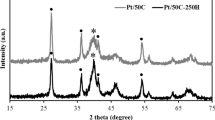
The oxide materials synthesized during the first step with route (i) were characterized by means of XRD. The analysis of the patterns for pure tin dioxides (blue line in Fig. 2a) showed that it has the typical tetragonal structure of SnO2 [space group: P42/mnm (no. 136)] with a unit cell parameter a = 4.739 Å that is slightly larger than that of rutile SnO2 (a = 4.737 Å), as was also reported in [50]. Earlier, we showed that a small amount of less than 1.5 at% of Cl is included in the as-prepared SnO2 powder [43] indicating the considerable oxygen deficiency of the as-prepared SnO2 powder. Moreover, chlorine has an ionic radius (1.83 Å) that is larger than that of oxygen (1.4 Å), and the substitution of a O2− site by a Cl− ion leads to high lattice strain, hence, the increase in the unit cell parameter.

XRD patterns of metal oxides and Pt catalysts on hybrid SnO2–C (a) and NiO–C (b) supports; TEM images of Pt (c), SnO2 d nanoparticles and NiO plates e synthesized via electrochemical oxidation and dispersion technique
The analysis of the patterns for pure nickel oxides (blue line on Fig. 2b) showed that the unit cell parameter of the as-prepared NiO is 4.194 Å. This is greater than the standard value of 4.17 Å (Fm3m, JCPDS 75-0269). As was shown earlier [45, 51], the increase in the unit cell parameter may be caused by the doping of NiO by Na+ during the synthesis to form a solid solution of Na x Ni 2+1−2x Ni3+O. The ionic radius of sodium (1.02 Å) is considerably larger than the ionic radius of nickel (0.6 Å), which means the substitution of a Ni2+ site by a Na+ ion results in lattice strain and increases the unit cell parameter. Thus, the used electrochemical technique allowed for the metal oxides with defect-induced structures.
The average crystal sizes calculated by using Scherrer equation are 15.6 and 9.1 nm for SnO2 and NiO, respectively. However, the SnO2 morphology can be described as individual particles, while the NiO morphology is a tightly packed array of nanoplates with a length of 500–2000 nm (Fig. 2d, e). Thus, SnO2 particles are slightly smaller but comparable to carbon black particles (30–40 nm), while NiO particles are significantly larger.
It should also be noted that the doping of the p-type semiconductor NiO by the cation Na+ and of the n-type semiconductor SnO2 by the anion Cl− leads to an additional increase in the nonstoichiometry; therefore, it can lead to an increase in the conductivities of the synthesized metal oxides. The conductivities of the synthesized NiO and SnO2 were 1 × 10−3 and 5 × 10−2 S cm−1, respectively, (Fig. 3a), and are some orders of magnitude higher than the room temperature conductivities of stoichiometric single crystals of NiO (10−9 S cm−1) and SnO2 (>10−4 S cm−1) [51, 52].

Conductivity (a) and BET specific surface area (b) of carbon black Vulcan XC-72, SnO2 and SnO2–C, NiO and NiO–C materials. Shaded area shows the interval of threshold percolation for different models of conductivity of strongly inhomogeneous media [53]
Although the conductivities of the as-prepared metal oxides are sufficiently large for a semiconductor, it is necessary to increase the conductivity by 2–3 order to use the metal oxides as supports for fuel cell catalysts. This is possible through the introduction of a good electronic conductor—carbon, thus creating a hybrid support “electronic conductor-semiconductor”. The dependence of the conductivity of such materials on the composition of the material is described by the percolation theory in strongly inhomogeneous media [53]. Based on this theory, we calculated the area of the metal oxide and carbon black loading where a percolation conductivity has to be observed, regardless of the particle morphologies of the two different components. The minimum value of the content of the conductive component in the material should be at least 65 v.v%. In Fig. 3a, the percolation area is highlighted by shading. The prepared materials have 68–70 and 87–88 v.v% of carbon black, which corresponds to 60 and 30 wt% of metal oxide. To eliminate the influence of the possible leaching of the less noble component (MO x ) during catalyst exploitation, we used the supports with the highest possible oxide loading. Figure 3a clearly shows that the prepared MO x –C hybrid supports have a high electronic conductivity, which increases with the carbon black loading, that is generally similar to the conductivity of carbon black (20 S cm−1). Thus, we determined the compositions that may be used as a hybrid support for the platinum catalysts of fuel cells.
The BET specific surface areas of the bare metal oxides and the hybrid support SnO2–C and NiO–C composites are shown in Fig. 3b. The as-prepared bare metal oxides have low specific surface areas of 82 and 79 m2 g−1 for tin dioxide and nickel oxide, respectively. However, when the carbon black Vulcan XC-7 was added, the specific surface area of the hybrid supports increased linearly. Even with a high loading of metal oxides (60 wt%), the specific surface areas of the hybrid supports are approximately 150 m2 g−1, which is sufficient to create an appropriate (proper) interface with the fuel cell’s catalytic layer.
The prepared Pt catalysts based on the MO x –C hybrid supports were also investigated by XRD. The XRD patterns of the Pt/C and Pt/MO x –C catalysts synthesized via the electrochemical oxidation and dispersion method are shown in Fig. 2a, b, and a TEM image of a Pt particle is shown in Fig. 2c. Three well-defined diffraction peaks at 39.6, 46.1 and 81.3° are assigned to the (111), (200) and (220) crystal planes, respectively, of the Pt face-centred cubic structure [space group: Fm3m (no. 225)]. In fact, the particle size along the [111] (D111) calculated from the Scherrer equation is 10.6 nm [46]. The metal oxide diffraction peaks display lower intensities, which increase with metal oxide content.
Scanning and transmission electron microscopy studies of the Pt/C and Pt/MO x –C catalysts showed that the platinum particles have a uniform distribution over the surface of the metals oxide as well as on the carbon surface (Fig. 4). Both Pt/NiO–C and Pt/SnO2–C catalysts have rather narrow size distributions of approximately 6–8 nm; however, as it is clear from the TEM images for Pt/SnO2–C, the presence of Pt agglomerates is more characteristic (Fig. 4e).

Scanning (a, d, g) and transmission (d, e, h) electron microscopy images; platinum particles size distribution (c, f, i) in Pt/C (a–c) and Pt/SnO2–C (d–f) and Pt/NiO–C (g–i) catalysts
3.2 Electrochemical measurements
3.2.1 ESA and features of CO electrooxidation on Pt/MO x –C catalysts
The ESA is the most important characteristic of the electrocatalytic materials. For the determination of the ESA, the CO-stripping method was used. The voltammetric curves corresponding to the CO-stripping from the Pt/MO x –C catalysts are presented in Fig. 5. The activity of Pt/C catalyst obtained by the same method is shown for comparison. The EAS of the Pt/C catalyst was 12 m2 g−1, while the presence of a metal oxide in the catalytic system leads to an increase in the specific surface area of the catalyst to 15 m2 g−1 for the Pt/NiO–C catalyst and 17 m2 g−1 for the Pt/SnO2–C catalyst (Table S1). The increase of the EAS in the presence of metal oxides can be due to a decrease of the Pt particles size and the better electrochemical performances of Pt on the surfaces of the metal oxides. In the presence of MO x , the electrical capacitance of the double electric layer of the Pt-based catalysts at 0.4–0.6 V decreases, as was shown in [54], and the hydrogen adsorption–desorption peaks are expressed more clearly.

CO-stripping on Pt/MO x –C catalysts compared to Pt/C catalysts in 0.5 M H2SO4 at scan rate 10 mV s−1. Line A onset potential of CO oxidation, line B potential of CO oxidation peak
It was observed that the Pt/C catalyst demonstrates an onset potential of CO oxidation at 0.55 V and a potential of the CO oxidation peak at 0.73 V (Fig. 5). In the presence of MO x and with increasing metal oxide content in the hybrid supports, the onset and peak potentials for CO oxidation are shifted in the cathodic direction, i.e. the overvoltage oxidation of CO adsorbed on the platinum decreases. In general, the overvoltage of the CO oxidation at Pt/SnO2–C is reduced by 90–140 mV, which is more than that at Pt/NiO–C (30–90 mV) in comparison with Pt/C catalyst (Table S1). This tendency is particularly expressed for Pt/SnO2–C, where the differences between the onset potentials of CO-stripping at the Pt/SnO2–C and Pt/C catalysts are 140 and 160 mV for Pt/SnO2–C (30 % SnO2) and Pt/SnO2–C (60 % SnO2), respectively. Although the presence of platinum agglomerates on the Pt/SnO2–C catalysts was observed by SEM and TEM (Fig. 4), this fact does not affect the value of the onset potential of CO oxidation on those catalysts. This may be attributed to a lack of influence of the aggregation effect on the onset potential and peak potential of CO oxidation [55].
The electrocatalytic behaviour of the Pt/MO x –C hybrid support is consistent with the well-known bifunctional mechanism of CO oxidation [11, 56]. Because OHad forms on the MO x surface at lower potentials than on Pt, CO oxidation occurs at the Pt/MO x –C catalysts at lower potentials than at Pt/C, reducing the anodic overvoltage and increasing the useful fuel cell voltage.
This enhanced effect of metal oxides on the stripping of CO adsorbed on the Pt surface was observed in previous studies [18, 54, 56, 57]. In [18], the CO stripping peak at Pt/NiO–C was shifted to the cathode side by 80 mV compared with that on Pt/C. The ESA of the catalysts in the presence of nickel oxide was higher than the value for the pure Pt/C catalyst. In [57], an analogous effect was observed for Pt/SnO2–C—the potential of the CO oxidation peak was observed at 0.62 V (RHE) compared with that at the Pt/C catalyst (0.76 V (RHE)). However, unlike [18] in [57], the ESA of catalysts in the presence of metal oxides was reduced more than ten times because of Pt particle agglomeration, which is due to the Pt selectively depositing onto the oxide sites [57]. It is very important to prepare catalysts with a good ratio of the sizes of the Pt and MO x particles and their loading in the catalyst, which allow for a three-phase boundary of platinum-metal oxide–carbon.
3.3 Electrochemical oxidation of alcohols
The as-prepared materials were tested for the electrooxidation of methanol and ethanol. The quasi steady-state polarization curves for the MOR and EOR at the Pt/MO x –C (M = Sn, Ni) catalysts are presented in Fig. 6. The Tafel slope for the methanol electrooxidation process is 60–63 mV for all of the investigated catalysts (Fig. 6a, b), as is usually observed on polycrystalline Pt and Pt/Pt electrodes [58]. The electrooxidation of ethanol is more difficult because of the slow rate of the adsorption step. The Tafel slopes in these cases are 152–132 mV [59], depending on catalyst composition, and they decrease with the increasing content of the oxide phase (Fig. 6c, d). In the Tafel region, the rate of MOR and EOR processes on the Pt/MO x –C catalysts are increased by 4–5 and 5–7 times, respectively, in comparison with Pt/C.

Tafel plots of Pt/SnO2–C catalysts (a, c) and Pt/NiO–C catalysts (b, d) compared to Pt/C catalysts in 0.5 M H2SO4 + 0.5 M CH3OH (a, b) and 0.5 M H2SO4 + 0.5 M C2H5OH (c, d) at scan rate 0.5 mV s−1
The CV curves of the Pt catalysts on the pure carbon (Vulcan XC-72) support Pt/C and the hybrid metal oxide–carbon supports Pt/SnO2–C, and Pt/NiO–C were measured in 0.5 M CH3OH and 0.5 M C2H5OH in acidic 0.5 M H2SO4 (Fig. S2) and alkaline 1 M NaOH (Fig. S3) solutions. The presence of the metal oxide in the support caused a cathodic shift of the onset and peak potential of the alcohol’s oxidation on both the forward and reverse scans (Fig. 7, black lines; Table S1). In particular, in the acidic medium, the MOR at Pt/C starts at nearly 0.65 V, while the onset potentials for Pt/SnO2–C (30 % SnO2) and Pt/SnO2–C (60 % SnO2) are 0.56 V and 0.58 V, respectively (Fig. 7a, black solid line). In the case of the EOR, the onset potential of the process at Pt/SnO2–C (30 % SnO2) and Pt/SnO2–C (60 % SnO2) is approximately 0.50 V and 0.55 V, respectively, which represent cathodic shifts of 170 and 120 mV, respectively, compared with the Pt/C catalyst (0.67 V) (Fig. 7b, black solid line). The tendency for the decrease of the onset potential of the MOR and EOR in the acid medium was also observed for the Pt/NiO–C catalysts (Fig. 7a, b, dash lines). The largest cathodic shift of the onset potential for alcohol oxidation of 110 mV was observed for the catalyst with a 60 % nickel oxide content in contrast to the Pt/SnO2–C catalysts in which the highest effect was demonstrated with a 30 % tin dioxide content (Table S1).

Influence of MO x loading on onset potential (solid lines) and specific surface activity at 0.6 V (dash lines) of Pt/SnO2–C catalysts (filled square) and Pt/NiO–C catalysts (filled circle) for 0.5 M CH3OH (a, c) and 0.5 M C2H5OH (b, d) oxidation in 0.5 M H2SO4 (a, b) and 1 M NaOH (c, d). Scan rate 5 mV s−1
The surface specific activity (SSA) of the as-prepared catalysts for alcohol oxidation at 0.6 V (on the forward scan) depends on the composition of the hybrid support. Therefore, in the acid medium, the SSA of tin dioxide containing materials has a maximum at 30 % SnO2. In this case, the SSA increases by approximately five times for the MOR and by more than seven times for the EOR compared with the SSA for Pt/C (Fig. 7 a, b, solid red lines). At the Pt/NiO–C catalyst, the influence of the metal oxide presence is less expressed—the maxima increases of the SSA are only approximately 2 and 4 times for the MOR and EOR, respectively, with 60 % NiO in the support (Fig. 7a, b, dash red lines). These data agree with the results under quasi steady-state conditions (Fig. 6).
In contrast to the oxidation in the acidic electrolyte, the presence of a metal oxide in the catalytic system caused a decrease in the overall rate and onset potential of the alcohols’ oxidation in alkaline media (Fig. 7c, d). In all of the cases, the onset potentials of the methanol and ethanol electro-oxidations in the presence of a metal oxide in the support are shifted towards more anodic potentials. In alkaline solutions, the specific surface activity of the Pt/C reference catalyst is higher than that for Pt/MO x –C (Table S1).
Discussing the results, we have to answer two main questions: why does the presence of a metal oxide in the catalyst composition accelerate the alcohol oxidation process in the acidic medium and inhibit the process in the alkaline medium? And why do the Pt catalysts on supports with different metal oxides show a different dependence of the catalytic activity on the oxide loading in the hybrid supports?
The forward scans of the alcohol oxidation curves measured in 0.5 M H2SO4 and 1 M NaOH at Pt/C and Pt/MO x –C are presented in Fig. 8. The MO x –C hybrid supports with 30 % SnO2 and 60 % NiO were selected for comparison as the most active catalytic materials. Lines A and B indicate the onset potential of the alcohol electrooxidation in acidic and alkaline solutions, respectively. The overall alcohol electrooxidation process at the pure Pt or Pt/C electrode, sometimes named the ‘dual pathway’, consists of parallel reactions, which can be formulated as shown in Fig. 1. For the realization of both of these pathways, a catalyst is required, and it should be able to (i) adsorb alcohol molecules and promote the dissociation of the C–H bond and C–C bond, leading to the formation of adsorbed intermediates (CH x , CHO, CO, etc.) [60] and (ii) facilitate the reaction of the adsorbed particles with some O-containing species to form CO2, HCOOH or CH3COOH. This means that on the surface of the catalyst has to be a certain balance between the adsorbed organic particles and oxygen-containing particles. The coverage of the catalyst’s surface by any type of adsorbate depends on the electrode potential. Therefore, the adsorption of alcohols at the surface of polycrystalline Pt can only begin at potentials near 0.2 V (RHE), but a strong interaction of water with the Pt surface is only possible at potentials above 0.45 V (RHE). This explains the fact that a high rate of oxidation at pure Pt occurs at potentials of ~0.7 V (RHE) in the acidic solution, which is too anodic for fuel cell applications [7].

Electrochemical oxidation of methanol (a, b and c) and ethanol (d, e and f) on Pt/C and Pt/MO x –C (M = Sn, Ni) catalysts in acidic (solid line) and alkaline (dash line) electrolytes, scan rate 5 mV sec−1
There are two ways to solve this problem. The first way is to increase the electrolyte pH, resulting in a change of the alcohol oxidation kinetics and better polarization characteristics of the alcohol oxidation on platinum compared with acidic media [61]. The difference between the onset potentials of the methanol and ethanol electrochemical oxidations at Pt/C in acidic and alkaline electrolytes is approximately 200 mV (RHE), because, in alkaline media, OHad formation is initiated at the Hupd potential region in contrast to that in acidic media [62]. Therefore, most electrocatalytic processes should be more facile in alkaline solutions than in acidic solutions [63]. The second way is using a two-component catalysts, for example, a Pt alloy with oxygen adsorbing metals or Pt supported on metal oxides, which allow for the creation of more favourable conditions for the alcohol electrooxidation process at Tafel region potentials. In this case, alcohols molecules are adsorbed with dehydration on the Pt surface, while the oxygen adsorbing metal provides the presence of oxygen-containing species in the reaction zone. On the other hand, in [64], it is suggested that the differences between the EOR in acidic and alkaline media mainly arise from the different reactivities of the adsorbed intermediate CH x species in the alkaline solution.
In the acid electrolyte, the presence of tin dioxide in the catalyst composition has a greater impact than the presence of nickel oxide. The maximum effect is observed for a catalyst with 30 % SnO2: the onset potential of the ethanol oxidation shifts toward the cathode by 170 mV (Fig. 8). The mechanism of the accelerating effect of the oxide component in the support for the MOR can be explained by the theory of bifunctional catalysis. [7, 65]. In the case of the EOR, electronic ligand effects are possible as well [56]. In the alkaline electrolyte, the presence of an oxide in the catalyst composition does not affect the process of ethanol oxidation (Fig. 8d–f), and negatively affects the oxidation of methanol. The onset potential of methanol oxidation in the presence of a nickel oxide is shifted to the anode side (Fig. 8a–c). Taking into account the high coverage of the Pt surface by OHad in alkaline media, sometimes the additional introduction of O-adsorbing metals can further enhance the surface concentration of OH, which induces negative effects, e.g. inhibits alcohol adsorption [7]. Thus, the coverage of OHad on the Pt surface plays an important role in the overall process rate: a promoting effect at low-surface coverage and an inhibiting effect at high surface coverage [62, 65]. It is likely that in the presence of oxides in the catalyst, the surface coverage of platinum by OHad species increases too strongly. This impedes the adsorption of alcohol molecules and reduces the rate of its oxidation.
By analysing the data of two-component catalysts with various chemical and physical methods, it should be noted that the approach proposed by us to introduce a second metal not into the active part of the catalyst (platinum or alloy, ad-atom, etc.) but in support and creation of a hybrid support, MO x –C provides higher catalytic effects for the alcohol oxidation reactions (Table S1).
As for the second question, why are the contents of the oxide in the more effective catalysts for electrochemical alcohols oxidation different—60 % for NiO and 30 % for SnO2? One of the reasons for may be the differences in the morphology of the oxides. The tin dioxide particles have a spherical shape and sizes of approximately 15–20 nm [43], [43], and the morphology of the prepared NiO is nanosheets of approximately 500–2000 nm in length and 20 nm in thickness [45], while the specific surface areas of the metal oxides are very similar: 82 and 79 m2 g−1 for tin dioxide and nickel oxide, respectively. Therefore, some part of the nickel oxide is essentially not available for contact with the platinum particles and cannot participate in an electrochemical reaction.
3.4 Durability tests
The stability of the catalyst and the fuel cell, in general, is largely dependent on the stability of the catalyst support. Because the catalysts work under very strict conditions and, in the presence of platinum, carbon oxidation occurs, the corrosion of the carbon support in the vicinity of the platinum nanoparticles causes a loss of its contact with the support and is thus out of the reaction zone [66]. One can expect that the introduction of a metal oxide in the support will have a positive impact on the catalyst’s durability.
We investigated the stability of the synthesized Pt/MO x –C (M = Sn, Ni) catalysts by cycling in 0.5 M H2SO4 over a potential range of 0.05–1.3 V versus RHE. Although the synthesized catalysts are designed for use as anodes of fuel cells (hence, their potential cannot rise above 0.7 V), we decided to test the catalysts under stricter oxidation conditions to definitively establish the differences in the stabilities of the carbon and hybrid supports.
Figure 9 shows the CV curves of the Pt/MO x –C catalysts in 0.5 M H2SO4 during cycling and the dependence of the charge of hydrogen desorption, which is normalized to the maximum amount of charge from the cycle numbers. It is clear that there are dramatic changes in the Pt/C catalyst CV curves under cycling. For the first 50 cycles, the values of the charge of hydrogen desorption increases (Fig. 9a, d) due to the purification of the platinum surface. However, after the 1000th cycle, the catalyst is nearly completely degraded. The peaks characteristic of platinum CVs (the reverse peaks of the desorption and adsorption of hydrogen and the irreversible peaks for the adsorption–desorption of oxygen) disappear. The oxidation peaks of the carbon support and the so-called “quinone” peak appear on the anode scan in the dual-layer potential region, and the “hydroquinone” peak appears at 0.4 V in the cathode scan with an increase in the current value of the last one. The behaviour of the Pt/NiO–C catalyst is the same, but the rate of catalyst degradation is somewhat higher than that of Pt/C (Fig. 9c, d) and is probably due to the solubility of NiO in the acidic medium. The catalyst based on the hybrid support containing tin oxide is significantly more stable (Fig. 9b, d, S4). The value of the charge of hydrogen desorption after 1500 cycles decreases to 60 %, while it has a tendency to stabilize for all of the catalysts on the SnO2–C support, independent of tin oxide loading (Fig. 9d). In the alkaline solution, the stability of the catalysts is very high for the Pt/C and Pt/NiO–C catalysts (Fig. S5).

CV curves of Pt/C (a), Pt/SnO2–C (30 %) (b), Pt/NiO–C (60 % NiO) (c) and normalized Q H (d) during cycling in 0.5 M H2SO4, scan rate 50 mV s−1. Comments in the text
Thus, the catalysts of the MO x –C hybrid supports demonstrated a higher catalytic activity in the oxidation of alcohols as well as Pt/SnO2–C catalysts demonstrated a higher stability than the Pt/C catalyst.
4 Conclusions
In summary, we propose an electrochemical technique for the preparation of hybrid MO x –C (M = Sn, Ni) supports via the electrochemical oxidation and dispersion of a metal under the influence of a pulsed alternating current. The proposed method is free from the drawbacks of other preparation methods, such as being multistage and complex, time-consuming, as well as having the necessity of using corrosive reagents and organic solvents and a calcination step at high temperatures. The hybrid MO x –C supports with an oxide loading of up to 60 % have good electrical conductivity and proper BET surface areas. In addition, we prepared and investigated Pt/MO x –C (M = Sn, Ni) catalytic systems with oxide contents of 30 and 60 % in hybrid supports and a Pt content of 25 % for the electrochemical oxidation of methanol and ethanol in acidic and alkaline solutions. The presence of tin dioxide in the hybrid support for catalysts decreases the onset potential of ethanol electrooxidation in acidic solution by 170 mV and increases the rate of the oxidation process by more than sevenfold at the low potential region that is important for direct alcohol fuel cell applications. In alkaline solutions, the presence of a metal oxide in catalysts is not effective, probably because of the high value of coverage of the platinum surface with OHad species. The difference in the optimum oxide content (30 % for SnO2 and 60 % for NiO) for the catalyst can be attributed to the different morphologies of the oxides. The Pt catalyst on the SnO2–C and NiO–C hybrid supports exhibited superior electrochemical stability in acidic and in alkaline media, respectively.
Thus, the proposed electrochemical method of producing hybrid supports systems may be extended to other metals, and we hope that this method will be useful to create electroactive hybrid materials for electrochemical energy conversion and storage devices.
References
Dodds PE, Staffell I, Hawkes AD, Li F, Grünewald P, McDowall W, Ekins P (2015) Hydrogen and fuel cell technologies for heating: a review. Int J Hydrog Energy 40:2065–2083. doi:10.1016/j.ijhydene.2014.11.059
Braunchweig B, Hibbitts D, Neurock M, Wieckowski A (2013) Electrocatalysis: a direct alcohol fuel cell and surface science perspective. Catal Today 202:197–209. doi:10.1016/j.cattod.2012.08.013
Cheng X, Shi Z, Glass N, Zhang L, Zhang J, Song D, Liu Z-S, Wang H, Shen J (2007) A review of PEM hydrogen fuel cell contamination: impacts, mechanisms, and mitigation. J Power Sources 165:739–756. doi:10.1016/j.jpowsour.2006.12.012
Mussatto SI, Dragone G, Guimaraes PM, Silva JP, Carneiro LM, Roberto IC, Vicente A, Domingues L, Teixeira JA (2010) Technological trends, global market, and challenges of bio-ethanol production. Biotechnol Adv 28:817–830. doi:10.1016/j.biotechadv.2010.07.001
Pedersen CM, Escudero-Escribano M, Velázquez-Palenzuela A, Christensen LH, Chorkendorff I, Stephens IEL (2015) Benchmarking Pt-based electrocatalysts for low temperature fuel cell reactions with the rotating disk electrode: oxygen reduction and hydrogen oxidation in the presence of CO (review article). Electrochim Acta 179:647–657. doi:10.1016/j.electacta.2015.03.176
Vigier F, Rousseau S, Coutanceau C, Leger J-M, Lamy C (2006) Electrocatalysis for the direct alcohol fuel cell. Top Catal 40:111–121. doi:10.1007/s11244-006-0113-7
Iwasita T (2002) Electrocatalysis of methanol oxidation. Electrochim Acta 47:3663–3674. doi:10.1016/S0013-4686(02)00336-5
Lipkovski J, Ross PN (1998) Electrocatalysis. Wiley, New York
Thomas JM, Thomas WJ (1967) Introduction to the principles of heteroreneous catalysis. Academic Press, London
Tian F, Jinnouchi R, Anderson AB (2009) How potentials of zero charge and potentials for water oxidation to OH(ads) on Pt(111) Electrodes vary with coverage. J Phys Chem C 113:17484–17492. doi:10.1021/jp905377d
Watanabe M, Motoo S (1975) Electrocatalysis by ad-atoms. J Electroanal Chem Interfacial Electrochem 60:267–273. doi:10.1016/S0022-0728(75)80261-0
Zheng Q-W, Fan C-J, Zhen C-H, Zhou Z-Y, Sun S-G (2008) Irreversible adsorption of Sn adatoms on basal planes of Pt single crystal and its impact on electrooxidation of ethanol. Electrochim Acta 53:6081–6088. doi:10.1016/j.electacta.2008.01.078
Smirnova NV, Petrii OA, Grzejdziak A (1988) Effect of ad-atoms on the electro-oxidation of ethylene glycol and oxalic acid on platinized platinum. J Electroanal Chem Interfacial Electrochem 251:73–87. doi:10.1016/0022-0728(88)80386-3
Bach Delpeuch A, Chatenet M, Rau MS, Cremers C (2015) Influence of H- and OH-adsorbates on the ethanol oxidation reaction: a DEMS study. Phys Chem Chem Phys 17:10881–10893. doi:10.1039/C5CP00132C
St. John S, Boolchand P, Angelopoulos AP (2013) Improved electrocatalytic ethanol oxidation activity in acidic and alkaline electrolytes using size-controlled Pt–Sn nanoparticles. Langmuir 29:16150–16159. doi:10.1021/la403704w
Antolini E (2011) An empirical model to evaluate the contribution of alloyed and non-alloyed tin to the ethanol oxidation reaction on Pt–Sn/C catalysts based on the presence of SnO2 and a Pt(1−x)Sn x solid solution: application to DEFC performance. Int J Hydrog Energy 36:11043–11047. doi:10.1016/j.ijhydene.2011.05.099
Smirnova NV, Kuriganova AB, Leont’eva DV, Leont’ev IN, Mikheikin AS (2013) Structural and electrocatalytic properties of Pt/C and Pt–Ni/C catalysts prepared by electrochemical dispersion. Kinet Catal 54:255–262. doi:10.1134/S0023158413020146
Ciapina EG, Santos SF, Gonzalez ER (2013) The electro-oxidation of carbon monoxide and ethanol on supported Pt nanoparticles: the influence of the support and catalyst microstructure. J Solid State Electrochem 17:1831–1842. doi:10.1007/s10008-013-2120-5
Figueiredo MC, Santasalo-Aarnio A, Vidal-Iglesias FJ, Solla-Gullón J, Feliu JM, Kontturi K, Kallio T (2013) Tailoring properties of platinum supported catalysts by irreversible adsorbed adatoms toward ethanol oxidation for direct ethanol fuel cells. Appl Catal B 140–141:378–385. doi:10.1016/j.apcatb.2013.04.038
Del Colle V, Souza-Garcia J, Tremiliosi-Filho G, Herrero E, Feliu JM (2011) Electrochemical and spectroscopic studies of ethanol oxidation on Pt stepped surfaces modified by tin adatoms. Phys Chem Chem Phys 13:12163–12172. doi:10.1039/C1CP20546C
Antolini E, Salgado JRC, Gonzalez ER (2006) The stability of Pt–M (M = first row transition metal) alloy catalysts and its effect on the activity in low temperature fuel cells: a literature review and tests on a Pt–Co catalyst. J Power Sources 160:957–968. doi:10.1016/j.jpowsour.2006.03.006
Baranova EA, Padilla MA, Halevi B, Amir T, Artyushkova K, Atanassov P (2012) Electrooxidation of ethanol on PtSn nanoparticles in alkaline solution: correlation between structure and catalytic properties. Electrochim Acta 80:377–382. doi:10.1016/j.electacta.2012.07.030
López-Cudero A, Solla-Gullón J, Herrero E, Aldaz A, Feliu JM (2010) CO electrooxidation on carbon supported platinum nanoparticles: effect of aggregation. J Electroanal Chem Interfacial Electrochem 644:117–126. doi:10.1016/j.jelechem.2009.06.016
Hitchcock AP, Berejnov V, Lee V, West M, Colbow V, Dutta M, Wessel S (2014) Carbon corrosion of proton exchange membrane fuel cell catalyst layers studied by scanning transmission X-ray microscopy. J Power Sources 266:66–78. doi:10.1016/j.jpowsour.2014.04.119
Dhanushkodi SR, Tam M, Kundu S, Fowler MW, Pritzker MD (2013) Carbon corrosion fingerprint development and de-convolution of performance loss according to degradation mechanism in PEM fuel cells. J Power Sources 240:114–121. doi:10.1016/j.jpowsour.2013.03.033
Li L, Qian Y, Yang J, Tan X, Dai Z, Jin Y, Wang H, Qu W, Chu Y (2016) A novel structural design of hybrid nanotube with CNTs and CeO2 supported Pt nanoparticles with improved performance for methanol electro-oxidation. Int J Hydrogen Energy 41:9284–9294. doi:10.1016/j.ijhydene.2016.04.069
Amin RS, Fetohi AE, Hameed RMA, El-Khatib KM (2016) Electrocatalytic activity of Pt–ZrO2 supported on different carbon materials for methanol oxidation in H2SO4 solution. Int J Hydrog Energy 41:1846–1858. doi:10.1016/j.ijhydene.2015.11.040
Comignani V, Sieben JM, Brigante ME, Duarte MME (2015) Carbon supported Pt–NiO nanoparticles for ethanol electro-oxidation in acid media. J Power Sources 278:119–127. doi:10.1016/j.jpowsour.2014.12.063
Wu M, Han M, Li M, Li Y, Zeng J, Liao S (2014) Preparation and characterizations of platinum electrocatalysts supported on thermally treated CeO2–C composite support for polymer electrolyte membrane fuel cells. Electrochim Acta 139:308–314. doi:10.1016/j.electacta.2014.07.029
Dou M, Hou M, Liang D, Lu W, Shao Z, Yi B (2013) SnO2 nanocluster supported Pt catalyst with high stability for proton exchange membrane fuel cells. Electrochim Acta 92:468–473. doi:10.1016/j.electacta.2013.01.070
Lv H, Cheng N, Peng T, Pan M, Mu S (2012) High stability platinum electrocatalysts with zirconia-carbon hybrid supports. J Mater Chem 22:1135–1141. doi:10.1039/C1JM14076K
Dou M, Hou M, Zhang H, Li G, Lu W, Wei Z, Shao Z, Yi B (2012) A highly stable anode, carbon-free, catalyst support based on tungsten trioxide nanoclusters for proton-exchange membrane fuel cells. ChemSusChem 5:945–951. doi:10.1002/cssc.201100706
Jiang Z-Z, Wang Z-B, Chu Y-Y, Gu D-M, Yin G-P (2011) Ultrahigh stable carbon riveted Pt/TiO2–C catalyst prepared by in situ carbonized glucose for proton exchange membrane fuel cell. Energy Environ Sci 4:728–735. doi:10.1039/C0EE00475H
Kim DB, Chun H-J, Lee YK, Kwon H-H, Lee H-I (2010) Preparation of Pt/NiO–C electrocatalyst and heat-treatment effect on its electrocatalytic performance for methanol oxidation. Int J Hydrog Energy 35:313–320. doi:10.1016/j.ijhydene.2009.10.037
Cui X, Guo L, Cui F, He Q, Shi J (2009) Electrocatalytic Activity and CO Tolerance Properties of Mesostructured Pt/WO3 composite as an anode catalyst for PEMFCs. J Phys Chem C 113:4134–4138. doi:10.1021/jp8079205
Shanmugam S, Gedanken A (2007) Carbon-coated anatase TiO2 nanocomposite as a high-performance electrocatalyst support. Small 3:1189–1193. doi:10.1002/smll.200600636
Feng Y-Y, Kong W-Q, Yin Q-Y, Du L-X, Zheng Y-T, Kong D-S (2014) Platinum catalysts promoted by In doped SnO2 support for methanol electrooxidation in alkaline electrolyte. J Power Sources 252:156–163. doi:10.1016/j.jpowsour.2013.12.008
Higuchi E, Takase T, Chiku M, Inoue H (2014) Preparation of ternary Pt/Rh/SnO2 anode catalysts for use in direct ethanol fuel cells and their electrocatalytic activity for ethanol oxidation reaction. J Power Sources 263:280–287. doi:10.1016/j.jpowsour.2014.04.056
Shen PK, Xu C, Zeng R, Liu Y (2006) Electro-oxidation of Methanol on NiO-Promoted Pt/C and Pd/C Catalysts. Electrochem Solid-State Lett 9:A39–A42. doi:10.1149/1.2139975
Simões FC, Olivi P (2010) Oxygen Reduction Reaction on Pt–NiO x /C, Pt–CoO x /C, and Pt–SnO2/C Electrodes in the Presence of Ethanol. Electrocatalysis 1:163–168. doi:10.1007/s12678-010-0026-x
Amin RS, Hameed RMA, El-Khatib KM, Youssef ME, Elzatahry AA (2012) Pt–NiO/C anode electrocatalysts for direct methanol fuel cells. Electrochim Acta 59:499–508. doi:10.1016/j.electacta.2011.11.013
Tripković AV, Popović KD, Lović JD, Jovanović VM, Kowal A (2004) Methanol oxidation at platinum electrodes in alkaline solution: comparison between supported catalysts and model systems. J Electroanal Chem 572:119–128. doi:10.1016/j.jelechem.2004.06.007
Kuriganova AB, Vlaic CA, Ivanov S, Leontyeva DV, Bund A, Smirnova NV (2016) Electrochemical dispersion method for the synthesis of SnO2 as anode material for lithium ion batteries. J Appl Electrochem 46:527–538. doi:10.1007/s10800-016-0936-2
Kuriganova AB, Smirnova NV (2014) Pt/SnO x –C composite material for electrocatalysis. Mendeleev Commun 24:351–352. doi:10.1016/j.mencom.2014.11.013
Leontyeva DV, Leontyev IN, Avramenko MV, Yuzyuk YI, Kukushkina YA, Smirnova NV (2013) Electrochemical dispergation as a simple and effective technique toward preparation of NiO based nanocomposite for supercapacitor application. Electrochim Acta 114:356–362. doi:10.1016/j.electacta.2013.10.031
Leontyev I, Kuriganova A, Kudryavtsev Y, Dkhil B, Smirnova N (2012) New life of a forgotten method: electrochemical route toward highly efficient Pt/C catalysts for low-temperature fuel cells. Appl Catal A 431–432:120–125. doi:10.1016/j.apcata.2012.04.025
Smirnova NV, Kuriganova AB, Novikova KS, Gerasimova EV (2014) The role of carbon support morphology in the formation of catalytic layer of solid-polymer fuel cell. Russ J Electrochem 50:899–903. doi:10.1134/S1023193514070143
Doronkin DE, Kuriganova AB, Leontyev IN, Baier S, Lichtenberg H, Smirnova NV, Grunwaldt J-D (2016) Electrochemically Synthesized Pt/Al2O3 Oxidation Catalysts. Catal Lett 146:452–463. doi:10.1007/s10562-015-1651-z
Brimaud S, Pronier S, Coutanceau C, Léger JM (2008) New findings on CO electrooxidation at platinum nanoparticle surfaces. Electrochem Commun 10:1703–1707. doi:10.1016/j.elecom.2008.08.045
Ayeshamariam A, Samy RP (2013) Synthesis, structural and optical characterizations of SnO2 nanoparticles. J Photonics Spintron 2:4–8
Makhlouf SA, Kassem MA, Abdel-Rahim MA (2009) Particle size-dependent electrical properties of nanocrystalline NiO. J Mater Sci 44:3438–3444. doi:10.1007/s10853-009-3457-0
Borges PD, Scolfaro LMR, Leite Alves HW, da Silva EF (2010) DFT study of the electronic, vibrational, and optical properties of SnO2. Theor Chem Acc 126:39–44. doi:10.1007/s00214-009-0672-3
Shklovskiĭ BI, Éfros AL (1975) Percolation theory and conductivity of strongly inhomogeneous media. Sov Phys Uspekhi 18:845. www.stacks.iop.org/0038-5670/18/i=11/a=R01
Guo D-J, You J-M (2012) Highly catalytic activity of Pt electrocatalyst supported on sulphated SnO2/multi-walled carbon nanotube composites for methanol electro-oxidation. J Power Sources 198:127–131. doi:10.1016/j.jpowsour.2011.10.017
Vidal-Iglesias FJ, Montiel V, Solla-Gullón J (2016) Influence of the metal loading on the electrocatalytic activity of carbon-supported (100) Pt nanoparticles. J Solid State Electrochem 20:1107–1118. doi:10.1007/s10008-015-2954-0
Moghaddam RB, Pickup PG (2012) Support effects on the oxidation of ethanol at Pt nanoparticles. Electrochim Acta 65:210–215. doi:10.1016/j.electacta.2012.01.042
Ruiz-Camacho B, Santoyo HHR, Medina-Flores JM, Álvarez-Martínez O (2014) Platinum deposited on TiO2–C and SnO2–C composites for methanol oxidation and oxygen reduction. Electrochim Acta 120:344–349. doi:10.1016/j.electacta.2013.12.055
Petry OA, Podlovchenko BI, Frumkin AN (1959) Lal H (1965) The behaviour of platinized-platinum and platinum-ruthenium electrodes in methanol solutions. J Electroanal Chem 10:253–269. doi:10.1016/0022-0728(65)85060-4
Podlovchenko BI, Petry OA, Frumkin AN (1959) Lal H (1966) The behaviour of a platinized-platinum electrode in solutions of alcohols containing more than one carbon atom, aldehydes and formic acid. J Electroanal Chem 11:12–25. doi:10.1016/0022-0728(66)80053-0
Melke J, Schoekel A, Gerteisen D, Dixon D, Ettingshausen F, Cremers C, Roth C, Ramaker DE (2012) Electrooxidation of ethanol on Pt. an in Situ and time-resolved XANES study. J Phys Chem C 116:2838–2849. doi:10.1021/jp206295h
Jing M, Jiang L, Yi B, Sun G (2013) Comparative study of methanol adsorption and electro-oxidation on carbon-supported platinum in acidic and alkaline electrolytes. J Electroanal Chem 688:172–179. doi:10.1016/j.jelechem.2012.10.028
Tripković AV, Popović KD, Momčilović JD, Dražić DM (1998) Kinetic and mechanistic study of methanol oxidation on a Pt(100) surface in alkaline media1. J Electroanal Chem 448:173–181. doi:10.1016/S0022-0728(97)00141-1
Spendelow JS, Wieckowski A (2007) Electrocatalysis of oxygen reduction and small alcohol oxidation in alkaline media. Phys Chem Chem Phys 9:2654–2675. doi:10.1039/B703315J
Lai SCS, Koper MTM (2009) Ethanol electro-oxidation on platinum in alkaline media. Phys Chem Chem Phys 11:10446–10456. doi:10.1039/B913170A
Tripković AV, Popović KD, Grgur BN, Blizanac B, Ross PN, Marković NM (2002) Methanol electrooxidation on supported Pt and PtRu catalysts in acid and alkaline solutions. Electrochim Acta 47:3707–3714. doi:10.1016/S0013-4686(02)00340-7
Leontyev IN, Leontyeva DV, Kuriganova AB, Popov YV, Maslova OA, Glebova NV, Nechitailov AA, Zelenina NK, Tomasov AA, Hennet L, Smirnova NV (2015) Characterization of the electrocatalytic activity of carbon-supported platinum-based catalysts by thermal gravimetric analysis. Mendeleev Commun 25:468–469. doi:10.1016/j.mencom.2015.11.024
Acknowledgments
The financial support by the Russian Science Foundation (Grant No. 14-23-00078) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kuriganova, A.B., Leontyeva, D.V., Ivanov, S. et al. Electrochemical dispersion technique for preparation of hybrid MO x –C supports and Pt/MO x –C electrocatalysts for low-temperature fuel cells. J Appl Electrochem 46, 1245–1260 (2016). https://doi.org/10.1007/s10800-016-1006-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-016-1006-5