
Abstract
The effect of the SnO2 nanoparticles content on the structure and electrochemical behavior of Pt/(SnO2/C) catalysts containing about 20 wt% Pt was studied. In this case, SnO2/C nanostructured materials were obtained by tin electrodeposition on the carbon particles in suspension. It was found that the optimal combination of stability and activity in the oxygen electroreduction reaction is demonstrated by materials containing 25–30% wt. SnO2. A comparative assessment of the durability and activity of Pt/(SnO2/C) catalysts and a commercial Pt/C catalyst with 20% Pt-loading has been carried out.
Graphical Abstractt

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Low-temperature fuel cells (LTFC) with a polymer membrane have numerous advantages: no polluting emissions into the environment, low noise, high energy conversion efficiency, and their autonomy. This made them promising for use in mobile and stationary power plants [1,2,3]. Despite the LTFC advantages, some economic and technical barriers hinder their large-scale commercialization. The search for effective electrocatalysts to be used in the oxygen reduction reaction (ORR), which combine high rates of activity and stability, is one of the actively developing areas of research in the field of LTFC. Platinum and its alloys with some d-metals, deposited in the form of nanoparticles on the surface of carbon supports, are the best ORR catalysts [4,5,6]. Carbon, which plays the role of an electrically conductive carrier, is susceptible to corrosion during LTFC operation, which leads to deterioration of platinum adhesion to the carrier surface and loss of electrical contact by individual platinum nanoparticles (NPs), having a decrease in the functional characteristics of the catalyst as a result [7,8,9]. In this regard, to optimize the composition of the catalysts or obtain new catalysts that exhibit high stability in the course of LTFC operation combined with high activity in ORR proves to be an urgent task.
Tin (IV) oxide, which has semiconducting properties, was proposed as a promising carrier material [10,11,12,13,14,15,16,17]. It was shown in previous studies [16, 17] that SnO2 particles combined with carbon can increase the electrocatalytic activity of Pt in ORR due to the electronic interaction between Pt and SnO2, which occurs during the formation of three-boundary structures: Pt-SnO-C. Recent studies have also shown that tin oxide is capable of increasing the platinum-containing catalyst stability with regard to CO, as well as increasing their activity in the reactions of alcohols electrooxidation [18, 19].
Tin oxide is most often obtained by hydrolysis of tin salts [16, 17, 20]. However, as the control over the surface morphology and purity of the resulting product are regarded, electrochemical methods of obtaining materials are considered to be more promising [21, 22]. Electrochemical methods make it possible to control the potential and the current in the synthesis process, which makes it possible to select conditions for obtaining materials with a certain composition/structure and, as a result, certain functional characteristics.
Earlier we have shown that the use of a SnO2/C composite material, containing 4% wt tin oxide as a carrier of platinum nanoparticles allows increasing the resistance of the catalyst to degradation while maintaining the activity in the oxygen reduction reaction at the level of commercial Pt/C material [23]. However, it remains unclear what the optimal composition of the Pt/(SnO2/C) catalyst is and how an increase in the mass fraction of tin dioxide affects the functional characteristics of materials.
This study is based on the hypothesis that the use of the SnO2/C nanostructured composite as a carrier will increase the activity of supported platinum electrocatalysts in ORR, as well as their corrosion-morphological stability. The aim of this work was to obtain a number of SnO2/C materials by the original method of electrolytic deposition, to synthesize Pt/(SnO2/C) catalysts based on them, and to study the structure and electrochemical behavior of the obtained platinum-containing catalysts in the ORR.
Experimental
Synthesis of the Nanostructured Sno2/C Materials
The preparation of the SnO2/C materials was carried out according to the original procedure described in detail in previous studies [23, 24]. The electrode space of the two-electrode electrochemical cell was filled with a suspension of a dispersed carbon material, a tin (II) chloride solution being uses as a basic electrolyte. The suspension was prepared by mixing Vulkan XC72 carbon black (Cabot Corporation, USA; specific surface area ~ 270 m2 g−1) and water solution of 1 M tin (II) chloride and 1 M H2SO4. The resulting mixture was dispersed in a Sonics ultrasonic homogenizer for 2 min; then, it was continuously stirred on a magnetic stirrer to maintain the homogeneity of the suspension. Electrodes were immersed in the suspension. A copper wire with a working surface area of 0.5 cm2 was used as a cathode, and a graphite rod was used as an anode. The electrolysis was carried out with vigorous stirring of the suspension, a direct current of 9 A being passed through it.
Microparticles of carbon, in contact with the cathode during mixing, actually became a part of the cathode. On such microparticles the process of the tin ion electroreduction took place, followed by the formation of the nanosized crystals of this metal. During electrolysis, many particles of the carbon support repeatedly meet the stationary cathode and nucleation/growth of tin particles occurred on their surface as a result. The constant renewal of carbon microparticles in the electron-conducting near-cathode layer, caused by artificial convection, led to a more or less uniform distribution of electrodeposited tin over the surface of carbon microparticles (see “Results and Discussions”). Apparently, already in the process of preparation, a part of the tin nanoparticles was oxidized by the dissolved oxygen and transformed into the nanoparticles of tin oxide (IV).
When electrolysis was over, the suspension was filtered and then washed repeatedly with the bidistilled water until the medium was neutral. Drying was carried out in a drying cabinet at 80 °C for a day. During filtration and drying, unoxidized tin NPs deposited on a carbon support were oxidized to form tin dioxide NPs.
Pt/(SnO2/C) Preparation
Pt/(SnO2/C) catalysts were synthesized by the chemical reduction of H2PtCl6 from the liquid phase, as described in Alekseenko et al. [25], in a suspension containing the particles of a composite SnO2-C support. The calculated mass fraction of Pt in Pt/(SnO2/C) was 20% wt. For this, the powdered composite obtained by the electrodeposition method was placed in 20 ml of ethylene glycol; after that, the required amount of H2[PtCl6]*6H2O was added. The resulting suspension was homogenized by ultrasound, then 0.5 ml of 37% wt. HCOH and 1 M NaOH solution in a water—ethylene glycol (1: 1) mixture were successively added until pH = 11. After standing for 2 h at 90 °C and spontaneous cooling of the stirred suspension, the mixture was filtered on a Buchner funnel, the product was washed 5–6 times with bidistilled water and ethyl alcohol, then dried at 80 °C to constant weight.
Measurement Methods and Techniques
X-ray phase analysis and thermogravimetry were used to determine the composition of the obtained SnO2/C and Pt/(SnO2/C) materials and the presence of crystallites of tin dioxide and platinum in them. Powder diffractograms of metal- and oxide-carbon materials were recorded on an automatic diffractometer ARL X′TRA (Thermo Fisher Scientific, Switzerland) (geometry according to Bregg-Brentano, radius 260 mm, vertical theta-theta goniometer) using copper K-alpha radiation (λ = 1.5418 Å). Typical settings: 40 kV, 30 mA, scan step 0.02°. The registration rate was varied from 2° per minute for simple X-ray phase analysis up to 0.5° per minute to clarify particular features of the structure. A regular plate of polycrystalline alpha quartz was used as a calibration standard.
The presence of tin oxide and platinum in the materials, as well as the average size (diameter) of crystallites (D) for both platinum and tin dioxide, was determined based on the obtained diffraction patterns using reflections (111) and (101), respectively. D was calculated with the Scherrer formula, the value of the peak width at a half maximum being substituted into it: D = Kλ/(FWHM cosθ), where λ is the wavelength of monochromatic radiation, Å; FWHM is the peak width at half maximum (in radians); D is the average crystallite size, nm; θ is the reflection angle, radians; and K = 0.89—Scherrer’s constant.
The mass fractions of tin oxide (ω(SnO2)) and platinum, in SnO2/C and Pt/(SnO2/C), respectively, were determined by thermogravimetry from the mass of the SnO2 or (Pt + SnO2) residue after carbon combustion (800 °C, 40 min).
The ratio of tin to platinum in the samples was determined by the method of X-ray fluorescence analysis on a spectrometer with total external reflection of X-ray radiation RFS-001 (Research Institute of Physics, Southern Federal University). The range of determined chemical elements according to the periodic table of chemical elements is from Al to U. The exposure time of the samples is 300 s. Registration and processing of X-ray fluorescence spectra was carried out using the UniveRS software (SFedU).
JEM-2100 microscope (JEOL, Japan) at a voltage of 200 kV and a resolution of 0.2 nm was used to obtain TEM photographs. For measurements, 0.5 mg of the catalyst was placed in 1 ml of isopropanol and dispersed by ultrasound; after that, the resulting suspension was applied to a copper grid covered with a layer of amorphous carbon and dried in air at room temperature for 20 min.
EDX method analysis of the elemental composition was carried out by identifying the positions of the characteristic lines of the elements on the energy scale in the secondary X-ray spectrum obtained at a given point (area) and by comparing them with the tabular data. Then, the concentrations of elements were calculated with the eZAF (MThin) software algorithm, implemented in the TEAM EDAX software, their intensity, and the cross section of the secondary X-ray emission for a given line of this element being taken into account. Phase distribution maps were obtained with the TEAM EDAX software by combining areas on the elemental composition map that have the same concentration values of the detected elements (within the specified values), on the image marked with a certain color as one phase.
Cyclic voltammetry was used to measure the platinum electrochemical active surface area (ECSA). A thin layer of Pt/(SnO2/C) material was applied to the end face of a rotating disk electrode. To obtain the catalysts suspension (catalytic “ink”), 900 μL of isopropyl alcohol and 100 μL of a 0.5% aqueous emulsion of Nafion® polymer were added to 0.0060 g of each sample. Then, the suspension was dispersed with ultrasound for 15 min. Under continuous stirring, an aliquot of “ink” 6 μL in volume was taken with a microdispenser and applied to the end face of a polished and degreased glassy carbon disk with an area of 0.196 cm2, the exact weight of the drop being recorded. To fix the porous layer of powder after drying, another 7 μL of 0.05% Nafion solution in isopropanol was applied; then, the electrode was dried for 15 min at room temperature. The potential sweep was set, and the current values were recorded with the AFCBP1 potentiostat (Pine Applied Instrumentation, United States).
Initially, to standardize the platinum surface and remove impurities, 100 potential sweep cycles were performed at the rate of 200 mV s−1 in the potential range from 0 to 1.26 V. Then, 2 cyclic voltammograms (CV) were recorded in the potential range 0.03 to 1.20 V at a sweep rate of 20 mV s−1. Next, based on the amount of electricity consumed for electrochemical adsorption and desorption of atomic hydrogen, ECSA was calculated using the 2nd CV, as described in Menshchikov et al. [26]. An Ag/AgCl/KCl electrode was used as a reference electrode. All potential values in this work are given relative to a reversible hydrogen electrode (RHE).
When determining the area of the platinum active surface by the amount of electricity spent on oxidation of a chemisorbed CO monolayer, CO was blown through the solution for 20 min at a potential of the investigated electrode of 0.1 V, and then argon was blown through for 30 min. After that, two CVs were recorded in the potential range of 0.03–1.20 V at a potential sweep rate of 20 mV s−1. The ECSA was calculated based on the amount of electricity consumed for the electrochemical desorption of CO, as described in Kuriganova et al. [21].
The catalyst activity in the ORR was measured in the electrolyte, O2 being passed through it for 1 h. Voltammogram recording with a linear potential scan towards high values was performed at a rate of 20 mV/s at four rotation speeds of the disk electrode: 400, 900, 1600, and 2500 rpm. To take into account the contribution of the ohmic voltage drop, the potential of the electrode under study was refined using the formula: E = Emeas-I ∗ R, where Emeas is the set value of the potential and I ∗ R is the ohmic potential drop equal to the product of the current strength and the resistance of the solution layer between the reference electrode and the investigated electrode, which was 25 ohms. This resistance value is in good agreement with the literature data [27]. To calculate the contribution of the processes occurring at the electrode in the deoxygenated solution (Ar atmosphere), a similar curve recorded at the same electrode during measurements in the Ar atmosphere was subtracted from the voltammogram: I(O2)-I(Ar), as described in Shinozaki et al. [28]. The catalyst activity in the ORR (kinetic current) was determined from the normalized voltammograms, the contribution of mass transfer under the conditions of using RDE being taken into account. The calculation of the kinetic current at a potential of 0.90 V (RHE) was carried out according to the Koutetsky-Levich equation [29]:
1/i = 1/ik + 1/id = 1/ik + 1/Zω0.5
Z = 0.62 nFD2/3 υ−1/6 c,
where i is the current at the disk electrode, A·cm−2; ik is the kinetic current, A·cm−2; id is the diffusion current, A·cm−2; ω is the rotation speed of the disk electrode (rad·s−1); n is the number of electrons involved in the electroreduction of an oxygen molecule; F is the Faraday constant, C·mol−1; D is the diffusion coefficient, cm2·s−1; υ is the kinematic viscosity of the electrolyte, cm2 s−1; c is the oxygen concentration in solution, mol·cm−3.
To assess the stability of electrocatalysts, the method of voltammetric cycling in a three-electrode cell in the potential range of 0.6–1.4 V with a potential sweep rate of 100 mV/s was chosen. Cycling was carried out in a 0.1-M HClO4 solution in an Ar atmosphere at 25 °C for 2000 cycles; in this case, after every 100 cycles, two cyclic voltammograms were recorded (at a potential sweep rate of 20 mV/s, the potential range was 0.03 to 1.20 V). The second CV was used to calculate the surface area (S) as described earlier. Stability was assessed by the change in the Sj/S0 ratio, where j is the number of stress test cycles, and by the S2000/S0 value.
Results and Discussions
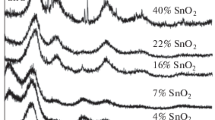
SnO2/C composites with tin mass fraction from 4 to 42% wt. were obtained by electrolytic deposition of tin on carbon black microparticles. The presence of a tin (IV) oxide phase in these materials was confirmed by the X-ray phase analysis (Fig. 1). An increase in the mass fraction of SnO2 in the materials leads to an increase in the intensity of characteristic peaks in the X-ray diffractogram (Fig. 1).

X-ray diffraction patterns of the carbon support Vulcan XC72 and SnO2/C materials with a mass fraction of tin oxide from 4 to 42% wt
To study the structure of the SnO2/C composite by transmission electron microscopy, the sample with the highest tin oxide content, 42% wt., was chosen. Analysis of TEM images (Fig. 2) showed that the nanoparticles of tin oxide were indeed formed on the microparticles of a carbon support, uniformly distributed over the surface of carbon particles, as a result of electrodeposition and subsequent oxidation of tin nanocrystals. The average size of tin dioxide NPs in a composite containing 42% wt. SnO2 was 2.9 ± 0.2 nm. Thus, the method of electrocrystallization of tin on carbon particles, which we developed for the preparation of SnO2/C materials, allowed the formation of deposited tin dioxide nanoparticles of a smaller size in comparison with the methods described in the literature [16, 17, 20,21,22]. Taking into account the results of Petrii [30], we believe that the formation of primary tin nanoparticles occurs mainly on the surface of carbon particles, and not in their pores.

a TEM micrograph of a SnO2/C material surface fragment and b a histogram of the size distribution of SnO2 nanoparticles. Mass fraction of SnO2—42%. Some tin dioxide NPs are circled in the photograph
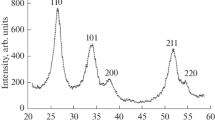
Then, platinum NPs were deposited on the SnO2/C composites, their mass fraction in the obtained materials being about 20%. X-ray diffractometry confirmed the presence of a platinum phase in all materials (Fig. 3). The average size of platinum crystallites was from 1.3 to 2.3 nm (Table 1).

X-ray diffraction patterns of Pt/(SnO2/C) samples, which contain about 20 wt% platinum
On the diffractogram corresponding to the S4P material (hereinafter, the number in the sample’s name corresponds to the mass fraction of tin dioxide in the material), reflections corresponding to SnO2 are not visible. This is most likely due to the low content of tin oxide compared to platinum. An increase in the mass fraction of tin oxide over 20% makes it possible to clearly identify the presence of the corresponding peaks in the diffraction patterns (Fig. 3). The intensity of these peaks increases with an increase in the mass fraction of tin dioxide in the catalyst.
In the process of platinum NP deposition on a composite carrier, they can be anchored on different parts of its surface. It can be assumed that the activity of platinum NPs fixed on carbon (Fig. 4 (a)) will not change in comparison with Pt/C. Platinum deposition on the surface of tin dioxide NPs (Fig. 4 (b)) can lead to a loss of electrical contact and, as a consequence, to a decrease in the catalyst activity in the current-forming reactions. According to the literature [16, 17], the most preferable one is the third version in the arrangement of platinum nanoparticles (Fig. 4 (b)). In this case, due to the simultaneous contact with the electrically conductive substrate and tin dioxide, platinum NPs can exhibit higher activity in current-forming reactions. Obviously, depending on the mass fraction of SnO2, the ratio of NPs with different versions of attachment to the surface of the SnO2/C substrate should change.

Schematic representation of versions for the location of platinum nanoparticles on the surface of SnO2/C carrier: a - Pt NP is located on the SnO2 surface, b - Pt NP is located on the carbon surface, c - Pt NP is in contact with both SnO2 and carbon (three-phase contact)
The micrographs of a Pt/(SnO2/C) surface fragment show nanoparticles of tin and platinum oxide (Fig. 5a). The spectral data obtained from the results of energy-dispersive X-ray spectroscopy (Fig. 5c) also confirm the presence of SnO2 on the catalyst surface. At the same time, the photomicrographs obtained in the reflected electron mode (Fig. 5b) indicate a uniform distribution of the platinum NPs, 2.3 nm in average size, over the catalyst surface (Fig. 5d). According to the photomicrographs, a significant part of the platinum NPs is in contact with both carbon and tin oxide NPs; i.e., it participates in the formation of the three-boundary zones described in previous literature [16, 17].

a - Photo of the S4P sample surface fragment obtained by transmission electron microscopy, b - STEM image of the S4P surface, c - result of the composition analysis by the method of energy dispersive transmission microscopy, and d - the histogram of the platinum NPs size distribution (For microscopic examination, the catalyst is deposited on the surface of a copper mesh, which causes a reflection of copper in b)
Elemental mapping of the surface showed that a significant proportion of platinum nanoparticles are located in the same areas as tin dioxide NPs (Fig. 6a–d).

Elemental mapping of the Pt/(SnO2/C) surface fragment of the S4P sample: STEM image of the mapped fragment of the surface a and maps of carbon b, tin c, and platinum d distribution
The CVs of Pt/(SnO2/C) electrodes after 100 cycles of potential scan (see the “Experimental” section) have a shape typical for platinum-containing materials (Fig. 7). Calculation of the Pt active surface area by the amount of electricity consumed for electrochemical adsorption and desorption of atomic hydrogen showed that the obtained catalysts have rather high ECSA values (Table 2). For all materials, the ECSA was about 90 m2 g−1, which is slightly inferior to the ECSA of the commercial Pt/C catalyst HiSPEC 3000 (~ 106 m2 g−1). The obtained ECSA values are also confirmed by the calculations based on the amount of electricity consumed for the electrochemical desorption of chemisorbed CO. This makes it possible to exclude the fact that SnO2 directly takes part in any electrochemical reactions that occur in the potential range of 0–0.4 V in regard to the RHE.

a - Catalyst cyclic voltammograms in the argon atmosphere. b - voltammograms of oxygen electroreduction with the linear sweep rate of potential O2 atmosphere. RDE rotation speed is 1600 rpm; c I−1- ω−0.5 dependence at 0.90 V (RHE) potential. A potential sweep rate is 20 mV • s−1; 0.1 M HClO4
Nevertheless, we note that the presence of tin oxide in the catalyst shifts the onset potential of CO oxidation and the maximum of the CO oxidation peak to more negative values (Fig. 8) Taking into account the literature data [16, 17, 21], this effect can be caused either by the bifunctional mechanism of catalysis or by the effect of the Pt/SnO2 contact on the electronic structure of platinum.

Cyclic voltammograms of the chemisorbed CO oxidation on the surface of the S29P catalyst and the commercial analogue HiSPEC 3000. Potential sweep rate is 20 mV s−1. Electrolyte is HClO4
Sample S25P, with the kinetic current calculated according to the Koutetsky-Levich equation (see the Technique section) at a potential of 0.90 V (RHE), being 1.3 times higher than that for the commercial Pt/C catalyst (Table 3). An increase in the ORR activity in the series of Pt/(SnO2/C) electrocatalysts occurs with an increase in the mass fraction of tin dioxide to 25–30% wt. (Table 3). This is probably due to an increase in the proportion of platinum NPs that simultaneously get in contact with both carbon and tin dioxide. Further increase in the mass fraction of tin dioxide up to 42% leads to a decrease in the ORR activity, which is apparently caused by partial electrical insulation of platinum due to an increase in the proportion of its nanoparticles, deposited on the surface of tin oxide particles without direct contact with carbon.
An accelerated voltammetric stress test in the potential range of 0.6–1.4 V, which corresponded to the so-called “carrier protocol” (see the “Experimental” section), was performed to assess the resistance of electrocatalysts to degradation. It is known that during the cycling of the potential in this range, the degradation of the electrocatalyst is largely associated with the corrosion of the carbon support, their oxidation being catalyzed by platinum NPs at the contact points [31, 32]. The test results showed that an increase in the mass fraction of tin oxide on the whole lead to an increase in the relative stability of electrocatalysts (Fig.9a), which was apparently due to an increase in the fraction of platinum NPs associated with SnO. Note that even a small addition of tin oxide (4% wt.) lead to an almost twofold increase in relative stability in regard to the commercial catalyst. As a result, the commercial material that showed the highest ECSA before the stress test had the lowest ECSA after the stress test (Fig. 9).

a- Relative and b - absolute changes in the area of the catalyst electrochemically active surface during the voltammetric stress test. Stress test potential range 0.6–1.4 V
Measurement of the catalyst activity in the ORR before and after stress testing showed that the most active samples after the stress test are samples S25P and S29P (Table 3). For these catalysts, the decrease in the half-wave potential due to degradation was 30 and 40 mV, respectively. Note that an increase in relative stability is observed with an increase in the mass fraction of tin dioxide, which follows from the comparison of values for both ECSA and kinetic currents. However, relatively low kinetic currents before and after the stress test do not allow the S42P sample to be considered the best one in the given series. This result correlates well with the data of Na Zhang et al. [17], who observed a positive effect of SnO2 on the stability of supported platinum electrocatalysts under “soft” stress testing conditions.
Thus, the results of this study showed that the deposition of platinum nanoparticles on a composite nanostructured support SnO2/C has a positive effect on the functional characteristics of electrocatalysts. The most promising materials are those containing 25–30% of tin dioxide, which is most likely associated with the largest fraction of platinum NPs involved in the formation of the three-boundary
 contacts. A further increase in the mass fraction of tin dioxide in the catalyst, although accompanied by an increase in stability, leads to a decrease in the catalyst activity in the ORR.
contacts. A further increase in the mass fraction of tin dioxide in the catalyst, although accompanied by an increase in stability, leads to a decrease in the catalyst activity in the ORR.
Conclusions
Tin nanoparticles, spontaneously oxidized to SnO2 by the components of the solution and by atmospheric oxygen, were formed on the surface of the carbon particles in suspension by the method of electrodeposition. As a result, composite SnO2/C materials were obtained, which contained from 4 to 40% of tin dioxide in the form of uniformly distributed small-sized NPs with an average diameter of less than 3 nm.
Pt/(SnO2/C) catalysts, in which platinum NPs maintain electrical contact with carbon due to the small size of the tin dioxide NPs, were synthesized by chemical reduction of Pt (IV) on the particles of electrolytically obtained SnO2/C support. Electrochemically active surface area of platinum in most Pt/(SnO2/C) catalysts containing 20 wt. Pt was close to 90 m2/g (Pt). Modification of the carbon support with the tin oxide nanoparticles made it possible to increase the corrosion-morphological stability of the catalysts under the voltammetric stress testing in the potential range of 0.6–1.4 V and positively affected the catalysts activity in ORR, when supports containing 25–29% wt. of SnO wt. of SnO V and positively affected the catalysts activity in ORR, when supports containing 25–29% wt. of SnO2 were used. The reason for the positive effect of SnO2 on the catalyst activity and stability is apparently due to the presence of the three-phase contacts, in which platinum nanoparticles get in touch with both tin oxide nanoparticles and the surface of the carbon support.y is apparently due to the presence of the three-phase contacts, in which platinum nanoparticles get in touch with both tin oxide nanoparticles and the surface of the carbon support.
References
C. Brady, E. Rees, G. Burstein, J. Power Sources 179, 17 (2008)
A. Schenk, C. Grimmer, M. Perchthaler, S. Weinberger, B. Pichler, C. Heinzl, C. Scheu, F.A. Mautner, B. Bitschnau, V. Hacker, J. Power Sources 266, 313 (2014)
A.B. Yaroslavtsev, Y.A. Dobrovolsky, N.S. Shaglaeva, L.A. Frolova, E.V. Gerasimova, E.A. Sanginov, Russ. Chem. Rev. 81, 191 (2012)
V.R. Stamenkovic, B.S. Mun, M. Arenz, K.J.J. Mayrhofer, C.A. Lucas, G. Wang, P.N. Ross, N.M. Markovic, Nat. Mater. 6, 241 (2007)
V.R. Stamenkovic, B. Fowler, B.S. Mun, G. Wang, P.N. Ross, C.A. Lucas, N.M. Markovic, Science 315, 493 (2007)
M. Oezaslan, F. Hasché, and P. Strasser, J. Electrochem. Soc. 159, (2012)
F. Ettingshausen, J. Kleemann, A. Marcu, G. Toth, H. Fuess, C. Roth, Fuel Cells 11, 238 (2011)
J.C. Meier, I. Katsounaros, C. Galeano, H.J. Bongard, A.A. Topalov, A. Kostka, A. Karschin, F. Schüth, K.J.J. Mayrhofer, Energy Environ. Sci. 5, 9319 (2012)
H. Schulenburg, B. Schwanitz, N. Linse, G.G. Scherer, A. Wokaun, J. Krbanjevic, R. Grothausmann, I. Manke, J. Phys. Chem. C. 115, 14236 (2011)
A.L. Santos, D. Profeti, P. Olivi, Electrochim. Acta 50, 2615 (2005)
T. Okanishi, T. Matsui, T. Takeguchi, R. Kikuchi, K. Eguchi, Appl. Catal. A 298, 181 (2006)
M. Nakada, A. Ishihara, S. Mitsushima, N. Kamiya, and K.I. Ota, Electrochem. Solid-State Lett. 10, (2007)
M. Dou, M. Hou, D. Liang, W. Lu, Z. Shao, B. Yi, Electrochim. Acta 92, 468 (2013)
D. Tripković, S. Stevanović, A. Gavrilović, J. Rogan, U. Lačnjevac, T. Kravić, V.M. Jovanović, Electrocatalysis 9, 76 (2017)
L.A. Frolova, Y.A. Dobrovolsky, N.G. Bukun, Russ. J. Electrochem. 47, 697 (2011)
K. Zhang, C. Feng, B. He, H. Dong, W. Dai, H. Lu, X. Zhang, J. Electroanal. Chem. 781, 198 (2016)
N. Zhang, S. Zhang, C. Du, Z. Wang, Y. Shao, F. Kong, Y. Lin, G. Yin, Electrochim. Acta 117, 413 (2014)
E. Antolini, E. Gonzalez, Solid State Ionics 180, 746 (2009)
S. Sharma, B.G. Pollet, J. Power Sources 208, 96 (2012)
J. Parrondo, F. Mijangos, B. Rambabu, J. Power Sources 195, 3977 (2010)
A.B. Kuriganova, D.V. Leontyeva, S. Ivanov, A. Bund, N.V. Smirnova, J. Appl. Electrochem. 46, 1245 (2016)
A.B. Kuriganova, N.V. Smirnova, Mendeleev Commun. 24, 351 (2014)
I.N. Novomlinskiy, V.E. Guterman, M.V. Danilenko, V.A. Volochaev, Russ. J. Electrochem. 55, 690 (2019)
V.E. Guterman, I.N. Novomlinskiy, L.M. Skibina, D.K. Mauer, RUS 2656914 19.09.2017
A.A. Alekseenko, V.E. Guterman, V.A. Volochaev, S.V. Belenov, Inorg. Mater. 51, 1258 (2015)
V. Menshchikov, A. Alekseenko, V. Guterman, A. Nechitailov, N. Glebova, A. Tomasov, O. Spiridonova, S. Belenov, N. Zelenina, O. Safronenko, Nanomaterials 10, 742 (2020)
D.V.D. Vliet, D.S. Strmcnik, C. Wang, V.R. Stamenkovic, N.M. Markovic, M.T. Koper, J. Electroanal. Chem. 647, 29 (2010)
K. Shinozaki, J. W. Zack, S. Pylypenko, B. S. Pivovar, and S. S. Kocha, J. Electrochem. Soc. 162, (2015)
M.K. Jeon, Y. Zhang, P.J. Mcginn, Electrochim. Acta 55, 5318 (2010)
O.A. Petrii, Russ. Chem. Rev. 84, 159 (2015)
Y.C. Park, K. Kakinuma, M. Uchida, H. Uchida, M. Watanabe, Electrochim. Acta 123, 84 (2014)
D.Y. Chung, K.J. Lee, Y.E. Sung, The Journal of Physical Chemistry C 120, 9028 (2016)
Acknowledgements
The authors are grateful to Dr. V.A. Volochaev for the help in registering XRD spectra.
Funding
This research was financially supported by the Ministry of Science and Higher Education of the Russian Federation (state assignment in the field of scientific activity no 0852-2020-0019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Novomlinskiy, I.N., Danilenko, M.V., Safronenko, O.I. et al. Influence of the Sn-Oxide-Carbon Carrier Composition on the Functional Characteristics of Deposited Platinum Electrocatalysts. Electrocatalysis 12, 489–498 (2021). https://doi.org/10.1007/s12678-021-00649-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-021-00649-8