
Abstract
In photosynthetic reaction centers of intact photosystem I (PSI) complexes from cyanobacteria, electron transfer at room temperature occurs along two symmetrical branches of redox cofactors A and B at a ratio of ~3 : 1 in favor of branch A. Previously, this has been indirectly demonstrated using pulsed absorption spectroscopy and more directly by measuring the decay modulation frequencies of electron spin echo signals (electron spin echo envelope modulation, ESEEM), which allows to determine the distance between the separated charges of the primary electron donor P700+ and phylloquinone acceptors A1A– and A1B– in the symmetric redox cofactors branches A and B. In the present work, these distances were determined using ESEEM in PSI complexes lacking three 4Fe–4S clusters, FX, FA, and FB, and the PsaC protein subunit (the so-called P700–A1 core), in which phylloquinone molecules A1A and A1B serve as the terminal electron acceptors. It was shown that in the P700–A1 core preparations, the average distance between the centers of the P700+A1– ion-radical pair at a temperature of 150 K in an aqueous glycerol solution and in a dried trehalose matrix, as well as in a trehalose matrix at 280 K, is ~25.5 Å, which corresponds to the symmetrical electron transfer along the A and B branches of redox cofactors at a ratio of 1 : 1. Possible reasons for the change in the electron transfer symmetry in PSI upon removal of the PsaC subunit and 4Fe–4S clusters FX, FA, and FB are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The reaction center (RC) of photosystem I (PSI) of cyanobacteria and green plants contains 11 redox cofactors, including six chlorophyll (Chl) molecules, two phylloquinone (PhQ) molecules, and three 4Fe–4S type iron–sulfur clusters – FX, FA, and FB [1]. Laser flash-induced electron transfer includes the oxidation of the primary electron donor chlorophyll dimer P700 and subsequent transfer through the primary chlorophyll acceptor A0 to the phylloquinone acceptors A1 and 4Fe–4S clusters FX, FA, and FB [2]. In the absence of exogenous electron acceptors, charge recombination between the reduced terminal 4Fe–4S [FA/FB]– clusters and the oxidized P700+ donor occurs in PSI RC. Previously, it was shown that sequential removal of the FB, FA, and FX clusters from PSI results in the backward electron transfer to P700+ from the preceding acceptors FA, FX, and A1, respectively [3-5].
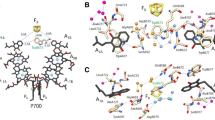
The PSI complex from cyanobacteria consists of 12 protein subunits, three of which (PsaA, PsaB, and PsaC) bind all electron transfer cofactors. These subunits also bind 79 out of 90 Chl molecules of the light-harvesting antenna. PsaA and PsaB subunits form a heterodimer that constitutes the central part of the complex and bind six Chl molecules, two PhQ molecules, and the 4Fe–4S cluster FX, while the PsaC subunit adjacent to the acceptor site of the PsaA/PsaB heterodimer binds the terminal iron–sulfur clusters FA and FB. Redox cofactors associated with PsaA/PsaB are located along the C2 axis of symmetry that passes through the geometric center of the Chl dimer P700 and the FX cluster and form two symmetrical branches (A and B) [1, 3]. The A branch contains the primary acceptor A0A (consisting of Chl molecules Chl2A and Chl3A) and the PhQ molecule A1A; the B branch contains the primary acceptor A0B (consisting of Chl molecules Chl2B and Chl3B) and the PhQ molecule A1B. Electron transfer occurs along both branches; it starts with the oxidation of P700 and ends at FX, where the branches converge and further transfer to the terminal acceptors [FA/FB] takes place (Fig. 1a).

Structure of the PSI RC: a) intact P700–FA/FB complex; b) P700–A1 core complex lacking 4Fe–4S clusters and PsaC subunit; c) distances between the spin density centers of P700+ and A1A–/A1B–. The paths of backward electron transfer are indicated by arrows.
The factors determining participation of both branches of redox cofactors in the electron transfer in PSI RCs have not been fully elucidated [3]. In the other two types of RCs studied, photosystem II (PSII) complexes and bacterial RCs, which also have two symmetrical branches of cofactors similar to the A and B branches, the transfer occurs only along one branch to the primary quinone acceptor QA, and then from QA– to the secondary acceptor QB with the formation of the stable semiquinone QB–. Excitation of such RCs by a second flash of light leads to complete reduction of QB– with the formation of protonated secondary quinone QBH2, which, in turn, is oxidized by the cytochrome bc1 complex (b6f in PSII). The transfer of electrons in PSI occurs from the PhQ molecules A1A and A1B to the iron–sulfur cluster FX, but not between the A1A and A1B. It can be assumed that the possibility of transfer along both branches of redox cofactors in PSI is an evolutionary adaptation that makes it possible to preserve an efficient reduction of low-potential electron acceptors, ferredoxin and NADP, in the case of random mutations near the primary acceptors in one of the branches.
The electron transfer in the RCs of cyanobacterial PSI occurs asymmetrically along the A and B branches at a ratio of ~3 : 1 in favor of the A branch [6]. In this case, the transfer from A0A to A1A and from A0B to A1B occurs with the same characteristic time τ ≈ 25 ps, while the transfer from A1A to FX is approximately an order of magnitude slower than from A1B to FX (τ ≈ 200 ns vs. τ ≈ 20 ns, respectively) [7-9]. The difference in the rates of the A1A→FX and A1B→FX reactions is mainly caused by the difference in the free energy (ΔG) values of these reactions, since the midpoint redox potentials (Em) of A1A and A1B differ by ~150 mV [10, 11]. It is possible that the recombination kinetics of the ion-radical pairs P700+A1B– and P700+A1A– in the PSI complexes lacking 4Fe–4S clusters (so called P700–A1 core, Fig. 1c) has different characteristic times (τ ≈ 20 and 200 µs, respectively) due to the difference between the ΔG values of the backward electron transfer reactions A1B–→P700+ and A1A–→P700+ [5, 12]. However, it should be noted that the removal of the PsaC subunit and iron–sulfur clusters can lead to changes in the Em values of A1A and A1B. Since the negatively charged clusters FA and FB are located asymmetrically with respect to the PhQ molecules A1A and A1B, the removal of these clusters should lead to a decrease in the difference between the redox potentials of A1A and A1B [10, 13].
Electron transfer asymmetry between the branches of redox cofactors in PSI continues to be a subject of discussion (see review [3]). As shown by Plato et al., the spin density center between the Chl1A and Chl1B monomers in the P700 dimer is significantly shifted towards Chl1B [14, 15]; as a result, the distances between the spin density centers of the P700+A1A– and P700+A1B– ion-radical pairs are ~26 Å and ~25 Å, respectively (Fig. 1c). Previously, EPR spectroscopy was used to register the modulation frequencies of the electron spin echo signal decay (electron spin echo envelope modulation, ESEEM), demonstrating that at cryogenic temperatures, the measured distance between the centers of the spin density in the ion-radical pair P700+A1– (~26 Å) in intact PSI complexes from cyanobacteria (P700–FA/FB) corresponds to the electron transfer along the A branch [16, 17]. The same distance was revealed by ESEEM for PSI complexes isolated from the cyanobacterial MenB mutant containing a high-potential quinone derivative (2,3-dichloronaphthoquinone) in the A1A and A1B binding sites. In these complexes, electron transfer to the 4Fe–4S clusters was not observed for thermodynamic reasons, and charge recombination occurred between A1– and P700+ [16]. The measured distance between P700+ and A1– was also ~26 Å, which corresponds to the predominant electron transfer along the A branch.
Another way to determine the degree of electron transfer asymmetry between the redox cofactor branches A and B is to measure the ratio of amplitudes of kinetic components corresponding to the forward electron transfer from A1A– and A1B– to FX. By measuring the electrochromic shift of carotenoid absorption band, it was shown that this ratio is 2 : 1 in favor of the A branch [18]. However, the carotenoid molecules in PSI RCs are oriented differently with respect to the PhQ molecules in the A1A and A1B sites. The P700–A1 core complexes were used to study the contribution of kinetic components corresponding to the charge recombination of the ion-radical pairs P700+A1A– and P700+A1B– on the one hand and to the electrochromic carotenoid shift on the other hand. Comparison of the relative amplitudes of these kinetic components showed that the contribution of the transfer along the A branch is underestimated by a factor of 2.33 compared to the contribution of the transfer along the branch [19]. Taking into account this coefficient, the ratio of forward electron transfer along the branches of redox cofactors should be ~4 : 1 in favor of the A branch. Since charge recombination from the PhQ molecules is not observed in the P700–FA/FB complexes, the kinetics of backward electron transfer from A1A– and A1B– to P700+ can only be measured if the forward transfer to the iron–sulfur clusters is arrested. Partial arrest of the forward electron transfer from Al to the 4Fe–4S clusters is observed with decreasing temperature and upon drying in a glassy trehalose matrix [20-22]. However, at room temperature in solution, charge recombination in the ion-radical pairs P700+A1A– and P700+A1B– can only be registered in the P700–A1 core complexes.
The study of the temperature dependence of the charge recombination kinetics between A1– and P700+ in the P700–A1 core complexes by pulse absorption spectrometry demonstrated that at 300 K, the ratio of the kinetic components contributions of the backward electron transfer from the reduced A1A– and A1B– acceptors to P700+ is ~60 : 40 in favor of the A branch. As the temperature drops to 150 K, these contributions become almost equal (50 : 50) [12].
The distances between the spin density centers in the P700+A1– ion-radical pairs were measured at cryogenic temperatures in water–glycerol solutions. At temperatures above 200 K, pulsed EPR spectroscopy could not be used reliably due to the fast spin relaxation. However, drying PSI complexes in a glassy trehalose matrix slows down spin relaxation and makes it possible to detect the formation of the P700+A1– ion-radical pairs using high-frequency EPR spectroscopy in the W and Q bands, as well as to determine the distance between the spin density centers in these pairs using ESEEM [17, 23, 24]. Previously, high-frequency EPR spectroscopy and ESEEM were used to study the spin-correlated P870+QA– radical pair in bacterial RCs in a dried trehalose matrix at 150 K and at room temperature [25]. It was shown that drying these preparations in a trehalose matrix does not change the molecular configuration of the P870+QA– radical pair. It can be assumed that measuring the distances between the spin density centers of the ion-radical pairs is also possible for other pigment–protein complexes in a dried trehalose matrix at room temperature. The use of dehydrated trehalose matrices for the long-term preservation of the activity of various protein complexes at room temperature was discussed in detail in a review by Möbius et al. [26].
Recently, we measured the distance between P700+ and A1– in the P700–FA/FB complexes in a dried trehalose matrix at cryogenic and room temperatures [23]. At 150 K and 220 K, this distance was ~26 Å and increased to ~27 Å at 280 K. However, this apparent increase in the distance (based on a decrease in the modulation frequency of the ESEEM signal), was explained by the acceleration of the A1– spin–lattice relaxation from 3 to 1 µs under the influence of the iron ions of the 4Fe–4S clusters. Therefore, it was concluded that the distance between the spin density centers of the P700+A1– pair in the trehalose matrix at room temperature remains unchanged and corresponds to the preferential electron transfer along the A branch.
In another study [24], we used the ESEEM method to determine the distance between P700+ and A1– in PSI complexes lacking the PsaC subunit and terminal 4Fe–4S clusters FA/FB (P700–FX core complexes). The obtained data demonstrated that this distance in the dried trehalose matrix (~25 Å) corresponded to the preferential electron transfer along the B branch [24].
In the present study, we measured the distance between P700+ and A1– in the P700–A1 core complexes of PSI in a water–glycerol solution and in a dried trehalose matrix at cryogenic temperatures, as well as in the trehalose matrix at 280 K.
MATERIALS AND METHODS
Preparation of P700–A1 core complexes. Trimeric complexes of PSI were isolated from cyanobacterium Synechocystis sp. PCC 6803 [27] by treating thylakoid membranes with dodecyl-β-D-maltoside, followed by centrifugation in a sucrose density gradient (5-20%) at 210,000g for 3 h using Beckman VTi 50 rotor (USA), obtained with the support from the Lomonosov Moscow State University Program of Development. To produce PSI complexes lacking the iron–sulfur clusters FX/FA/FB (P700–A1 core, see Fig. 1b), intact PSI complexes from Synechocystis sp. PCC 6803 were incubated in a medium containing 50 mM Tris-HCl (pH 8.0) and 6.8 M urea for 90 min at room temperature in the dark. The final detergent concentration did not exceed ~0.05% (m/V). To remove urea, the PSI suspension was washed with 50 mM Tris-HCl buffer (pH 8.0) down to the urea concentration of ~0.03%. After the suspension was concentrated, the Chl concentration in the PSI samples was ~2 mg Chl/ml; at this stage, the terminal FA/FB clusters had been removed from the complex.
The obtained samples were incubated in a medium containing 50 mM Tris-HCl (pH 8.0), 3.4 M urea, and 5 mM potassium ferricyanide for 2 h at room temperature in the dark. To remove urea and ferricyanide, the PSI suspension was repeatedly washed with a buffer containing 50 mM Tris-HCl (pH 8.0) to the final concentrations of urea and ferricyanide of ~0.03% and ~0.005% (m/V), respectively. After concentrating the P700–A1 core suspension, the Chl concentration in RCs was ~1.5 mg Chl/ml (see [28] and references therein for the method description).
To obtain trehalose matrices, a solution containing PSI complexes and trehalose at a molar ratio of 1 : 40,000 (PSI : trehalose) was dehydrated in the presence of saturated aqueous solution of LiCl [21] (under these conditions, a relative air humidity above the salt solution was maintained at 11%). Dehydration of the PSI solution for 14 days in a desiccator with a saturated LiCl solution allowed to achieve an equilibrium between the protein in the trehalose matrix and air vapor. The water–glycerol solutions of the PSI complexes contained 50% glycerol (of the total volume).
ESEEM of P700–A1 core complexes. To find the distance between the separated charges in RCs, we employed the pulse primary electron spin echo method. The measurements were carried out in the Q-band (34.03 GHz) with a Bruker Elexsys E580 EPR spectrometer (USA) using an EN 5107D2 resonator with a photoexcitation window that introduced the laser beam directly to the sample and Bruker ER 4131VT nitrogen cryostat (USA). YAG:Nd3+ laser Quantel Brilliant B (France–USA) was used for photoexcitation (wavelength, 532 nm; energy, 1 mJ; a repetition rate, 10 Hz). Measurements were performed in quartz capillaries with an outer diameter of 1.2 mm and an inner diameter of 1 mm sealed on one side. Dried trehalose matrices were placed in the capillaries in an atmosphere of inert gas. The experimental protocol is shown in Fig. 2 and is described in more detail in [23]. The Fourier transform of the signals was performed with the Bruker Xepr software package (USA) using the fast Fourier transform algorithm.

Experimental protocol for measuring the modulation frequency of the primary spin echo signal caused by the dipole–dipole interaction of the ions-radicals of the charge-separated state in PSI RC.
In ESEEM experiments, laser pulse was used to induce charge separation in the PSI RC. After some delay, a sequence of two microwave pulses generated the primary electron spin echo signal. The dependence of the echo signal amplitude on the time interval τ between the two microwave pulses was recorded. The duration of the microwave π/2 pulse was 40 ns. During the experiment, ESEEM was observed that was caused by the spin–spin dipole–dipole interaction between the separated charges P700+A1– in the PSI RC [29]. The frequency of the observed modulation is equal to the frequency of the spin–spin dipole–dipole interaction of the separated charges, which is defined as:
where r is the distance between the separated charges; g1, g2 are the g-factors of the separated charges; β is the Bohr magneton; ħ is the Planck constant; θ is the angle between the radius vector r and the direction of the induction vector of the constant magnetic field B0.
Equation (1) demonstrates that the dipole frequency ωd–d takes on different values with a certain distribution, depending on the angle θ. Orientations of the vector r with θ = π/2 have the greatest statistical weight, i.e., the largest contributions to the observed modulation of the echo signal come from RCs with θ = π/2, for which the dipole frequency is equal to:
The EPR spectrum of the spin-correlated pair P700+A1– obtained by integrating the echo signal with a fixed value of τ = 300 ns, is shown at Fig. 3. The ESEEM signals were measured for the resonant value of the magnetic field corresponding to the signal from P700+ of the ion-radical pair P700+A1–. Signal modulation was observed for only one magnetization component of the echo signal [30].

EPR spectrum of the P700+A1– ion-radical pair in the P700–A1 core PSI complexes incorporated into the trehalose matrix at 280 K. The spectrum was obtained by integrating the echo signal for various magnetic field values; the time interval τ in a two-pulse sequence was 300 ns.
RESULTS
Figure 4 shows the ESEEM signals of the P700–A1 core PSI complexes at room and cryogenic temperatures; Fig. 5 demonstrates the corresponding Fourier spectra.

ESEEM signals of P700+ in the P700–A1 core PSI complex obtained in water–glycerol solution at 150 K (black curve), in trehalose matrix at 150 K (red curve), and in trehalose matrix at 280 K (blue curve); green curves represent the fitting of the experimental data according to Eq. (3). The curves are arbitrarily shifted along the y-axis.

Fourier spectrum of P700+ ESEEM signals obtained for the P700–A1 core PSI complex (a) and its main peak (b). The spectra are shown for the complexes in a water–glycerol solution at 150 K (black curve), in the trehalose matrix at 150 K (red curve), and in the trehalose matrix at 280 K (blue curve).
Figure 5 shows that the main contribution to the modulation of the echo signal is due to the frequency of ~3.2 MHz. Figure 5b reveals the features of the Fourier spectra in the region around 3.2 MHz at an enlarged scale, which makes it possible to discern the modulation frequency shift by ~60 kHz upon temperature increase from 150 to 280 K for the sample in the trehalose matrix.
The most probable values of the dipole frequency for the studied systems and the distance between electron spins (separated charges) were found according to Eq. (2), based on the obtained Fourier spectra. Figure 5 shows that at 150 K, the Fourier spectra maxima in trehalose and glycerol coincide.
To verify the obtained estimates, the echo signal modulation curves V were calculated according to the following equation (see [23, 31]), based on the distance between the spins:
where ‹cos(ωd–d τ)› = ½ ∫cos(Ω(1–3cos2ϑ) sinϑ dϑ) is the mean dipole–dipole interaction orientation, and p is the probability of spin flip by the microwave pulse at the time moment τ (see Fig. 2).
The obtained distances between the cation and anion in the P700+A1– ion-radical pair for various PSI complexes and temperatures are presented in the table. The distances for intact PSI and P700–FX core complex lacking terminal 4Fe–4S clusters FA/FB taken from our previously published works [23, 24], are also provided.
Modulation frequencies of the observed echo signal and the corresponding distances between the separated charges P700+ and A1– for the PSI complexes in the dried trehalose matrix
PSI complex type | Modulation frequency, MHz | Distance, Å | ||
|---|---|---|---|---|
calculated from ESEEM signal according to (3) | calculated from Fourier spectrum of ESEEM signal | |||
P700–FA/FB | – | 2.98 | 26 ± 0.1 [23] | |
P700–FX core | – | 3.35 | 25 ± 0.1 [24] | |
P700–A1 core | 150 K | 3.15 | 3.17 | 25.5 ± 0.1 |
280 K | 3.10 | 3.11 | 25.6 ± 0.1 | |
Analysis of the data for the P700–A1 core presented in the table leads to the two observations. Firstly, an increase in the temperature by 130 K caused an increase in the distance between the cofactors by only 0.1 Å. Secondly, the distance between the separated charges P700+ and A1– in the P700–A1 core complexes calculated from the modulation frequency of the observed ESEEM signal, has an intermediate value between the distances for P700+A1A– and P700+A1B– pairs in intact PSI.
When interpreting the results obtained for the P700–A1 core complexes, we should take into account that the observed ESEEM signal modulation frequency may be lower than the frequency of the dipole–dipole interaction according to the Eq. (1), due to the low-frequency shift caused by the spin–lattice relaxation of the cofactor A1– spins in intact PSI (as shown previously by us in [23, 24]). In this case, a significant contribution to the spin–lattice relaxation is made by the interaction with paramagnetic iron–sulfur clusters. In the previous studies [23, 24], we estimated the distances between the separated charges for the P700–FA/FB and P700–FX core complexes as ~26 Å and ~25 Å, respectively, by taking into account the above-mentioned low-frequency ESEEM frequency shift. Comparing these results with the X-ray data (Fig. 1), we came to the conclusion that the charge separation occurs predominantly along the A branch in the P700–FA/FB complexes, while in the P700–FX core complexes, the separation occurs along the B branch of the PSI RC.
In this work, we studied the P700–A1 core complexes, in which the rate of spin–lattice relaxation of separated charges is much lower than the spin–lattice relaxation rate for the P700–FA/FB and P700–FX core complexes of PSI. Therefore, in the case of P700–A1 core complexes, the low-frequency shift of the modulation frequency due to the spin–lattice relaxation is negligibly small, and the ESEEM frequency in this system is equal to the dipole frequency (1). The echo signal modulation frequency observed for the P700–A1 core can be represented as the average value between the signal modulation frequencies in the ESEEM experiments for the intact P700–FA/FB and P700–FX core complexes (here, the modulation frequency for the intact RC was obtained at T = 150 K, when the low-frequency shift of the dipole modulation frequency is negligible):
To interpret this phenomenon, we have to assume that both A and B cofactor branches of the RC are involved in the charge separation in the P700–A1 core complexes, with dipole frequencies of 2.98 MHz and 3.35 MHz, respectively. From empirical modeling, it can be concluded that Fourier transform of the observed signal of the separated charges in the PSI RC can produce one broadened bell-shaped curve instead of a curve with two resolved maxima provided that following two conditions are met. Firstly, the phase relaxation times of spins should be <1 μs, and, secondly, the distances between the spins in two subensembles of pairs should differ by no more than 1 Å. Both these conditions are valid for the putative signals of the P700+A1A– and P700+A1B– ion-radical pairs. In this case, it can be expected that the observed ESEEM signal is the sum of the contributions of two RC subensembles with separated charges in the A and B branches [see Eq. (3)]:
where p is the probability of spin flip by the microwave pulse at time moment τ [µs].
Two cases can be recognized when the observed signal evolution in time according to (5) leads to the appearance of oscillations with the average frequency (4). Firstly, for arbitrary statistical weights of the above-described subensembles [an arbitrary value of f in (5)], the average frequency (4) appears if the electron can jump quickly enough between A1A and A1B, namely, the frequency of electron jumps should be equal to the modulation frequency difference, i.e., 3.35 – 2.98 = 0.37 MHz. We consider this scenario unlikely.
Secondly, Eq. (5) results in the modulation frequency (4) for the case of equiprobable contribution of A and B branches to the charge separation in the RC, e.g., in the case f = 1/2. The modulation frequency is also equal to the average frequency (4) as a good approximation when the two branches have comparable contributions to the charge separation. We believe that this is the situation realized in the system under study. To explain the decrease in the modulation frequency with increasing temperature (see table), we must assume that the fraction f of the separated pairs of charges in the A branch should increase upon heating: f = 0.49 at T = 150 K, f = 0.65 at T = 280 K.
DISCUSSION
The distance between the spin density centers of the ion-radical pair P700+A1– in the P700–A1 core PSI complexes from the cyanobacterium Synechocystis sp. PCC 6803 (25.5 Å) almost coincides with the average value between the distances in the pairs P700+A1A– (~26 Å) and P700+A1B– (~25 Å), which corresponds to the symmetrical electron transfer from the Chl dimer P700 along the redox cofactor branches A and B. Analogous conclusion was made earlier on the basis of less direct experiments [12, 20]. Similar estimates of the distance in the P700+A1A– pair (26 Å) were obtained earlier for intact PSI complexes and PSI complexes containing high-potential quinone derivatives in the A1 site [16]. Similar estimates for the distances between the spin density centers of P700+A1– radical ion pairs (26 and 25 Å for branches A and B, respectively) were also obtained in the study of PSI mutants with the blocked transfer along one of the branches [17] for the complexes in a water–glycerin mixture at low temperature.
In [32], the spectra of the ion-radical pairs assigned by the authors to P700+A1A– and P700+A1B– states were recorded using high-frequency time-resolved EPR spectroscopy (frequency, 130 GHz) for deuterated PSI complexes isolated from the cyanobacterium Synechococcus leopoliensis. Similar to the earlier works conducted in PSI from the cyanobacterium Synechocystis sp. PCC 6803 [12, 16, 17, 33], the authors, who used intact P700–FA/FB complexes, observed formation of the P700+A1A– pair only at low temperatures. In authors’ opinion, this result could be explained by the fact that under the used experimental conditions the forward electron transfer to the 4Fe–4S clusters proceeded rapidly and predominantly along the B branch and the period in a series of laser pulses was too short for the backward electron transfer to occur. As a result, only the P700+A1A– pair was observed. However, the electron transfer exclusively along the A branch was previously found in the PSI complexes containing high-potential naphthoquinone derivatives in the A1A and A1B binding sites [16]. Since electron transfer to the 4Fe–4S clusters was not observed in these PSI complexes, it was concluded that the predominant electron transfer to A1A was not caused by the rapid and almost irreversible transfer along the B branch to the 4Fe–4S clusters, but was related to a higher redox potential value of A1A as compared to A1B. It was shown [32] using P700–A1 score complexes that the resulting EPR spectrum can be approximated by the sum of two spectra characteristic of P700+A1B– and P700+A1A– states at a ratio of 56 : 44.
In the P700–A1 core complexes from Synechocystis sp. PCC 6803, the contribution ratio of recombinations of the P700+A1A– and P700+A1B– pairs in solution at room temperature was 60 : 40 in favor of the A branch [19]. In the dried trehalose matrix, however, this ratio was inverted to 60 : 40 in favor of the B branch. In the P700–FX core complexes lacking terminal 4Fe–4S clusters FA/FB and PsaC, the ratio between the branches A and B was 65 : 35 in favor of the A branch, while in the trehalose matrix, it was 75 : 25 in favor of the B branch. Charge recombination from phylloquinone acceptors A1A– and A1B– was observed in 40-45% of RCs in these samples due to the fact that the functionally active FX cluster was present only in 55-60% of RCs in this fraction [19].
In another study [12], the temperature dependence of the recombination kinetics between A1– and P700+ was measured in the PSI complexes isolated from the mutant strain RubA that lacked the 4Fe–4S clusters and were functionally equivalent to the P700–A1 core preparations. It was shown that at room temperature, the ratio of the contributions of the electron transfer along the redox cofactor branches was 60 : 40 in favor of the A branch; when the temperature was lowered to 150 K, the contributions of the A and B branches became equal (50 : 50). In the P700–FX core complexes, the ratio of backward electron transfer along the A and B branches was 60 : 40 at room temperature, but increased to 77 : 23 in favor of the A branch at 150 K [20].
Therefore, our earlier evaluation of the charge recombination kinetics of the ion-radical pairs P700+A1A– and P700+A1B– revealed that the backward electron transfer in the P700–A1 core complexes occurs along both cofactor branches with an approximately equal contributions (50 ± 10%), at room temperature, as well as in a water–glycerol solution at 150 K and at room temperature in a dried trehalose matrix. At the same time, in the P700–FX core complexes, the contribution of the A and B branches was (62.5 ± 2.5) : (37.5 ± 2.5) at room temperature in solution and increased to 75 : 25 in favor of the A branch at 150 K and in dried trehalose matrix. These results show that the presence of the FX cluster promotes asymmetric electron transfer along the A branch, and that the degree of asymmetry increases both with decreasing temperature and upon drying in a glassy trehalose matrix, i.e., when the conformational mobility of the protein is restricted.
The question arises on why the distance between the spin density centers in the P700+A1– ion-radical pair measured in the P700–A1 core complexes using ESEEM, reflects an approximately equal contribution of the A and B branches, while in the P700–FX core complexes, this distance corresponds to the predominant formation of the P700+A1B–. There are several possible explanations for this experimental observation:
1) Electron transfer in the P700–FX core complexes occurs along the A branch, but under conditions of limited protein mobility (low temperature, drying in a trehalose matrix) and at a laser flash frequency of 10 Hz, accumulation of the reduced acceptors FX– and A1A– takes place, as these acceptors do not have enough time to recombine with P700+. At the same time, the backward electron transfer from the lower-potential A1B– to P700+ occurs faster (characteristic time, 10-20 µs); as a result, the distance characteristic of the P700+A1B– pair is registered in the ESEEM experiment.
2) The balance of the redox potentials (Em) of A1A and A1B changes in these complexes in the trehalose matrix, making electron transfer to quinone A1B more thermodynamically preferable than to A1A.
3) Drying in a trehalose matrix disrupts the cluster of intraprotein water molecules near the A1A site (but not near the A1B site), since this cluster is less isolated from the protein outer surface [19].
There is also the question why is the distance corresponding to the ion-radical pair P700+A1A– measured in intact P700–FA/FB complexes? We assume that the electron transfer in these complexes, as well as in the P700–FX core, proceeds predominantly along the A branch, but the accumulation of the reduced A1A– state does not occur under experimental conditions with successive laser flashes, due to a larger capacity of the acceptors (three 4Fe–4S cluster versus one in the P700–FX core complexes).
Further experiments with the PSI containing different number of 4Fe–4S clusters and exogenous quinones of various structure and redox potentials inserted into the A1A and A1B sites can test the validity of these hypotheses.
Abbreviations
- A1 :
-
phylloquinone, secondary electron acceptor
- Chl:
-
chlorophyll
- Em :
-
midpoint redox potential
- ESEEM:
-
electron spin echo envelope modulation
- P700 :
-
chlorophyll dimer, primary electron donor
- PhQ:
-
phylloquinone
- PSI:
-
photosystem I
- RC:
-
reaction center
References
Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W., and Krauß, N. (2001) Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution, Nature, 411, 909-917, https://doi.org/10.1038/35082000.
Brettel, K., and Leibl, W. (2001) Electron transfer in photosystem I, Biochim. Biophys. Acta, 1507, 100-114, https://doi.org/10.1016/s0005-2728(01)00202-x.
Srinivasan, N., and Golbeck, J. H. (2009) Protein–cofactor interactions in bioenergetic complexes: The role of the A1A and A1B phylloquinones in Photosystem I, Biochim. Biophys. Acta Bioenerg., 1787, 1057-1088, https://doi.org/10.1016/j.bbabio.2009.04.010.
Parrett, K. G., Mehari, T., Warren, P. G., and Golbeck, J. H. (1989) Purification and properties of the intact P-700 and Fx-containing Photosystem I core protein, Biochim. Biophys. Acta, 973, 324-332, https://doi.org/10.1016/S0005-2728(89)80439-6.
Brettel, K., and Golbeck, J. H. (1995) Spectral and kinetic characterization of electron acceptor A1 in a Photosystem I core devoid of iron-sulfur centers FX, FB and FA, Photosynth. Res., 45, 183-193, https://doi.org/10.1007/BF00015559.
Makita, H., and Hastings, G. (2015) Directionality of electron transfer in cyanobacterial photosystem I at 298 and 77 K, FEBS Lett., 589, 1412-1417, https://doi.org/10.1016/j.febslet.2015.04.048.
Joliot, P., and Joliot, A. (1999) In vivo analysis of the electron transfer within photosystem I: Are the two phylloquinones involved? Biochemistry, 38, 11130-11136, https://doi.org/10.1021/bi990857c.
Schlodder, E., Falkenberg, K., Gergeleit, M., and Brettel, K. (1998) Temperature dependence of forward and reverse electron transfer from A1-, the reduced secondary electron acceptor in photosystem I, Biochemistry, 37, 9466-9476, https://doi.org/10.1021/bi973182r.
Guergova-Kuras, M., Boudreaux, B., Joliot, A., Joliot, P., and Redding, K. (2001) Evidence for two active branches for electron transfer in photosystem I, Proc. Natl. Acad. Sci. USA, 98, 4437-4442, https://doi.org/10.1073/pnas.081078898.
Ptushenko, V. V., Cherepanov, D. A., Krishtalik, L. I., and Semenov, A. Y. (2008) Semi-continuum electrostatic calculations of redox potentials in photosystem I, Photosynth. Res., 97, 55-74, https://doi.org/10.1007/s11120-008-9309-y.
Ishikita, H., and Knapp, E.-W. (2003) Redox potential of quinones in both electron transfer branches of photosystem I, J. Biol. Chem., 278, 52002-52011, https://doi.org/10.1074/jbc.M306434200.
Cherepanov, D. A., Milanovsky, G. E., Gopta, O. A., Balasubramanian, R., Bryant, D. A., et al. (2018) Electron–phonon coupling in cyanobacterial photosystem I, J. Phys. Chem. B, 122, 7943-7955, https://doi.org/10.1021/acs.jpcb.8b03906.
Ishikita, H., Stehlik, D., Golbeck, J. H., and Knapp, E.-W. (2006) Electrostatic influence of PsaC protein binding to the PsaA/PsaB heterodimer in photosystem I, Biophys. J., 90, 1081-1089, https://doi.org/10.1529/biophysj.105.069781.
Plato, M., Krauß, N., Fromme, P., and Lubitz, W. (2003) Molecular orbital study of the primary electron donor P700 of photosystem I based on a recent X-ray single crystal structure analysis, Chem. Phys., 294, 483-499, https://doi.org/10.1016/S0301-0104(03)00378-1.
Lubitz, W. (2006) EPR studies of the primary electron donor P700 in photosystem, in Photosystem I, Dordrecht, Springer Netherlands, pp. 245-269, https://doi.org/10.1007/978-1-4020-4256-0_17.
Mula, S., Savitsky, A., Möbius, K., Lubitz, W., Golbeck, J. H., et al. (2012) Incorporation of a high potential quinone reveals that electron transfer in Photosystem I becomes highly asymmetric at low temperature, Photochem. Photobiol. Sci., 11, 946-956, https://doi.org/10.1039/c2pp05340c.
Savitsky, A., Gopta, O., Mamedov, M., Golbeck, J. H., Tikhonov, A., et al. (2010) Alteration of the axial met ligand to electron acceptor A0 in Photosystem I: effect on the generation of P700+ A1− radical pairs as studied by W-band transient EPR, Appl. Magnetic Resonance, 37, 85-102, https://doi.org/10.1007/s00723-009-0052-0.
Sun, J., Hao, S., Radle, M., Xu, W., Shelaev, I., Nadtochenko, V., et al. (2014) Evidence that histidine forms a coordination bond to the A0A and A0B chlorophylls and a second H-bond to the A1A and A1B phylloquinones in M688HPsaA and M668HPsaB variants of Synechocystis sp. PCC 6803, Biochim. Biophys. Acta Bioenerg., 1837, 1362-1375, https://doi.org/10.1016/j.bbabio.2014.04.004.
Kurashov, V., Gorka, M., Milanovsky, G. E., Johnson, T. W., Cherepanov, D. A., et al. (2018) Critical evaluation of electron transfer kinetics in P700–FA/FB, P700–FX, and P700–A1 Photosystem I core complexes in liquid and in trehalose glass, Biochim. Biophys. Acta Bioenerg., 1859, 1288-1301, https://doi.org/10.1016/j.bbabio.2018.09.367.
Milanovsky, G., Gopta, O., Petrova, A., Mamedov, M., Gorka, M., et al. (2019) Multiple pathways of charge recombination revealed by the temperature dependence of electron transfer kinetics in cyanobacterial photosystem I, Biochim. Biophys. Acta Bioenerg., 1860, 601-610, https://doi.org/10.1016/J.BBABIO.2019.06.008.
Malferrari, M., Savitsky, A., Mamedov, M. D., Milanovsky, G. E., Lubitz, W., et al. (2016) Trehalose matrix effects on charge-recombination kinetics in Photosystem I of oxygenic photosynthesis at different dehydration levels, Biochim. Biophys. Acta Bioenerg., 1857, 1440-1454, https://doi.org/10.1016/j.bbabio.2016.05.001.
Shelaev, I., Gorka, M., Savitsky, A., Kurashov, V., Mamedov, M., et al. (2017) Effect of dehydrated trehalose matrix on the kinetics of forward electron transfer reactions in photosystem I, Zeitschr. Physikal. Chemie, 231, 325-345, https://doi.org/10.1515/zpch-2016-0860.
Sukhanov, A. A., Mamedov, M. D., Möbius, K., Semenov, A. Y., and Salikhov, K. M. (2018) The decrease of the ESEEM frequency of P700+A1- ion-radical pair in Photosystem I Embedded in trehalose glassy matrix at room temperature can be explained by acceleration of spin–lattice relaxation, Appl. Magnetic Resonance, 49, 1011-1025, https://doi.org/10.1007/s00723-018-1017-y.
Sukhanov, A. A., Mamedov, M. D., Möbius, K., Semenov, A. Y., and Salikhov, K. M. (2020) Impact of iron–sulfur clusters on the spin–lattice relaxation rate and ESEEM frequency of the oxidized primary donor P700+ and reduced phylloquinone acceptor A1−· in radical pairs in Photosystem I embedded in trehalose glassy matrix, Appl. Magnetic Resonance, 51, 909-924, https://doi.org/10.1007/s00723-020-01210-4.
Savitsky, A., Malferrari, M., Francia, F., Venturoli, G., and Möbius, K. (2010) Bacterial photosynthetic reaction centers in trehalose glasses: coupling between protein conformational dynamics and electron-transfer kinetics as studied by laser-flash and high-field EPR spectroscopies, J. Phys. Chem. B, 114, 12729-12743, https://doi.org/10.1021/jp105801q.
Möbius, K., Savitsky, A., Malferrari, M., Francia, F., Mamedov, M. D., et al. (2020) Soft dynamic confinement of membrane proteins by dehydrated trehalose matrices: high-field EPR and fast-laser studies, Appl. Magnetic Resonance, 51, 773-850, https://doi.org/10.1007/s00723-020-01240-y.
Shen, G., Zhao, J., Reimer, S. K., Antonkine, M. L., Cai, Q., et al. (2002) Assembly of photosystem I. I. Inactivation of the rubA gene encoding a membrane-associated rubredoxin in the cyanobacterium Synechococcus sp. PCC 7002 causes a loss of photosystem I activity, J. Biol. Chem., 277, 20343-20354, https://doi.org/10.1074/jbc.M201103200.
Vassiliev, I. R., Jung, Y. S., Mamedov, M. D., Semenov, A. Yu., and Golbeck, J. H. (1997) Near-IR absorbance changes and electrogenic reactions in the microsecond-to-second time domain in Photosystem I, Biophys. J., 72, 301-315, https://doi.org/10.1016/S0006-3495(97)78669-7.
Salikhov, K. M. (1976) Electron Spin Echo and Its Applications [in Russian] (Semenov, A. G., and Tsvetkov, Y. D., eds) Nauka, Novosibirsk, Siberian branch.
Salikhov, K. M., Kandrashkin, Y. E., and Salikhov, A. K. (1992) Peculiarities of free induction and primary spin echo signals for spin-correlated radical pairs, Appl. Magnetic Resonance, 3, 199-216, https://doi.org/10.1007/BF03166790.
Salikhov, K. M., Khairuzhdinov, I. T., and Zaripov, R. B. (2014) Three-pulse ELDOR theory revisited, Appl. Magnetic Resonance, 45, 573-619, https://doi.org/10.1007/s00723-014-0541-7.
Poluektov, O. G., Niklas, J., and Utschig, L. M. (2019) Spin-correlated radical pairs as quantum sensors of bidirectional ET mechanisms in Photosystem I, J. Phys. Chem. B, 123, 7536-7544, https://doi.org/10.1021/acs.jpcb.9b06636.
Dashdorj, N., Xu, W., Cohen, R. O., Golbeck, J. H., and Savikhin, S. (2005) Asymmetric electron transfer in cyanobacterial Photosystem I: charge separation and secondary electron transfer dynamics of mutations near the primary electron acceptor A0, Biophys. J., 88, 1238-1249, https://doi.org/10.1529/BIOPHYSJ.104.050963.
Acknowledgments
This work is dedicated to the memory of the outstanding scientist in the field of primary photosynthetic reactions – Vladimir Anatolyevich Shuvalov. V. A. Shuvalov made an invaluable contribution to the study of the mechanisms of primary reactions in PSI, including the mechanisms of the phylloquinone acceptor A1 reduction. Three co-authors of this article (M.M., G.M., and A.S.) were co-authors of many works by V. A. Shuvalov over the past 15 years and gratefully recall the period of their collaboration with V. A. Shuvalov as exceptionally interesting and productive.
Funding
The study was supported by the Russian Science Foundation and by the Cabinet of Ministers of the Republic of Tatarstan within the framework of the scientific project no. 22-23-20165 (https://rscf.ru/project/22-23-20165/).
Author information
Authors and Affiliations
Contributions
M. D. Mamedov and G. E. Milanovsky isolated PSI complexes; A. A. Sukhanov and K. M. Salikhov recorded EPR spectra; A. Yu. Semenov developed the concept, supervised the study, and wrote the article; M. D. Mamedov, G. E. Milanovsky, A. A. Sukhanov, K. M. Salikhov, and A. Yu. Semenov discussed the results and edited the manuscript.
Corresponding author
Ethics declarations
The authors declare no conflict of interest in financial or any other sphere. This article does not contain description of studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Sukhanov, A.A., Mamedov, M.D., Milanovsky, G.E. et al. Changes in the Electron Transfer Symmetry in the Photosystem I Reaction Centers upon Removal of Iron–Sulfur Clusters. Biochemistry Moscow 87, 1109–1118 (2022). https://doi.org/10.1134/S0006297922100042
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922100042