
Abstract
The midpoint redox potentials (E m ) of all cofactors in photosystem I from Synechococcus elongatus as well as of the iron–sulfur (Fe4S4) clusters in two soluble ferredoxins from Azotobacter vinelandii and Clostridium acidiurici were calculated within the framework of a semi-continuum dielectric approach. The widely used treatment of proteins as uniform media with single dielectric permittivity is oversimplified, particularly, because permanent charges are considered both as a source for intraprotein electric field and as a part of dielectric polarizability. Our approach overcomes this inconsistency by using two dielectric constants: optical ε o = 2.5 for permanent charges pre-existing in crystal structure, and static ε s for newly formed charges. We also take into account a substantial dielectric heterogeneity of photosystem I revealed by photoelectric measurements and a liquid junction potential correction for E m values of relevant redox cofactors measured in aprotic solvents. We show that calculations based on a single permittivity have the discrepancy with experimental data larger than 0.7 V, whereas E m values calculated within our approach fall in the range of experimental estimates. The electrostatic analysis combined with quantum chemistry calculations shows that (i) the energy decrease upon chlorophyll dimerization is essential for the downhill mode of primary charge separation between the special pair P700 and the primary acceptor A0; (ii) the primary donor is apparently P700 but not a pair of accessory chlorophylls; (iii) the electron transfer from the A branch quinone QA to the iron–sulfur cluster FX is most probably downhill, whereas that from the B branch quinone QB to FX is essentially downhill.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chlorophyll (Chl)-protein complexes of photosystem I (PS I) from cyanobacteria, algae, and higher plants, in which low-potential iron–sulfur clusters (FA/FB) are terminal electron acceptors, belong to the photosynthetic reaction centers (RC) of type I. PS I mediates photoinduced transfer of an electron from plastocyanin (Pc) to ferredoxin (Fd).
The three-dimensional structure of PS I from the thermophilic cyanobacterium Synechococcus elongatus has been resolved by X-ray diffraction with resolution 2.5 Å (Jordan et al. 2001). The monomeric complex (∼300 kD) contains 12 different protein subunits. The core of the complex consists of two large subunits (products of expression of genes PsaA and PsaB) which bind 96 molecules of Chl a, 22 molecules of β-carotene, two molecules of phylloquinone, and an interpolypeptide iron–sulfur center FX. Most Chl a molecules serve as antenna pigments. Terminal iron–sulfur clusters FA/FB (Fe4S4 centers) are bound to the peripheral stromal subunit PsaC with molecular weight ∼9 kD.
The scheme of mutual arrangement of electron transport cofactors and subunits PsaA, PsaB, and РsaC constituting the PS I core complex is shown in Fig. 1. The primary electron donor P700 comprises two molecules of Chl (Chl1A/Chl1B), the porphyrin planes of the molecules being parallel to one another (interplane distance, 3.6 Å) and perpendicular to the membrane plane. The X-ray diffraction analysis of crystals of the PS I complex also revealed two domains of high electron density, which were attributed to Chl a molecules in positions corresponding to accessory bacteriochlorophyll molecules in RC of purple bacteria (Chl2A/Chl2B), two Chl a molecules in positions corresponding to bacteriopheophytin molecules in RC of purple bacteria (Deisenhofer et al. 1985; Allen et al. 1987) (Chl3A and Chl3B), and two phylloquinone molecules (QA and QB).

The structure of PS I and redox cofactors. Only peptide backbone of the three main subunits PsaA, PsaB, and PsaC is shown
Pairs of Chl and phylloquinone molecules are located in nearly symmetric electron-transport branches A and B and bound to subunits PsaA and PsaB, respectively. Branch A incorporates Chl molecules Chl1A, Chl2A, Chl3A, and phylloquinone QA, whereas branch B incorporates Chl molecules Chl1B, Chl2B, Chl3B, and phylloquinone QB. The PS I electron-transport chain includes P700, A0 (one or two pairs of Chl molecules denoted as Chl2A/Chl3A and Chl2B/Chl3B), A1 (one or the two molecules of phylloquinone (QA/QB), and iron–sulfur clusters FX, FA, and FB. The problem of involvement of one or the two symmetric branches of cofactors in electron transfer from P700 to FX remains insufficiently understood (Guergova-Kuras et al. 2001; Xu et al. 2003).
A diagram of the electron-transport reactions in PS I with lifetimes of direct and back reactions as well as midpoint redox potential (E m ) values for the respective redox cofactors are shown in Fig. 2. Upon absorbing a light quantum, the primary electron donor P700 (the oxidation redox potential E m = +430 to +460 mV, Mamedov et al. 1996; Nakamura et al. 2005) turns into the excited state P700*, which is followed by the primary act of charge separation between P700* and primary acceptor A0 forming within <20 ps the intermediate state P+A0 −. After that electron is transferred within 50 ps to the phylloquinone A1 and then to the clusters FX and FA/FB within ≤200 ns. Furthermore, electron transfer to the natural acceptor Fd occurs within the time range of 0.5–100 μs. In the absence of the exogenous acceptor of electron, the kinetics of the back reactions of the electron from FX − and (FA/FB)− to P700 + are characterized by lifetime 0.5–5 and 30–100 ms, respectively. In the presence of the native secondary donor Pc, fast reduction of photooxidized P700 prevents charge recombination between FX − or (FA/FB)− and P700 + (for review, see Brettel and Leibl 2001).

Diagram of direct and back reactions in PS I with characteristic reaction times and midpoint redox potentials (E m ) of the electron transferring cofactors
The redox potentials of the electron transfer cofactors other than the primary electron donor P700 (see above) are not sufficiently determined. The oxidation potential of the excited state P700* could be estimated by subtracting the energy of quantum with the wavelength of 700 nm (1770 meV) from the E m value of P700 that gives a figure of about −1320 mV. The reduction potential of A0 was estimated to be in the range from −1070 mV (Kleinherenbrink et al. 1994) to −1290 mV (Shuvalov, 1976), while that of A1 falls within the range from −700 to −800 mV (see Brettel and Leibl 2001 and references therein). The E m values of FX, FA, and FB (−700, −540 and −590 mV) were determined by low temperature EPR spectroscopy (for review see Brettel and Leibl 2001). However, there are some indications (Golbeck et al, 1987; Jordan et al. 1998) that at room temperature these potentials are 50–100 mV positively shifted.
The knowledge of the redox properties of all the cofactors is necessary for elucidation of electron transfer mechanisms. Therefore, theoretical calculations of the redox-potentials of cofactors in PS I seem to be very important.
There are two main approaches to the theoretical analysis of protein electrostatics (for some reviews see Honig and Nicholls 1995; Warshel and Papazyan 1998; Schaefer et al. 1998; Simonson 2001, 2003; Bashford 2004). The first approach is purely microscopic: it explicitly considers various atomic interactions in protein and surrounding solution; macroscopic properties are acquired in this approach by statistical averaging using Molecular Dynamics or Monte Carlo simulations. In principle, such methods might be rather rigorous but, due to an enormous volume of computations and ambiguities in force-filed parameterization, their practical implementation requires many approximations and simplifications. The alternative and much more widespread approach is semi-microscopic one (it can be also called semi-macroscopic or semi-continuum). In this method, the protein molecule is described as a system of atomic partial charges imbedded in a continuum dielectric medium. There are also some methods combining these two approaches (e.g., Georgescu et al. 2002).
The main difficulty of the semi-microscopic method is certain ambiguity in the choice of the medium dielectric constant because the definition of macroscopic dielectric permittivity at microscopic distances is theoretically not entirely clear (Papazyan and Warshel 1997), though for proteins it works often surprisingly well (Simonson 2003).
Another problem of the semi-macroscopic approach is that the partial charges of atoms in protein exert the effects of two kinds: (i) the partial charges of atoms give rise to intraprotein electric fields, and (ii) the shift of their equilibrium positions in response to an electric field constitutes the inertial part of the total dielectric response, i.e., a substantial part of the protein’s static dielectric constant (analogue of orientational polarization of common dielectrics). Therefore, calculating intraprotein electric field with the usage of single dielectric constant, one accounts twice for the effect of partial charges: as a source of the field and as a self-screening polarization (King et al. 1991). This contradiction was resolved by Krishtalik et al. (1997). It was shown that correct analysis of the charge transfer energetics in such kind of media within the framework of the semi-continuum formalism requires separate consideration of two physically different energy components. The first component is the energy of charge insertion to a definite position within protein where some electric field pre-exists owing to charges of the protein. If the coordinates of protein atoms are resolved in microscopic three-dimensional structure, the dielectric screening of the pre-existing field does not include shifts of the atoms. Thus, at known positions of atoms, only electronic polarization contributes to the pre-existing field, which should be calculated using the optical dielectric constant ε o . The second component is the dielectric response energy (or Bornian charging energy) that accounts for interaction of the inserted charge with all kinds of polarization induced by this charge, including the shifts of protein atoms. The latter component should be calculated using the static dielectric constant ε s (Mertz and Krishtalik 1999). This approach was successfully applied to the analysis of the active site of α-chymotrypsin, but strong hydrogen bonding of imidazole in water made the continuum calculations in this case less accurate (Krishtalik 2005). The use of two dielectric constants was proposed also by Simonson et al. (1999) who calculated the charging energy with the static dielectric constant and the pre-existing field with the dielectric constant close to 1, neglecting thereby the electronic polarization of the medium.
The electrostatic calculations of redox potentials for some components of the electron transfer chain in PS I were described in the literature (Torres et al. 2003; Ishikita and Knapp 2003; Ishikita et al. 2006a, b). Similar calculations were also made for iron–sulfur clusters in other proteins (Stephens et al. 1996, Banci et al. 1996, Beck et all. 2001, Torres et al. 2003). In these works, a conventional algorithm of semi-continuum calculations was employed, in particular, a single static dielectric constant was used for all electrostatic effects, but also some ad hoc hypotheses were implemented to achieve quantitative agreement with experimental values (see Discussion for detailed analysis of the cited works).
In this work, we describe electrostatic calculations for all redox centers in PS I and two ferredoxin molecules using a unified semi-continuum approach. Our analysis of the problem offers three important novel elements.
First, according to the consideration given above, we analyze the effects of pre-existing field and dielectric response using the optical and static dielectric constants, respectively. It is shown that only this approach, in contrast to the traditional algorithm with the single dielectric constant, leads in all cases to a reasonable agreement with the experimental data.
Second, we use a variable dielectric permittivity for different parts of protein rather than ascribing a single value of the static dielectric constant to the whole protein. This assumption is based on the experimental data on electrogenic effects in photosystems, particularly in PS I (see Semenov et al. 2006 for review). These studies demonstrated that, while the dielectric constant in the hydrophobic core is low, it increases substantially in the vicinity of quinones and Fe4S4 clusters. Unexpectedly, this increase is non-monotonic, and the surroundings of some deeper-buried centers happen to be more polarizable than of the outer ones. In the Results and discussion section, we provide a detailed analysis of these data. The variation of the static dielectric permittivity exerts a considerable effect on the Bornian charging energy of components in the electron transfer chain.
Third, we perform a detailed analysis of the experimental data on the midpoint potentials (E m ) of the relevant redox couples in aprotic solvents versus standard hydrogen electrode (SHE). These data are used as reference values for the subsequent electrostatic calculations in proteins. As described in the Results and discussion section, the non-trivial corrections in the reference values have to be introduced based on the newest results of studies of ions solvation energies.
Methods
The calculation of the midpoint redox potentials of cofactors in PS I was based on the experimental potentials of the respective compounds in aprotic solvents (see Results and discussion). The shift of the redox potentials upon the transfer of the corresponding redox couple from the solvent into the definite site in the protein was calculated within the framework of the semi-continuum dielectric model. Two main components of the transfer energy were considered:
-
(1)
The change in the dielectric response energy (analogue of the Bornian solvation energy for liquid solvents); the applicability of continuum electrostatics to ions substantially larger than solvent molecules has been demonstrated recently (Bunakova et al. 2004) for metallocene ions (radius of ∼3.7 Å) in aprotic solvents with the dielectric constants almost as low as those of proteins (up to 7.2).
-
(2)
The effect of the pre-existing electric field created at the given site within the protein by all its permanent dipoles (atoms’ partial charges) and free charges; their coordinates were taken from the corresponding PDB file.
For the first component, protein was considered as a continuum medium, for the second one—as a set of discrete charges imbedded in a dielectric.
In all these calculations, the PS I dielectric heterogeneity, as revealed from electrometric experiments, was taken into account (see Results and discussion).
Coordinates
Atomic coordinates were taken from the crystal structure of cyanobacterial PS I at 2.5 Å resolution (PDB code 1JB0) (Jordan et al. 2001). Polar hydrogen atoms were generated by CHARMM (Brooks et al. 1983), with the positions of all non-hydrogen atoms being fixed. Histidines that are ligands of chlorophyll were treated as neutral ones. Crystallographic water molecules were removed from the structure, except two water molecules serving as chlorophyll ligands and 20 water molecules in the vicinity of FX, whose hydrogen atom positions were determined as providing maximum hydrogen bonds to the neighboring polar groups. The coordinates of these water molecules are given in supplementary material.
Atomic partial charges
Partial charges for amino acid residues in all our calculations were taken from the semi-empirical parameterization scheme PARSE that was adjusted to describe solvation energies (Sitkoff et al. 1994). The dipole moment of one peptide monomer in α-helix calculated by this scheme is 5.0 D, whereas commonly used schemes CHARM19 (Brooks et al. 1983) and AMBER94 (Cornell et al. 1995) give the average dipole moment of 3.7 and 4.0 D, respectively. In solution the average dipole moment of monomer in α-helix has been found about 6 D (Wada 1976) so PARSE is more consistent with the experimental findings. Krishtalik (2005) has recently employed a scaling factor of 1.3 introduced in the charging scheme AMBER94 to account for the field screening by the internal dielectric constant ε i , and the results obtained with the corrected scheme were found similar to those obtained with PARSE. Atomic partial charges for Fe4S4 clusters in the reduced and oxidized states were taken from (Torres et al. 2003); for quinones and chlorophyll molecules the partial charges were calculated ab initio with GAMESS (Schmidt et al. 1993) using the effective core potential (ECP) basis SBKJC (Stevens–Basch–Krauss–Jasien–Cundari) that is 31G double-split basis set (Stevens et al. 1992; Cundari and Stevens 1993). The partial charges calculated by three different methods (according to Mulliken, according to Lowdin, and electrostatic potential fitted charges) were essentially different but led to practically similar (within 5%) results. The most regular behavior was found for Lowdin charges that were used for electrostatic calculations.
Calculation of solvation energies changes of cofactors in PS I and ferredoxins
The change of redox potentials as compared to these potentials in solution is determined by the difference in energy of transfer of the oxidized and reduced cofactor from solution into protein. The transfer energy is determined by the energy of the medium dielectric response (the Bornian solvation energy), and by the energy of interaction of the charge transferred with the electric field created by all partial and full charges of the protein’s atoms.
The Bornian contribution was calculated in two steps (e.g., Krishtalik et al. 1993; Zhou 1994) by using the program DelPhi (Nicholls and Honig 1991). The numerical solution of the Poisson–Boltzmann equation (PBE) was done in a three-dimensional grid with the space interval of 1 Å and the margins of 16 Å, the error of solution was less than 10 mV. The energy of transfer of the redox center (with the internal dielectric constant ε i = 2.5) from DMF (εDMF = 37, ionic strength 0.05 M) into a given site of the protein with varying static dielectric constant ε s surrounded by an aqueous solution (ε sw = 81, ionic strength 0.2 М) was calculated. As noted in Introduction, PS I is a dielectrically heterogeneous system; the electrometric studies allowed its consideration as a multilayer dielectric. In our calculations, we used the dielectric permittivity distribution that follows from these experimental data; this distribution correlates well with the variation of the medium polarity (see the Results and discussion section).
The internal dielectric permittivity of the cofactors (ε i ) was always taken equal 2.5. In the case of Fe4S4 clusters, we considered as the functional unit the complex Fe4S4(SC)4, which structure was taken directly from the PDB file 1JB0. In the case of menaquinone, the active unit included 14 atoms (C1–C11, C2M, O1, O4). The coordinates of the chlorophyll rings were taken from 1JB0 structure without phytol chain. The van der Waals radii of atoms were taken in accordance with the parameterization PARSE (Sitkoff et al. 1994), the probe radius of solvent was 2.0 Ǻ and 1.4 Ǻ for DMF and water, respectively, the partial charges of the cofactors were assigned as described above (the charging energy was only slightly dependent on the details of the charge distribution, typical variations were less than 1%).
Calculation of electrostatic fields in PS I
According to the reasons given in the Introduction, we calculated the pre-existing electrostatic field of permanent charges using the optical dielectric constant of the protein, ε o, and the field created by new charges using the static dielectric constant of the protein, ε s . The value of ε o was accepted equal to 2.5 (this figure was obtained from the typical value ε o ≈ 2 for organic liquids including amides corrected for higher protein density according to the Clausius–Mosotti equation, e.g., Mertz and Krishtalik 2000). Different values of ε s in different parts of PS I were taken into account in accordance with electrometric measurements and protein polarity. The pre-existing (permanent) charges included protein backbone, uncharged amino acid residues, chlorophyll ligands and water molecules, whereas ionizable residues (Arg+, Lys+, His+, Glu− and Asp−) and cofactors were treated as newly created charges. The latter choice comes to mind because (i) charged residues are normally arranged at flexible loops on the protein surface and their conformation and electric states in crystal and solution might be essentially different; and (ii) the redox states of the cofactors in crystal are usually uncertain. The solution of PBE was obtained by DelPhi in a grid with the spacing of 0.5 Å using all other parameters as described above.
Calculation of E m values for chlorophyll dimers
Midpoint redox potential of dimer differs from that of monomer because of the direct quantum interaction of counterparts in the dimer. The wave function of two neighboring cofactors is altered due to their conjugation resulting in decrease of the system energy. We calculated the shift of the electronic energy, and hence of midpoint potential for three pairs of chlorophyll dimers in PS I (P700 and two pairs of neighboring accessory chlorophyll rings) using the program GAMESS (Schmidt et al. 1993). The modeled portion of chlorophyll ring included 28 heavy atoms and 12 hydrogen atoms comprising the conjugated π-electron system. The geometry of each dimer was taken from the PDB structure, the hydrogen atoms were built using the program HyperChem. The calculations were performed using effective core potential (ECP) basis SBKJC (Stevens et al. 1992; Cundari and Stevens 1993). The energies of neutral and oxidized dimer were compared with the respective energies of monomer taking into account the basis set superposition error (BSSE).
The conjugation energy in vacuum was found to be 140 meV for P700 and 120 meV for a “loose dimer” (Chl2A/3A, Chl2B/3B). The effect of asymmetric preexisted electric field on the dimer conjugation was calculated by the Hückel approximation according to (Artz et al. 1997). Due to the asymmetry the conjugation energies decreased to 123 meV, 68 meV and 71 meV for P700, Chl2A/3A and Chl2B/3B, correspondingly. An additional decrease of the conjugation due to dielectric polarization of the surrounding in response to the charge redistribution within the dimer is determined by the inertial part of the medium polarization only (Kuznetsov 1997). In order to account for this energy shift, we calculated, also within the framework of multilayer dielectric model, the difference of the dielectric response energies in the optical (ε ο ) and static (ε s ) media for dimer with the charge localized at one of the monomers and that delocalized at both molecules. This effect decreases the stabilization energy by 28 meV for P700 and 25 meV for other dimers. The final conjugation corrections included in Table 3 were 95, 43, and 46 meV for P700, Chl2A/3A and Chl2B/3B, respectively.
Calculation of protonation states of ionizable residues
The protonation states of ionizable residues (in total, 325 residues: 70 Arg, 73 Lys, 28 His, 65 Glu, and 89 Asp) were obtained by the Monte Carlo sampling method using the program Karlsberg (Hansmann 1997; Rabenstein et al. 1998), which operates in combination with the PBE solver MEAD (Bashford and Karplus 1990). The PBE was solved by a three-step grid-focusing procedure using 2.0, 1.0 and 0.25 Ǻ spacing and homogeneous media with an averaged uniform dielectric constant of PS I ε p = 4.5 in iron–sulfur clusters region inside and ε w = 80 outside, respectively. The protonation states of ionizable residues found at pH 7 and temperature 25°C were used then in the multi-dielectric model has described above.
Results and discussion
In this section, the basic experimental data on redox potentials in solution and distribution of the dielectric constant in PS I as revealed by electrometric measurements are analyzed and then the results of electrostatic calculations of redox potentials in proteins are discussed.
Reference redox potentials
Calculations of redox potentials in PS I were performed using the experimental midpoint potentials in aprotic solvent dimethylformamide (DMF) and applying the methods of continuum electrostatics to calculate the energy changes of ion transfer from this solvent into protein. The key problem is the conversion of the non-aqueous redox potentials in DMF to the aqueous standard hydrogen electrode (SHE). This conversion becomes possible only under an extrathermodynamic assumption. This step can be illustrated by the example of the redox potential of the Fe4S4(SCH3)4 −2/−3 couple. DePamphilis et al. (1974) measured the electromotive force (e.m.f.) of the following cells:
Here, the An/An−1 redox couple represents iron–sulfur or ferrocene (Fc) couples, TPAP is tetra-n-propylammonium perchlorate, the vertical bars separate different phases, and the components of the same phase are separated by commas. The left half-cell is the redox electrode in DMF, the right half-cell is the aqueous saturated calomel electrode (SCE). Experimentally, the following e.m.f. were obtained: +0.481 V for Fc+/0 couple and −1.294 V for Fe4S4(SCH3)4 2/3 couple.
The measured e.m.f. of the cell (1) is not equal to the difference between the two electrode potentials, because the cell involves one additional phase boundary, the boundary between electrolyte solutions in DMF and H2O. At this boundary, a liquid junction potential (l.j.p.) inevitably arises that also contributes to the measured potential difference. The l.j.p. is determined by nonequivalent distribution of cations and anions between two phases due to the differences in their solvation energies.
If the difference of two e.m.f. measured for Fc and FeS couples is taken, the l.j.p cancels out, and the value of e.m.f. for the cell without l.j.p. is found to be:
Here, dashed vertical bar separates two solutions in the same solvent with the same supporting electrolyte (0.05 M TPAP). At this boundary, no potential difference arises. Therefore, the cell (2) gives the redox potential of the Fe4S4(SCH3)4 −2/−3 couple versus the internal standard − Fc+/0 electrode in the same solvent; this potential is equal to −1.775 V.
In order to relate this potential to the aqueous SHE it is necessary to use an extrathermodynamic assumption. A widely accepted assumption is that the potential of the Fc/Fc+ couple is the same in all solvents (for review see Marcus 1985). Besides, the change in the dielectric response energy of the Fc+ cation upon its transfer from water to DMF has to be taken into account using the Born equation; this correction increases Fc/Fc+ potential in DMF by 0.03 V. While Born equation describes well Fc+ solvation energy in aprotic solvents, it cannot be applied quantitatively to aqueous solutions: due to its 3D hydrogen bond network, water displays an anomalously strong dielectric response to an ionic charge. The effect of this anomaly on Fc+/0 potential in water was determined as equal to ∼0.07 V (Khanova et al. 2006). Another effect is the pre-existing intraphase potential. According to the molecular dynamic simulations, there is a deviation from the strictly random orientation of water dipoles around a neutral solute molecule (e.g., Ashbaugh 2000; Grossfield 2005). Due to a more extended electronic shell of oxygen atoms as compared to those of hydrogen their repulsion from the solute is stronger. Therefore, on the average, H atoms are somewhat closer to the dissolved particle than O. This leads to the appearance of a positive potential on the neutral molecule. This effect shifts the redox potential of Fc/Fc+ couple to positive values. According to recent estimates, in water this shift is greater by ∼0.07 V than in DMF (Krishtalik 2008). The total increase of the Fc/Fc+ potential in DMF as compared to water is 0.03 + 0.07 – 0.07 = +0.03 V. In the cell III, the vertical double bar shows that the l.j.p. is eliminated using the extrathermodynamic assumption; the arbitrary supporting electrolyte CA in both half-cells is the same and of the same concentration.
The Fc/Fc+ potential in water relative to the standard hydrogen electrode is +0.40 V. Hence, to reduce the potential of the couple of interest to the standard hydrogen electrode, it should be corrected by +0.4 + 0.03 = +0.43 V. The final figure for Fe4S4(SCH3)−2/−3 is −1.345 V. Though the potentials here and later are given up to the third decimal, it should be mentioned that the accuracy of these figures hardly exceeds 20 mV.
The potentials of quinone couples were determined in a cell without l.j.p. where not aqueous SCE but SCE in the same solvent, viz. DMF, was used (Prince 1983)
Here, C is glassy carbon electrode, TBTF is tetrabutylammonium tetrafluoroborate. Due to a great excess of KCl, the diffusion potential at the boundary of two solutions can be neglected. The measured potential for Q/Q•− (menaquinone-10) is −0.709 V. However, this potential cannot be related directly to aqueous SHE because electrode potentials of SCE in water and DMF are undoubtedly different due to the difference in the chloride solvation energies. In this case, we also can go over to the internal standard because the Fc+/0 potential against SCE (DMF) has been determined in the same work as equal to +0.524 V. Hence, the Q/Q•− potential versus Fc+/0 in DMF (as in cell III) is −1.233 V. Similarly to considerations given above, this potential can be reduced to aqueous SHE as −1.233 + 0.43 = −0.803V. Ishikita and Knapp (2003) and Ishikita et al. (2006) have not accounted for the difference between the SCE potentials in water and DMF and, therefore, obtained a too high value of −0.463 V.
The experimental redox potential of the Chl a/Chl a − couple in DMF against aqueous SHE was found equal to −0.86 ± 0.02 V (Kiselev et al. 1970; Wasielewski et al. 1980), the corresponding value for Chl a/Chl a + couple is +0.835 ± 0.005 V (Saji and Bard 1977; Wasielewski et al. 1980). These data should be corrected for the l.j.p. that was determined by Krishtalik et al. (1991) under experimental conditions similar to those used in the works cited above; taking into account the corrections considered before it is equal to 0.30 V. The final values accepted in further calculations are −1.16 V for Chl a/Chl a − and +0.535 V for Chl a/Chl a + versus SHE.
In the literature, attempts were made to calculate the redox potentials by yet another method using the following procedure. The ionization potential of the reduced form is calculated quantum—chemically, and the difference of solvation energies of reduced and oxidized forms is added to this quantity (the latter energies are calculated electrostatically). This sum is compared with the so-called “absolute electrode potential” of the aqueous SHE (4.44 V). Such calculations were performed by Mouesca et al. (1994) and Torres et al. (2003) for iron–sulfur clusters, and by Hasegawa and Noguchi (2005) for chlorophyll. The principal drawback of this approach is that the “absolute electrode potential” involves the real solvation energy of hydrogen ion that includes a component due to the water surface potential (Pleskov 1987) rather than the chemical solvation energies (that were calculated for the redox couples investigated). The latter value is estimated as +0.14 V (Krishtalik 2008), so that the correction is substantial. Another difficulty in these calculations is that the final result is sensitive to the error in ab initio calculation of a large component, namely ionization potential.
Distribution of dielectric permittivity (ε) in PS I
PS I is a large protein complex that consists of 12 protein subunits and more than hundred cofactors, comprising in total more than 24000 atoms. The structure has a membrane-embedded hydrophobic core composed of more than 30 trans-membrane α-helices and two opposite water-protruding hydrophilic portions. Electrometric data revealed a notable variation of dielectric properties of PS I in the direction normal to the membrane plane: the polarity was found to be lower in the middle of the membrane than at its edges (Semenov et al. 2003). The variation of dielectric permittivity was estimated from the electrometric data by using a simple model of planar capacitor that includes several layers of dielectric media with different dielectric constants. If the electron transfer occurs in two steps denoted as A → B → C, the relationship between the respective dielectric constants εAB and εBC could be acquired from the respective electrogenic amplitudes ΔψAB and ΔψBC using the equation: \( \varepsilon _{{{\text{AB}}}} /\varepsilon _{{{\text{BC}}}} = (d_{{{\text{AB}}}} /d_{{{\text{BC}}}} ) \cdot (\Updelta \psi _{{{\text{BC}}}} /\Updelta \psi _{{{\text{AB}}}} ), \)where d AB and d BC are the projections of the A → B and B → C distance vectors onto the membrane normal, respectively. The available experimental electrometric data on ET reactions in PS I, the respective distances and calculated permittivites were summarized in recent review (Semenov et al. 2006). For the hydrophobic core of the complex, the low dielectric constant equal to 3.5 is accepted. It should be emphasized, however, that the reliability of the obtained estimates of the dielectric constant is restricted by (i) the accuracy of the measured electrogenic amplitudes and (ii) the general applicability of the planar capacitor model to the real non-planar protein complex protruding far out of the membrane. The restriction (i) is significant for fast ET reactions between donor P700 and quinone acceptor A1 whose kinetics could not be resolved by electrometric measurements because the risetime of voltage generation in this system was much slower than its relaxation decay owing to ion diffusion currents (Trissl et al. 1987; Hecks et al. 1994; Leibl et al. 1995). The restriction (ii) relates to the terminal ET reactions with soluble proteins Pc and especially Fd (the electrogenic contribution of the latter reaction was not resolved, probably, because FB is located at the protein surface of the water-protruding portion of PS I). Consequently, we did not consider the electrometric data for Fd and estimated the magnitude of permittivity basing on the polarity of PS I molecular structure.
The pattern of dielectric permittivity obtained by the electrometric data correlates strongly with the polarity of PS I molecular structure. According to the theory of polar media, the value of microscopic dielectric permittivity is determined by the concentration of polar groups (dipoles) and their rotational mobility (Fröhlich 1958). Figure 3a shows the distribution of polar atoms in PS I (excluding protein backbone) along the axis normal to the membrane plane. It is obvious that the number of polar atoms is smaller in the membrane core part of the complex and higher in the water-protruding parts. This figure shows, however, the total number of atoms rather than their density, which affects directly the value of dielectric permittivity. For this reason, we calculated the volume density of polar atoms along the axis normal to the membrane plane by averaging the density within 4 Å thin layers (Fig. 3b, blue line). The microscopic polarity has a wide minimum in the middle of the membrane around the accessory chlorophyll A0 and gradually increases toward both edges of the complex. In the acceptor part of PS I, the pattern of polarity reveals a maximum between FX and FA clusters and a local minimum between FA and FB clusters (the density profile outside FB is less reliable due to a restricted amount of atoms involved). The profile of protein polarity is in qualitative agreement with the dielectric profile (red line in Fig. 3b) obtained by electrometric measurements (Semenov et al 2006). The correlation between both types of the data could be used for estimates of the dielectric constant distribution in other membrane proteins of known structures in the absence of direct electrometric data.

Distribution of polarity and dielectric permittivity in PS I. a—distribution of oxygen and nitrogen atoms of polar amino acid side chains (red) and crystallographic water (blue) in relation to the redox cofactors in PS I complex; b—distribution of the average protein polarity (blue) and the effective dielectric permittivity (red) across the membrane. Open circles along the “distance” axis mark the places of cofactors localization. Polarity is the number of polar atoms within a 4 Å thin layer parallel to the membrane plane divided by the whole number of atoms within this layer. Dielectric permittivity is determined from the electrometric experiments (Semenov et al. 2003) as explained in “Distribution of dielectric permittivity (ε) in PS I” of the Results and discussion section
Thus, we considered heterogeneous distribution of static dielectric permittivity (ε) within the PS I protein complex using the electrometric data summarized in Table 1. Namely, we divided PS I in five planar layers of uniform dielectric mediums parallel to the membrane plane. Each amino acid residue in the complex was attributed to the given layer by the position of the N atom of backbone. Heteroatoms of non-protein cofactors were ascribed to the respective dielectric layers by their positions. The respective dielectric permittivity profile is shown in Fig. 3b (red line).
Electrostatic effects of protein environment on the redox transition of Fe4S4 complex in PS I and soluble ferredoxins
PS I contains three Fe4S4 complexes named FX, FA and FB, respectively. Each complex is fastened in the protein matrix by four cystein residues, more than ten hydrogen bonds with protein backbone (amid groups) and several other hydrogen donor groups. FX is located in the middle of the acceptor part of PS I (approximately at the rotation axis of A/B heterodimer) between QA and QB phylloquinones. FA and FB are arranged asymmetrically in the C subunit very close to the surrounding aqueous solution at the stromal side of thylakoid membrane (see Fig. 1). Structurally the subunit C of PS I is similar to soluble ferredoxin from Clostridium acidiurici (Kamlowski et al. 1997). We also modeled a small well-characterized soluble ferredoxin from Azotobacter vinelandii (Chen et al. 2000).
The main redox reaction of Fe4S4 in PS I and soluble ferredoxins is Fe4S4 2+/1+ transition. Since the complex is coordinated by four cystein residues carrying four negative charges, the reduction of the complex Fe4S4Cys4 2−/3− creates in total three negative charges in the protein matrix. The redox potential of Fe4S4(SCH3)4 2−/3− transition in DMF versus the standard hydrogen electrode is −1.345 V (see subsection “Reference redox potentials”). This value can be used as reference point for Fe4S4Cys4 2−/3− transition in protein. Due to the low dielectric permittivity of protein as compared with DMF (4–8 for protein and 37 for DMF), creation of three negative charges in protein is energetically unfavorable due to the Born-charging penalty, which is as great as 0.7–1.3 eV. In all cases considered, the protein environment compensates this energy penalty with a great pre-existing positive electrostatic field. This compensation, which is especially great in the case of FX, includes the following effects: (a) a large number of hydrogen bonds (from 8 to 11) with amid groups of protein backbone whose positive ends are oriented toward sulfur atoms of the clusters, (b) the presence of several Arg residues near FX, and (c) large number of water molecules in several intraprotein cavities near FX. The total of these factors compensate the great Born-charging penalty of the Fe4S4Cys4 2−/3− transition.
We begin consideration of PS I by using a simple phenomenological electrostatic approach that treats protein as a homogeneous dielectric medium surrounded by aqueous solution with static dielectric permittivities, ε p and ε w , respectively. The influence of protein environment on the redox transitions of cofactors includes the effects of (i) permanent charges, (ii) charged amino acid residues and (iii) changes of dielectric response (Bornian charging) energy on redox potentials of cofactors. Table 2 summarizes the midpoint redox potential values of Fe4S4 complexes in PS I and two ferredoxins from C. acidiurici and A. vinelandii, and the main electrostatic contributions calculated using the same set of model parameters at the protein dielectric permittivity ε p = 4.
It can be concluded from Table 2 that the use of a single macroscopic dielectric permittivity ε p is fundamentally inconsistent with the experimental midpoint potentials of Fe4S4 complexes measured in vivo. In all five cases of Fe4S4 clusters, the effect of electric field and the change in solvation energy almost compensate each other, so that the sum of the two main components is about zero, and the calculated potentials fall in the range from −1.20 to −1.55 V that is shifted by 0.7–1.0 V toward negative values with respect to the experimental data. Since both the components are proportional to the value of dielectric constant, their sum remains small at any value of the parameter ε p , and it is evident that straightforward adjustment of ε p cannot eliminate the systematic deviation of the theory from the experimental E m values. Moreover, consideration of the effects of dielectric heterogeneity in PS I would not improve the agreement.
As described in introduction, the treatment of electric field in protein with resolved three-dimensional atomic structure within the framework of phenomenological macroscopic approach using single value of dielectric permittivity is theoretically inconsistent. We attempt in this work to allow for the usage of both optical and static values of dielectric constant and for the microscopic heterogeneity of dielectric properties of PS I as represented in Table 1. Figure 3b shows the dielectric permittivity profile along the normal to the membrane plane in PS I. In particular, the permittivity demonstrates non-monotonous behavior near FX, FA and FB clusters: it is essentially higher in the vicinity of FX and lower near FA and FB. Structurally, such behavior could be due to a large amount of water molecules bound in the vicinity of FX cluster. Functionally, the varying dielectric permittivity might tune the redox potential of iron–sulfur clusters in a way that makes the electron go energetically downhill when transferred sequentially via a chain of cofactors from A1 to FA. The calculations demonstrate that although the midpoint potential of P700, A0 and A1 depend only slightly on the value of protein dielectric permittivity, the redox potential values of highly charged Fe4S4 clusters change substantially (up to 0.1–0.15 V) even at 10% variation of ε p .
Calculation of E m values for redox cofactors in PS I and ferredoxins
Table 3 shows the midpoint redox potential values of cofactors in PS I and two soluble ferredoxins calculated with the multilayer dielectric model using optical dielectric constant ε o = 2.5 for pre-existing fields and the variable static dielectric constant ε p as given in Table 1 for newly created fields. Calculations were performed both for equilibrium and functional midpoint potential values (they differ by the redox states of other cofactors). The Table 3 shows also the experimental values both in DMF solution and in protein (if known), and the electrostatic contributions of solvation energy change, permanent charges of backbone and neutral side chains, chargeable protein residues, and chargeable cofactors. The results are represented for both branches of cofactors in PS I because the problem of their operation mode remains unsolved. It also remains unknown how strong is the electronic coupling between chlorophyll monomers in the primary dimer P700 and primary acceptor A0, so that Table 3 shows both monomeric and dimeric modes of their operations.
Solvation energy change
The sign of the change in the solvation energy of the cofactor depends on the sign of its electric charge. Therefore, this shift is positive for P700/P700 + redox couple and negative for all other cofactors. The absolute magnitude of this effect is much greater for Fe4S4 clusters because of their high electric charge (−2/−3 transition). Among other singly charged cofactors the greater shift was found for A1, which is placed in the most hydrophobic part of the complex and has the smallest van der Waals volume.
Permanent charges
The most significant contribution to the field created by permanent charges originates from the protein backbone dipoles. It was positive in all cases, in accordance with data by Gunner et al. (2000), but its magnitude rises regularly from the donor side of PS I (<0.1 V near P700) to the acceptor side (up to +1.6 V for Fe4S4 clusters). For chlorophyll molecules a notable contribution comes from the axial ligands: His in the neutral state (about −230 mV) and Met or water molecule (from −90 to −110 mV). For quinone molecules QA and QB and the FX cluster remarkable contributions arise from bound intraprotein water molecules.
Charged amino acids
Charged amino acids are located mainly at the protein boundary and in the internal protein cavities between FX and FA filled with crystallographic water (Fig. 3a). As a result, charged amino acid residues contribute most significantly to the redox potentials of the cofactors FX, QA and QB (up to +0.5 V), whereas for other cofactors the contribution is small.
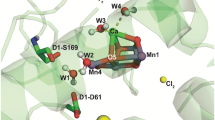
The iron–sulfur cluster F X is surrounded by a large amount of chargeable amino acid residues (Fig. 4): in total, more than 40 residues have the interaction energy greater than kT and the largest contributions (with absolute values greater than 0.1 eV) are provided by the residues Arg-B674, Arg-B712, Lys-A555, Arg-A728, Glu-B679 and Asp-A593. It follows from Fig. 4 that charged residues have a stratiform arrangement around FX: the layers formed by several positively charged residues alternate the layers with negatively charged residues. The integral effects of positive and negative residues are about +2.0 V and −1.5 V, respectively; thus, the total effect, being a difference of large quantities, has to be rather sensitive to the choice of model parameters (van der Waals volumes of atoms, the probe radius of protein environment, microscopic heterogeneity of protein) which could considerably affect calculations of the ionization states for chargeable residues and the magnitude of their electrostatic interactions.

The integral electrostatic potential ϕ at the FX cluster induced by charged amino acid residues located at a given distance R from the cluster center. The residues are ranged by their distance to the cluster (the most important residues are labeled); the difference between two neighboring points along the ordinate axis shows the electrostatic potential induced by the more distant residue
The iron–sulfur cluster F A is surrounded by ∼30 chargeable amino acid residues with the interaction energy greater than kT. The main contributions (greater than 0.1 eV) are provided by the residues Glu-C54, Arg-A583, Lys-C51, Asp-A579, Glu-D62, Arg-C52, but the positive and negative residues essentially compensate each other (Table 3). The number of charged residues around F B is ∼10, but their contributions are smaller than kT.
The quinones Q A and Q B are located in the vicinity of the same strongly charged protein region as FX: the number of strongly interacting residues is about 30 and 35 for QA and QB, respectively. The most important residues for QA are Arg-A694, Arg-A728, Lys-B542, Arg-B712, Asp-B580, Glu-A699, and for QB are Arg-B674, Lys-A555, Arg-B712, Arg-A728, Glu-B679, Asp-A593, respectively. The resulting effects for both quinones are positive and have almost similar magnitude as that for FX.
Chlorophyll molecules are located in a hydrophobic portion of PS I far from charged residues, so that their individual and total effects are very small except for Chl3A and Chl3B. In the latter case relatively strong contributions (approx. +120 mV) are provided by Lys-B542 and Lys-A555, respectively.
Charged redox cofactors
This contribution arises from other charged cofactors. For example, after the primary charge separation electron resides at the primary acceptor A0, it becomes exposed to the electric fields of newly created cation P700 + and of anions of FX, FA and FB. When the electron is transferred via the chain of carriers, the positive effect of P700 + decreases, while the negative effect of Fe4S4 clusters increases.
Compensation effects
Transfer of a charged particle from a polar solvent into a low-dielectric protein medium causes some loss of the dielectric response energy (Bornian solvation energy). As can be seen from Table 3, the corresponding change of redox potential is compensated to a substantial extent by the effect of the intraprotein electric field. It is notable that when the dielectric response energy shifts the redox potential toward positive values, the intraprotein electric field is negative (P700), while for other cofactors the situation is reverse.
The compensation of the loss of the dielectric response energy by the proper intraprotein electric field (in particular, the field of permanent charges) has been proposed as a general feature of proteins that makes possible the charge transfer processes inside low-dielectric proteinaceous media (Krishtalik and Topolev 1984). The results of the present work provide another corroboration of this general idea.
Comparison of our results with previous studies
Iron–sulfur clusters
The redox potentials of iron–sulfur clusters in several proteins including PS I were calculated by Torres et al. (2003). As mentioned in the “Reference redox potentials” subsection, the midpoint potentials of Fe4S4(SCH3)4 −3/−2 transition in situ were obtained in this work by conversion of the absolute ionization potential found by ab initio DFT calculations in vacuum to the reference values versus the standard hydrogen electrode using corrections obtained by solution of Poisson–Boltzmann equation. However, this approach is not sufficiently precise. The simple case of liquid DMF Torres et al., analyzed with a homogeneous dielectric model using a single dielectric constant ε = 37 and calculated the potential of Fe4S4(SCH3)4 −3/−2 transition from −1.01 to −1.26 V, that is 0.1–0.3 V more positive than the experimental value of −1.345 V (see subsection “Reference redox potentials”). The more challenging case of protein environment Torres et al. treated by using a model with three dielectric layers: the protein itself with the permittivity ε p = 4, the surrounding solution with the permittivity ε w = 80, and a layer of amide groups hydrogen bonded to iron–sulfur cluster with the permittivity εamide = 1. The latter choice was substantiated as follows: “The physical idea underlying this model is that hydrogen bonds to the cluster tend to inhibit protein mobility at this interface, so there is little protein dielectric screening and εs = 1 is an appropriate dielectric constant over the region of the hydrogen bonded amides.” However, this suggestion is only an implausible ad hoc hypothesis. First, the absence of the dipoles mobility means only the absence of orientational polarization rather than both orientational and electronic polarizations; the latter is characterized by the optical constant 2.5. Second, there is no reason why the mobility of amide groups hydrogen bonded to sulfur atoms should be reduced compared to the mobility of such groups in α-helices or β-sheets forming the core of protein structure. Moreover, because the average length of hydrogen bonds with sulfur atoms is 3.5 Å, which is ∼0.8 Å longer than normal hydrogen bonds, it seems reasonable that their mobility is even higher than the respective mobility in protein. Thus, because the ab initio DFT calculations of iron–sulfur cluster were performed in vacuum (ε = 1) without consideration of neighboring amide groups, Torres et al. overestimated the field of amides adjacent to the cluster. For the FX cluster in PS I this overestimation is as great as 0.5 V. Together with the liquid junction potential (l.j.p.) correction, which was not taken into account by Torres et al., this gives an error of ∼0.7 V. Such great disagreement with experimental data seems to be an inevitable feature of any model based on the use of single dielectric constant (cf. Tables 2 and 3). The large overestimation of the effect of adjacent groups is partially compensated in the cited work by underestimation of the field of other permanent charges, which was calculated with εs = 4instead of εo = 2.5. The final estimate for the potential of FX in the three-layer model by Torres et al. is −0.98 V that is 0.3–0.35 V below the experimental value.
Quinone acceptors
The calculation of the redox potentials of quinones in PS I was performed by Ishikita and Knapp (2003). As discussed in the subsection on the reference redox potentials, they had not taken into account the correction for l.j.p., and, therefore, overestimated the reference potential by 240 mV. This led them to a conclusion that the electron transfer from QA to FX should be substantially uphill. If recalculated with the correct reference potential, the calculations by Ishikita and Knapp would give redox potentials of −771 mV for QA and −926 mV for QB showing that both electron transfer reactions are definitely downhill. These corrected values are 100/82 mV more negative than our estimates. Perhaps, this discrepancy might be caused by less positive potential induced by permanent charges due to higher (static instead of optical) dielectric constant used in our calculations. In both works, virtually the same difference in redox potentials of two quinones was obtained: the relative values are less sensitive to variation of parameters because the corresponding errors in both potentials largely compensate each other. In spite of a substantial error in absolute values of these redox potentials, Ishikita and Knapp have properly calculated a large difference between two quinones.
Chlorophyll moieties
Ishikita et al. (2006a) calculated the redox potential of P700 starting with the experimental value for Chl a determined in CH2Cl2, namely E m = +800 mV against aqueous SHE. The correction for l.j.p. was not introduced. The data for CH2Cl2 are not as reliable as the data for DMF to introduce this correction. The change of Bornian solvation energy for Chl a transfer from CH2Cl2 to DMF can be calculated by solution of Poisson–Boltzmann equation as 90 meV. This value gives the uncorrected reference potential for Chl a oxidation in DMF equal to +710 mV. As described above, the potential corrected for l.j.p. is +535 mV. Thus, if the corrected potential was used in the calculations of Ishikita et al., the values of Chl1A and Chl1B would be 175 mV lower, i.e., 412 mV and 424 mV, respectively. This is by 132 and 156 mV lower than our data for the monomer forms of Chl1A and Chl1B. However, the resulting potential of +420 mV has to be decreased by the energy of quantum conjugation of monomers, which is as great as 95 mV according to our estimation. This points to a noticeable discrepancy with the experimental value of approx. +450 mV.
Possible sources of uncertainty of the model
Analysis of various approaches to E m calculation
As noted in the introduction, our electrostatic calculations include two novel elements: use of the optical dielectric constant for calculation of the field of permanent charges and consideration of different local static dielectric constants for calculation of the dielectric response energy and the field of chargeable groups. If a single dielectric constant is used (e.g., typical value ε s = 4), considerable errors arise in calculation of the main energy components (cf. Tables 2 and 3 and the discussion above). For the cases of P700, A0 and A1, where the intraprotein electric field induced by permanent charges is rather small, this error does not exceed 50 mV, so that the traditional approach with a single dielectric constant in this particular case gives the potentials of a reasonable order of magnitude. The iron–sulfur clusters represent, however, a striking exception: the effect of permanent charges in this case is large and the conventional approach underestimates substantially their contribution, so that the resulting potentials are 0.7–1.0 V more negative than experimental values.
The most straightforward way to rationalize such great discrepancy is the usage of optical dielectric constant for the effects of permanent charges. It should be noted that the value of ε o close to 2.5 gives in all cases a good compensation of the Bornian penalty. If ε o ∼ 1 is used (as supposed by Simonson et al., 1999), the field of permanent charges increases more than twofold, which leads to a large discrepancy with the experimental data.
Another rationale might be polarization of 8–12 amide groups hydrogen bonded to the Fe4S4 clusters. Such polarization might arise due to the direct chemical influence of negatively charged sulfur atoms at extremely low reduction potential. If such “chemical” interaction has magnitude of ∼0.1 eV per hydrogen bond, this effect would provide a considerable contribution to the salvation energy and might essentially reduce the discrepancy mentioned above. Extensive ab initio calculations of the iron–sulfur clusters together with neighboring amide groups are required to estimate the non-electrostatic contributions to the solvation energy.
Uncertainties of the suggested dielectric model
The final effect of the protein environment on the shift of redox potential values in PS I results from large and oppositely directed contributions (Table 3 and Fig. 4). For this reason, the results of calculations are sensitive to uncertainties in the model parameters, such as atomic coordinates, partial charges, effective dielectric constants, or unknown state of charged amino acid residues and cofactors in the X-ray crystal structure. Another error arises because the treatment of chargeable groups in our approach is somewhat challenging: their ionization states in crystal are unknown, so that the use of the static dielectric constant for chargeable residues could not be unequivocally proven. Variation of the mentioned parameters in the range of 10% would result in alteration of the midpoint potentials for chlorophylls and quinones of less than 50 mV and for iron–sulfur clusters of about 150 mV. These variations are, however, not independent of each other: alteration of global parameters such as protein partial charges or optical dielectric constant would lead to proportional changes of E m values, so that the differences between E m values of cofactors would be substantially smaller. In order to specify the possible errors of the approach suggested in this work more quantitatively, these approximations require further elaboration.
Correlation between the calculated and experimental E m values and consequences for electron transfer mechanisms
Analysis of the calculated midpoint redox potentials (E m ) of the cofactors in PS I presented in Table 3 shows several remarkable peculiarities.
P 700 . The calculated oxidation potential value of the special pair chlorophyll (Chl) P700 (P700/P700 +) is close to the experimentally obtained E m value of ∼450 mV. This value takes into account the stabilization energy of the charged state due to the chlorophyll dimerization. It should be noted that consideration of P700 as a monomer gives the E m values that are ∼120–150 mV higher than the experimental data. This result indicates that the charge density distribution over the dimer is not completely asymmetrical. EPR and ENDOR studies showed significantly asymmetrical distribution of spin density between the two halves of the P700 dimer (Kass et al. 2001). On the other hand, FTIR data showed that the charge distribution between PA + and PB + ranges from 1:1 to 2:1 in favor of PB (Breton et al. 2002). It was recently shown using semiempirical quantum chemistry calculations and the available coordinates that, due to spin polarization, the spin asymmetry was significantly larger than any charge asymmetry in the P700 + dimer (Plato et al. 2003). Though the real charge distribution value is not well defined, it is clear that the asymmetry degree is not very high. Our calculations based on the model of Artz et al. (1997) showed that the charge distribution was 1.6:1 (in favor of Chl1A). Thus, our calculations, which demonstrate strong quantum conjugation and electrostatic interaction of monomers, are generally in line with the experimental data.
A 0 . The nature of the primary electron acceptor A0 in PS I is not completely understood. In the X-ray structure of PS I there are two pairs of Chl molecules associated with the PsaA and PsaB subunits of heterodimer. It was previously believed that either one or both distal to P700 Chl molecules (Chl3A and Chl3B) play the role of A0. However, mutual arrangement (almost parallel planes of porphyrin rings) and relatively close distance between central Mg2+ atoms of Chl2A and Chl3A, and Chl2B and Chl3B (8.7 and 8.2 Å, respectively) imply the possibility of significant interaction between the pairs of Chl molecules. This fact allows considering A0 as a Chl2A/Chl3A (Chl2B/Chl3B) dimer. The distances between the monomers in this dimer are longer than in P700, and the asymmetry in the energy of monomers interaction with surrounding protein is much higher than in P700. Therefore, the dimer can be considered as looser. The asymmetry of charge distribution among the monomers was calculated to be as large as 7:1 for both pairs (in favor of Chl3A and Chl3B). In total, this effect produces a ∼45 mV positive shift of the reduction potential of A0/A0 − pair in both A and B chains.
The energy of 700 nm quanta is 1.770 mV. Thus, the redox potential of P700* in excited state is −1.320 mV. Therefore, if A0 is represented by Chl3A or Chl3B, the ΔG value between P700* and A0/A0 − lies in the range of −5 to −50 mV. On the other hand, if A0 is considered as a dimer, the energy gap increases up to −50 to −90 mV. This value is essentially lower, but closer to the 250 mV estimation of the value of ΔG between P700* and A0/A0 − based on delayed fluorescence measurements (Kleinherenbrink et al. 1994). Probably, the latter value is larger because of more developed dielectric response due to longer time of these measurements. Our calculations show that consideration of A0 as a dimer results in more realistic value of its reduction potential. It should be noted that consideration of monomeric Chl2A/2B as a primary acceptor is hardly possible because its reduction potential is ∼100 mV more negative, than that of the P700* excited state.
A 1 . The A1 acceptor is represented by one or two phylloquinone molecules (QA and QB) belonging to the A and B branches of cofactors. The E m value of A1 was estimated as −800 mV (Vos and Van Gorkom 1990), or at least less than −700 mV (see Brettel and Leibl 2001). The data obtained by Joliot and Joliot (1999) and Guergova-Kuras et al. (2001) indicated that both quinone molecules participate in the electron transfer to the next electron acceptor—the iron–sulfur cluster FX.
As it is discussed above, Ishikita and Knapp (2003) suggested that QA → FX electron transfer is uphill in energy by ∼120–170 mV, while QB → FX transfer is almost equipotential, and ascribed the former reaction to the slow phase and the latter to the fast phase of A1 → FX electron transfer observed in (Guergova-Kuras et al. 2001; Joliot and Joliot 1999). On the other hand, our calculations show that both QA → FX and QB → FX electron transfer are exergonic, although the energy gap for the former reaction is not very high (∼85 mV). The energy gap of ∼170 mV between QB and QA is greater than that between QA and FX. This raises the question why the electron transfer proceeds from QB - to FX only and not to QA also. The plausible main reason for this is that the edge-to-edge distance between QB and QA (15.2 Å) is considerably greater than the distance between QB and FX (6.7 Å). Calculations based on the Moser–Dutton rule (Moser et al. 1992) show that the rate constant of QB → FX electron transfer is about 5 orders of magnitude greater than that of QB → QA. This estimation clearly shows why the latter reaction seems highly unlikely. Besides, the calculated energy gap between QB and QA shows that QB → FX electron transfer should be ∼25 times faster than QA → FX (K = 10−1/2(ΔG/2.3kT) = 28), which corresponds to the experimentally obtained difference between the fast and the slow phase kinetics of these reactions (cf. τ values of 10 and 270 ns, obtained by Guergova-Kuras et al. 2001; Joliot and Joliot 1999).
Iron–sulfur clusters. The E m value of the closest to A1 cluster FX is −585 mV, which is ∼65–115 mV more positive than the experimental values obtained by Parrett et al. (1989), Chamorovsky and Cammack (1982), and calculated by Shinkarev et al. (2000). However, these values were obtained by equiluibrium redox titration and, therefore, should be compared with the calculated equilibrium redox-potential of FX, which is −654 mV. This calculated value falls within the range of the experimentally obtained values (−650 to −700 mV, see Refs above).
Redox titrations of low-temperature EPR signals ascribed to FA and FB clusters lead to potentials of −540 mV and −590 mV, respectively (see Brettel 1997 for review). However, redox titration of the recombination kinetics at room temperature yielded midpoint potentials of approx. −500 and −550 mV (Golbeck et al. 1987), and −440 and −465 mV for FA and FB clusters (Jordan et al. 1998). In both cases, the accuracy of the obtained E m values is rather poor. In fact, in these experiments the measured value was the extent of the slow (∼70 ms) kinetic phase, which is ascribed to the overall back-reaction from FA/FB clusters. Besides, the clear distinction of two waves in redox-titration curve separated by 25 mV (in the latter case) is hardly possible. Nevertheless, it is most likely that the E m values of FA/FB clusters at room temperature are more positive than at liquid helium temperatures. It should be also noted that the low-temperature EPR redox titrations were performed under dark conditions, when the primary donor P700 was reduced, while room temperature measurements of the flash-induced recombination kinetics were performed for oxidized P700. The effect of P700 + on the FA/FB cluster results in the positive shift of E m by ∼20 mV.
Our calculations give equilibrium potentials of −480 mV for FA and −540 mV (−585 mV for operative potential) for FB. These values are approximately in the range of experimental values. It worth noting that the difference of Gibbs energies between operative potentials of FX and FB is too small (36 mV) to account for the effective electron transfer to FB.
However, as mentioned above, the accuracy of our calculations in the case of Fe4S4 clusters is lower than for chlorophyll and quinone cofactors. This is because the contribution of permanent charges of the protein and Born charging (responsible for the bringing of the cluster from water into the protein) for the Fe4S4 clusters are rather high, but have opposite effects. Another factor that decreases precision in this case is a high sensitivity of the Born penalty to the experimentally obtained values of static dielectric constant ε S (note that the Born-constituent values in the case of Fe4S4 clusters is considerably greater than in other cases). Thus, the resulting E m value represents the algebraic sum of relatively high contributions with different signs. Nevertheless, the calculated values show that the E m of FB is more negative than the E m of FA, which is consistent with the experimental estimation and the lower ε s value in the vicinity of FB as compared to that of FA. As mentioned above, these ε s values are also consistent with the unusual character of distribution of charged amino acids and water molecules around FA and FB. The E m value of FB also decreases because of less positive potential created by permanent charges.
Ferredoxins. In addition to PS I cofactors, we also calculated the E m values of Fe4S4 clusters for two soluble ferredoxin molecules from Azotobacter vinelandii and Clostridium acidiurici. The structures of these ferredoxins are quite similar to that of the PS I PsaC subunit, which carries the terminal FA and FB clusters (Kamlowski et al. 1997). The static ε value of 5.5 was taken for these proteins. It is close to the average value for (FeS)4 cofactors. However, this is not quite convincing because, in contrast to PS I, the content of water molecules and polar groups in these proteins is not particularly high. On the other hand, this value seems to be reasonable by analogy with other small globular proteins. It is known from molecular dynamic simulations that there is distinct increase in the dielectric permittivity of these proteins near their surface. This increase is due to the increased mobility of outer amino acid residues (Simonson and Perahia 1995, and the literature cited therein). Semi-continuum calculations of pK of the α-chymotrypsin active site also showed that the effective ε s was close to 5 (Krishtalik 2005).
Consideration of Chl2A/2B monomers as primary donor species
Recently, Holzwarth et al. (2006) suggested that the Chls of the PA and PB pair (Chls1A/1B), constituting primary electron donor P700, are not oxidized in the first electron transfer step and serve as a secondary electron donor. They proposed that the accessory Chls (designated as Chl2A and Chl2B) function as primary electron donor(s) and Chls 3A and 3B (also called A0) are the primary electron acceptor(s). In order to check this hypothesis, we calculated the oxidation midpoint potentials of Chl2A and Chl2B species. The E m values appeared to be highly positive (+815 to −825 mV for Chl2A and Chl2B, respectively (see Table 4). Taking into account the 700 nm quantum energy of 1770 mV, the redox potential of the Chl2A(2B)* excited state(s) would be approx. −950 mV. This value is ∼60 mV less negative than the values of reduction potentials of the hypothetical primary acceptor(s) Chl3A and Chl3B (see Table 4). Thus, according to our calculations, the electron transfer between Chl2A(2B) and Chl3A(3B) is thermodynamically unfavorable, and, therefore, the functioning of accessory Chls 2A(2B) as primary donor seems unlikely.
Conclusions
Our electrostatic calculations of PS I and ferredoxins employ two main novel elements, namely, use of the optical dielectric permittivity to account for pre-existing field induced by permanent charges, and use of the variable static dielectric constant (derived from electrometric measurements) for solvation energy changes and for potential set by charged residues. This approach provides an opportunity to estimate the midpoint redox potentials of cofactors with a reasonable agreement with known experimental data.
Our calculations show (or confirm) the following peculiarities of the electron transfer in PS I:
-
(1)
Chl1A and Chl1B molecules constituting the primary donor P700 strongly interact and function as a dimeric Chl molecule;
-
(2)
Chls 2A/2B essentially interact with Chls 3A/3B and serve as a primary electron acceptor A0, although the Chl2A/Chl3A (or Chl2B/Chl3B) dimers are looser than P700;
-
(3)
The electron transfers from both QB and QA to FX are, most probably, downhill. The E m value of quinone acceptor QB is approx. −170 mV more negative than that of QA. This implies that the electron transfer from QB to FX should be ∼25 times faster than that from QA to FX, which is in agreement with the difference between two kinetic phases of Q- oxidation observed by Joliot and Joliot (1999);
-
(4)
The functioning of monomeric Chls 2A/2B as primary donor(s) and monomeric Chls 3A/3B as primary acceptor(s) seems unlikely, since in this case the electron transfer would be thermodynamically unfavorable.
Abbreviations
- Chl:
-
Chlorophyll
- Chl1A and Chl1B:
-
The same as PA and PB, the monomer chlorophyll molecules of P700
- Chl2A and Chl2B:
-
The “accessory chlorophyll” molecules, probable redox cofactors, localized between Chl1A and Chl3A and between Chl1B and Chl3B, respectively
- Chl3A and Chl3B:
-
The primary electron acceptor chlorophyll molecules, redox cofactors in chains A and B, respectively
- DMF:
-
Dimethylformamide
- E m :
-
Midpoint redox potential of a redox pair
- e.m.f.:
-
Electromotive force
- ENDOR:
-
Electron-nuclear double resonance spectroscopy
- EPR:
-
Electron paramagnetic resonance spectroscopy
- ET:
-
Electron transport
- FX, FA, FB :
-
Clusters of Fe4S4 type, redox cofactors at the acceptor side of PS I
- Fc:
-
Ferrocene
- Fd:
-
Ferredoxin, FeS containing protein, electron acceptor for PS I
- FTIR:
-
Fourier transform infrared spectroscopy
- l.j.p.:
-
Liquid junction potential
- P700 :
-
The same as Chl1A/1B dimer, the primary electron donor in PS I
- PA and PB :
-
The same as Chl1A and Chl1B
- PBE:
-
Poisson–Boltzmann equation
- Pc:
-
Plastocyanin, electron donor for PS I
- PS I:
-
Photosystem I
- QA and QB :
-
Phylloquinone molecules, redox cofactors in chains A and B, respectively
- RC:
-
(bacterial) Photosynthetic reaction center
- SCE:
-
The standard calomel electrode
- SHE:
-
The standard hydrogen electrode
- TBTF:
-
Tetrabutylammonium tetrafluoroborate
- TPAP:
-
Tetra-n-propylammonium perchlorate
References
Allen JP, Feher G, Yeates TO, Komiya H, Rees DC (1987) Structure of the reaction center from Rhodobacter sphaeroides R-26; the cofactors. Proc Natl Acad Sci USA 84:5730–5734
Artz K, Williams JC, Allen JP, Lendzian F, Rautter J, Lubitz W (1997) Relationship between the oxidation potential and electron spin density of the primary electron donor in reaction centers from Rhodobacter sphaeroides. Proc Natl Acad Sci USA 94:13582–13587
Ashbaugh HS (2000) Electrostatic contribution to the excess chemical potential of the clusters using the TIP3P water model. J Phys Chem B 104:7235–7238
Banci L, Bertini I, Savellini GG, Luchinat C (1996) Individual reduction potentials of the iron-ions in Fe2S2 and high potential Fe4S4 ferredoxins. Inorg Chem 35:4248–4253
Bashford D (2004) Macroscopic electrostatic models for protonation states in proteins. Front Biosci 9:1082–1099
Bashford D, Karplus M (1990) pK of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry 29:10219–10225
Beck BW, Xie Q, Ichiye T (2001) Sequence determination of reduction potentials by cysteinyl hydrogen bond and peptide dipoles in [4Fe–4S] ferredoxins. Biophys J 81:601–613
Brettel K (1997) Electron transfer and arrangement of the redox cofactors in photosystem 1. Biochim Biophys Acta 1318:322–373
Brettel K, Leibl W (2001) Electron transfer in Photosystem I. Biochim Biophys Acta 1507:100–114
Breton J, Xu W, Diner BA, Chitnis P (2002) The two histidine axial ligands of the primary electron donor chlorophylls (P700) in photosystem I are similarly perturbed upon P700 + formation. Biochemistry 41:11200–11210
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
Bunakova LV, Topolev VV, Khanova LA, Krishtalik LI (2004) Energy of transfer of ions into a low-permittivity solvent. Russ J Electrochem 40:794–801
Chamorovsky SK, Cammack R (1982) Direct determination of the midpoint potential of the acceptor X in chloroplast photosystem I by electrochemical reduction and ESR spectroscopy. Photobiochem Photobiophys 4:195–200
Chen K, Hirst J, Camba R, Bonagura CA, Stout CD, Burgess BK, Armstrong FA (2000) Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature 405:814–817
Chen KS, Bonagura CA, Tilley GJ, McEvoy JP, Jung YS, Armstrong FA, Stout CD, Burgess BK (2002) Crystal structures of ferredoxin variants exhibiting large changes in [Fe-S] reduction potential. Nat Struct Biol 9:188–192
Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117:5179–5197
Cundari TR, Stevens WJ (1993) Effective core potential methods for the lanthanides. J Chem Phys 98(7):5555–5565
Deisenhofer J, Epp O, Miki K, Huber R, Michel H (1985) Structure of the protein subunits in the photosynthetic reaction center of Rhodopseudomonas viridis at 3 Å resolution. Nature 318:618–624
DePamphilis BV, Averill BA, Herskovitz T, Que L, Holm Jr, Holm RH (1974) Synthetic analogs of the active sites of iron-sulfur proteins. VI. Spectral and redox characteristics of the tetranuclear clusters [Fe4S4(SR)4]2−. J Am Chem Soc 96:4159–4167
Fröhlich H (1958) Theory of dielectrics: dielectric constant and dielectric loss. Clarendon Press
Georgescu RE, Alexov EG, Gunner MR (2002) Combining conformational flexibility and continuum electrostatics for calculating pKa’s in proteins. Biophys J 83:1731–1748
Golbeck JH, Parrett KG, McDermott AE (1987) Photosystem I charge separation in the absence of centers A and B. III. Biochemical characterization of a reaction center particle containing P700 and FX. Biochim Biophys Acta 893:149–160
Grossfield A (2005) Dependence of ion hydration on the sign of the ion’s charge. J Chem Phys 122:02450601–024506010
Guergova-Kuras M, Boudreaux B, Joliot A, Joliot P, Redding K (2001) Evidence for two active branches for electron transfer in photosystem I. Proc Natl Acad Sci USA 98:4437–4442
Gunner M, Saleh M, Cross E, A. ud-Doula, Wise M (2000) Backbone dipoles generate positive potentials in all proteins: origins and implications of the effect. Biophys J 78:1126–1144
Hansmann UHE (1997) Parallel tempering algorithm for conformational studies of biological molecules. Chem Phys Lett 281:140–150
Hasegawa K, Noguchi T (2005) Density functional theory calculation on the dielectric-constant dependence of the oxidation potential of chlorophyll: Implication for the high potential of P680 in photosystem II. Biochemistry 44:8865–8872
Hecks B, Wulf K, Breton J, Leibl W, Trissl HW (1994) Primary charge separation in photosystem I: a two-step electrogenic charge separation connected with P700 +A0 − and P700 +A1 − formation. Biochemistry 33:8619–8624
Holzwarth AR, Muller MG, Niklas J, Lubitz W (2006) Ultrafast transient absorption studies on photosystem I reaction centers from Chlamydomonas reinhardtii. 2: Mutations near the P700 reaction center chlorophylls provide new insight into the nature of the primary electron donor. Biophys J 90:552–565
Honig BH, Nicholls A (1995) Classical electrostatics in biology and chemistry. Science 268:1144–1149
Ishikita H, Knapp EW (2003) Redox potential of quinones in both electron transfer branches of photosystem I. J Biol Chem 278:52002–52011
Ishikita H, Saenger W, Biesiadka J, Loll B, Knapp EW (2006a) How photosynthetic reaction centers control oxidation power in chlorophyll pairs P680, P700, and P870. Proc Natl Acad Sci USA 103:9855–9860
Ishikita H, Stehlik D, Golbeck JH, Knapp EW (2006b) Electrostatic influence of PsaC protein binding to the PsaA/PsaB heterodimer in Photosystem I. Biophys J 90:1081–1089
Iwaki M, Itoh S (1994) Reaction of reconstituted acceptor quinone and dynamic equilibration of electron transfer in the photosystem I reaction center. Plant Cell Physiol 35:983–993
Joliot P, Joliot A (1999) In vivo analysis of the electron transfer within photosystem I: are the two phylloquinones involved? Biochemistry 38:11130–11136
Jordan P, Nessau U, Schlodder E (1998) Charge recombination between the reduced iron-sulfur clusters and P700. In: Garab G (ed) Photosynthesis: mechanisms and effects, vol 1. Kluwer Academic Publishers, The Netherlands, pp 663–666
Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauss N (2001) Three-dimensional structure of cyanobacterial photosystem I at 2.5 angstroms resolution. Nature 411:909–917
Kamlowski A, van der Est A, Fromme P, Krauss N, Schubert WD, Klukas O, Stehlik D (1997) The structural organization of the PsaC protein in Photosystem I from single crystal EPR and X-ray crystallographic studies. Biochim Biophys Acta 1319:199–213
Kass H, Fromme P, Witt HT, Lubitz W (2001) Orientation and electronic structure of the primary donor radical cation P700 + in photosystem I: a single crystal EPR and ENDOR study. J Phys Chem B 105:1225–1239
Khanova LA, Topolev VV, Krishtalik LI (2006) Effect of the aqueous-organic solvent structure on the cobalticenium-cobaltocene redox-potential. The redox couple as a basis for determination of the single ion transfer energies. Chem Phys 326:33–42
Kharkats YuI, Krishtalik LI (1985) Medium reorganization energy and enzymatic-reaction activation-energy. J Theor Biol 112(2):221–249
King G, Lee FS, Warshel A (1991) Microscopic simulations of macroscopic dielectric constants of solvated proteins. J Chem Phys 95:4366–4377
Kiselev BA, Kozlov YuN, Evstigneev VB (1970) Chlorophyll polarography. Biofizika 15:594–601
Kleinherenbrink FAM, Hastings G, Wittmershaus BP, Blankenship RE (1994) Delayed fluorescence from Fe-S type photosynthetic reaction centers at low redox potential. Biochemistry 33:3096–3105
Krishtalik LI (2005) Continuum electrostatics of proteins: experimental test with model solvents and the method of the proteins pK calculations. Chem Phys 319:316–329
Krishtalik LI (2008) The solvent’s surface potential and the intraphase pre-existing potential. Russ J Electrochem 44:48–55
Krishtalik LI, Topolev VV (1984) Vnutriglobulyarnoe electrostaticheskoe pole fermenta (intraglobular electrostatic field of an enzyme). Molekulyarnaya Biologiya (M) 18:712–718
Krishtalik LI, Alpatova NM, Ovsyannikova EV (1991) Electrostatic ion-solvent interaction. Electrochim Acta 36:435–445
Krishtalik LI, Tae G-S, Cherepanov DA, Cramer WA (1993) The redox properties of cytochromes-b imposed by the membrane electrostatic environment. Biophys J 65(1):184–195
Krishtalik LI, Kuznetsov AM, Mertz EL (1997) Electrostatics of proteins: description in terms of two dielectric constants simultaneously. Proteins: Struct Funct Genet 28:174–182
Kuznetsov AM (1997) Charge transfer in chemical reactions kinetics. Lausanne, Presses polytechniques et universitaires romandes
Leibl W, Toupance B, Breton J (1995) Photoelectric characterization of forward electron transfer to iron-sulfur centers in photosystem I. Biochemistry 34:10237–10244
Mamedov MD, Gadjieva RM, Gourovskaya KN, Drachev LA, Semenov AY (1996) Electrogenicity at the donor/acceptor sides of cyanobacterial photosystem I. J Bioenerg Biomembr 28:517–522
Marcus Y (1985) Ion solvation. Wiley, Chichester
Mertz EL, Krishtalik LI (1999) Free energy of charge transfer and intraprotein electric field: method of calculation depends on the charge state of protein at a given structure. Bioelectrochem Bioenerg 48:397–405
Mertz EL, Krishtalik LI (2000) Low dielectric response in enzyme active site. Proc Natl Acad Sci USA 97:2081–2086
Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL (1992) Nature of biological electron transfer. Nature 355:796–802
Mouesca J-M, Chen JL, Noodleman L, Bashford D, Case DA (1994) Density Functional/Poisson-Boltzmann calculations of redox potentials for iron-sulfur clusters. J Am Chem Soc 116:11898–11914
Nakamura A, Suzawa T, Kato Y, Watanabe T (2005) Significant species-dependence of P700 redox potential as verified by spectroelectrochemistry: comparison of spinach and Thermosynechococcus elongatus. FEBS Lett 579:2273–2276
Nicholls A, Honig B (1991) A rapid finite-difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J Comupt Chem 12:435–445
Papazyan A, Warshel A (1997) Continuum and dipole-lattice models of solvation. J Phys Chem B 101:11254–11264
Parrett KG, Mehari T, Warren PG, Golbeck JH (1989) Purification and properties of the intact P700 and Fx-containing photosystem I core protein. Biochim Biophys Acta 973:324–332
Plato M, Krauss N, Fromme P, Lubitz W (2003) Molecular orbital studies of the primary electron donor P700 of photosystem I based on a recent X-ray single crystal analysis. Chem Phys 294:483–499
Pleskov YuV (1987) The absolute potential of a standard hydrogen electrode: a new estimate. Comments. J Phys Chem 91:1691–1692
Prince RC, Dutton PL, Bruce JM (1983) Electrochemistry of ubiquinones: menaquinones and plastoquinones in aprotic solvents. FEBS Lett 160:273–276
Rabenstein B, Ullmann GM, Knapp EW (1998) Calculation of protonation patterns in proteins with structural relaxation and molecular ensembles – application to the photosynthetic reaction center. Eur Biophys J 27:626–637
Saji T, Bard AJ (1977) Electrogenerated chemiluminescence. 29. The electrochemistry and chemiluminescence of chlorophyll a in N,N-dimethylformamide solutions. J Am Chem Soc 99:2235–2240
Schaefer M, Vlijmen HW, Karplus M (1998) Electrostatic contributions to molecular free energies in solution. Adv Protein Chem 51:1–57
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic-structure system. J Comput Chem 14:1347–1363
Semenov AY, Mamedov MD, Chamorovsky SK (2003) Photoelectric studies of the transmembrane charge transfer reactions in photosystem I pigment-protein complexes. FEBS Lett 553:223–228
Semenov AY, Mamedov MD, Chamorovsky SK (2006) Electrogenic reactions associated with electron transfer in photosystem I. In: Golbeck JH (ed) Photosystem I: the light-driven, plastocyanin:ferredoxin oxidoreductase. Kluwer Academic Publishers, pp 319–338
Shinkarev V, Vasiliev I, Golbeck JH (2000) A kinetic assessment of the sequence of electron transfer from FX to FA and further to FB in photosystem I: the value of the equilibrium constant between FX and FA. Biophys J 78:363–372
Shuvalov VA (1976) The study of the primary photoprocesses in photosystem I of chloroplasts recombination luminescence, chlorophyll triplet state and triplet-triplet annihilation. Biochim Biophys Acta 430:113–121
Simonson T (2001) Macromolecular electrostatics: continuum models and their growing pains. Curr Opin Struct Biol 11:243–252
Simonson T (2003) Electrostatics and dynamics of proteins. Rep Prog Phys 66:737–787
Simonson T, Perahia D (1995) Internal and interfacial dielectric properties of cytochrome c from molecular dynamics in aqueous solution. Proc Nat Acad Sci USA 92:1082–1086
Simonson T, Archontis G, Karplus M (1999) A Poisson-Boltzmann study of charge insertion in an enzyme active site: the effect of dielectric relaxation. J Phys Chem B 103:6142–6156
Sitkoff D, Sharp KA, Honig B (1994) Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem 98:1978–1988
Soriano GM, Cramer WA, Krishtalik LI (1997) Electrostatic effects on electron-transfer kinetics in the cytochrome f plastocyanin complex. Biophys J 73(6):3265–3276
Stephens PJ, Jollie DR, Warshel A (1996) Protein control of redox potentials of iron-sulfur proteins. Chem Rev 96:2491–2513
Stevens WJ, Krauss M, Basch H, Jasien PG (1992) Relativistic compact effective potentials and efficient, shared-exponent basis-sets for the 3rd-row, 4th-row, and 5th-row atoms. Can J Chem 70(2):612–630
Stombaugh NA, Sundquist JE, Burris RH, Orme-Johnson WH (1976) Oxidation-reduction properties of several low potential iron-sulfur proteins and of methylviologen. Biochemistry 15:2633–2641
Torres RA, Lovell T, Noodleman L, Case DA (2003) Density functional and reduction potential calculations of Fe4S4 clusters. J Am. Chem Soc 125:1923–1936
Trissl HW, Leibl W, Deprez J, Dobek A, Breton J (1987) Trapping and annihilation in the antenna system of photosystem I. Biochim Biophys Acta 893:320–332
Vos MH, Van Gorkom HJ (1990) Thermodynamical and structural information on photosynthetic systems obtained from electroluminescence kinetics. Biophys J 58:1547–1555
Wada A (1976) The alpha-helix as an electric macro-dipole. Adv Biophys 9:1–63
Warshel A, Papazyan A (1998) Electrostatic effects in macromolecules: fundamental concepts and practical modeling. Curr Opin Struct Biol 8:211–217
Wasielewski MR, Smith RL, Kostika AG (1980) Electrochemical production of chlorophyll a and pheophytin a excited states. J Am Chem Soc 102:6923–6928
Xu W, Chitnis P, Valieva A, van der Est A, Brettel K, Guergova-Kuras M, Pushkar J, Zech SG, Stehlik D, Shen G, Zybaylov B, Golbeck JH (2003) Electron transfer in cyanobacterial photosystem I: II. Determination of forward electron transfer rates of site-directed mutants in a putative electron transfer pathway from A0 through A1 to FX. J Biol Chem 278:27876–27887
Zhou H-X (1994) Effects of mutations and complex formation on the reduction potentials of cytochrome c and cytochrome c peroxidase. J Am Chem Soc 116:10362–10375
Acknowledgments
The authors are grateful to Dr. Sergey Chamorovsky and Constantin Chamorovsky for critical reading of this manuscript, and to Dr. Igor Ouporov for help in programs compilation. This study was supported by Grants from the Russian Foundation for Basic Research (05-04-48557 and 06-04-48672) and from Russian Federal Agency for Science and Innovation (02.512.11.2085).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ptushenko, V.V., Cherepanov, D.A., Krishtalik, L.I. et al. Semi-continuum electrostatic calculations of redox potentials in photosystem I. Photosynth Res 97, 55–74 (2008). https://doi.org/10.1007/s11120-008-9309-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-008-9309-y