
Abstract
In photosystem I (PSI) complexes at room temperature electron transfer from A1– to FX is an order of magnitude faster on the B-branch compared to the A-branch. One factor that might contribute to this branch asymmetry in time constants is TrpB673 (Thermosynechococcus elongatus numbering), which is located between A1B and FX. The corresponding residue on the A-branch, between A1A and FX, is GlyA693. Here, microsecond time-resolved step-scan FTIR difference spectroscopy at 77 K has been used to study isolated PSI complexes from wild type and TrpB673Phe mutant (WB673F mutant) cells from Synechocystis sp. PCC 6803. WB673F mutant cells require glucose for growth and are light sensitive. Photoaccumulated FTIR difference spectra indicate changes in amide I and II protein vibrations upon mutation of TrpB673 to Phe, indicating the protein environment near FX is altered upon mutation. In the WB673F mutant PSI samples, but not in WT PSI samples, the phylloquinone molecule that occupies the A1 binding site is likely doubly protonated following long periods of repetitive flash illumination at room temperature. PSI with (doubly) protonated quinone in the A1 binding site are not functional in electron transfer. However, electron transfer functionality can be restored by incubating the light-treated mutant PSI samples in the presence of added phylloquinone.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In oxygen-evolving photosynthetic organisms solar energy is captured and converted in two large membrane-spanning protein complexes called photosystem I (PSI) and II (PSII) (Walker 1993). In both photosystems, light energy is captured in large pigment arrays, which are designed to rapidly transfer the excitation energy to a reaction center (RC) core where the photochemistry takes place (Fromme et al. 2001). Upon light excitation of special pigments in the RC, electrons are transferred via a series of protein-embedded acceptors, across a biological membrane (thylakoid membrane) (Walker 1993). In this manuscript we focus on PSI, where the cofactors operate in a highly reducing regime (Brettel 1997).
The organization of the pigments in PSI, and especially the pigments involved in electron transfer (ET), are highly conserved in higher plants, algae, and cyanobacteria (Antoshvili et al. 2018; Jordan et al. 2001; Malavath et al. 2018; Mazor et al. 2017; Qin et al. 2019; Suga et al. 2019). The PSI complex consists of 11–12 protein subunits, with 3 of them (PsaA, PsaB, and PsaC) binding the ET cofactors (Jordan et al. 2001; Malavath et al. 2018; Mazor et al. 2017). The cofactors that participate in ET are termed P700, A−1, A0, A1, FX, FA, and FB (Fromme et al. 2001). P700 is a special pair of chlorophyll a and chlorophyll a’ (Chl a and Chl a’) molecules that are often termed PB and PA, respectively. Chl a’ is a 132-epimer of Chl a (Fromme et al. 2001). A−1 and A0 are monomeric Chl a species. A1 is a phylloquinone molecule (2-methyl-3-phytyl-1,4-naphthoquinone, PhQ), or a close analogue (Jordan et al. 2001; Ozawa et al. 2012).
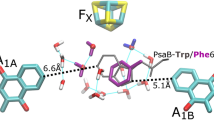
PSI RCs contain two nearly symmetrical branches of ET cofactors, termed the A- and B-branches (Fig. 1a). Cofactors in each branch are denoted by an A or B subscript (e.g. A1A and A1B). The two branches emanate from P700 and re-converge at FX. FX, FA and FB are [4Fe-4S] clusters. FX is bound to both PsaA and PsaB, while FA and FB are bound to the PsaC stromal protein subunit. The geometry of the ET cofactors (minus FA and FB) are outlined in Fig. 1a.

a Structural organization of the ET cofactors in PSI. TrpB673 is also shown. Image was derived from 2.5-Å X-ray crystal structure of PSI isolated from Thermosynechococcus elongatus (PDB 1JB0) (Jordan et al. 2001). The terminal acceptors FA and FB are not shown. Hydrocarbon tails of PhQ and Chl a/a’ are truncated for clarity. Edge-to-edge distances from TrpB673 to A1A, A1B, and FX are included (dotted) along with the edge-to-edge distances from FX to A1A and A1B. b Structure showing select amino acid residues and water molecules around A1A, A1B, and FX. Oxygen atoms of water molecules are enlarged for visualization purpose. Possible H-bonding interactions, ranging from 2.7 to 3.4 Å, are depicted as dotted lines. c Alternative view of the structure in B. Atom coloring: dark grey: carbon atoms of A1, light khaki: all other carbon atoms, red: oxygen, blue: nitrogen, light green: magnesium, yellow: sulfur, dark yellow: iron, magenta: water molecules behind Ser/Leu/Ala chains on both branches, orange: water molecules between A1A and FX, and light blue: water molecules between A1B and FX
Following the primary charge separation events, the P700+A1– radical pair is formed within ~ 50 ps of light excitation of the PSI RC complex (Hastings et al. 1994, 1995). The P700+A1– radical pair state is further stabilized by ET from A1– to FX. At RT, the ET rates are branch specific, with A1A– → FX occurring in ~ 300 ns at RT, and A1B– → FX occurring in ~ 15 ns (Agalarov and Brettel 2003; Guergova-Kuras et al. 2001; Joliot and Joliot 1999; Makita et al. 2015; Setif and Brettel 1993). This large difference in the time constants associated with ET down either branch is difficult to rationalize given that the same pigment occupies the A1A and A1B binding sites, and that the environment surrounding the pigments on both branches are near identical. Kinetic models that explain the biphasic nature of the A1 ET in terms of energetic asymmetries have been proposed previously, however (Agalarov and Brettel 2003; Santabarbara et al. 2019, 2005).
From FX–, an electron is further transferred to the terminal electron acceptors FA and FB on a sub-µs timescale (Byrdin et al. 2006).
In isolated PSI complexes, the P700+FA/B− radical pair state recombines in 50–100 ms (Jordan et al. 1998; Makita and Hastings 2016a; Shinkarev et al. 2002). This recombination proceeds via repopulation of the A1– state. This conclusion follows from the observation of a change in recombination rate upon altering the pigment that occupies the A1 binding site (Lüneberg et al. 1994; Makita and Hastings 2016a, 2017; Shen et al. 2002; Shinkarev et al. 2002).
When the A1 cofactor is disabled or is no longer functional in its ability to perform forward ET, the P700+A0− radical pair state recombines on a few tens of ns. In a portion of the complexes a decay pathway that involves the P700 triplet state (3P700) is apparent (Brettel 1997; Schlodder et al. 2001; Webber and Lubitz 2001). 3P700 forms in ~ 10 to 100 ns, and decays in 10–20 µs at RT (Makita et al. 2015; Schlodder et al. 2001). The formation of 3P700 is thought to occur through a spin flipping mechanism, where the singlet spin configuration of P700+A0– evolves to a triplet configuration via hyperfine interactions (Brettel 1997).
At cryogenic temperatures (~ 120 K and below), ET in PSI becomes heterogeneous. In ~ 50% of the PSI complexes the P700+FA/B− radical pair state forms irreversibly (Makita and Hastings 2015; Schlodder et al. 1998), and does not contribute in repetitive flash measurements. In ~ 40% of PSI complexes P700+A1– recombines in ~ 360 µs, and occurs predominantly down the A-branch (Makita and Hastings 2015; Schlodder et al. 1998). In the remaining 10%, P700+FX– forms and then decays in a few ms (Makita and Hastings 2015; Schlodder et al. 1998). In PSI complexes in which the pigment in the A1 binding site is not functional, 3P700 forms, and at 77 K decays in ~ 200 to 300 µs (Makita and Hastings 2018; Makita and Hastings 2019; Schlodder et al. 2001).
In addition to the functionality of the quinone in the A1 binding site, recent studies have started to reveal how structural properties may relate to the thermodynamic and energetic properties of the pigment in the A1 binding site (Cherepanov et al. 2018; Makita and Hastings 2017; Milanovsky et al. 2017; Santabarbara et al. 2019). However, there are many unresolved questions on how different pigment-protein interactions may modulate the functionality of the quinones as ET cofactors in the A1 binding site (Srinivasan and Golbeck 2009). Figure 1b and c show two views of the environment surrounding the quinones in the A1A and A1B binding sites. Amino acid numbering in this manuscript is for Thermosynechococcus elongatus (T. elongatus) (PDB 1JB0) (Jordan et al. 2001).
Several molecules in Fig. 1b and c are noteworthy for their potential impact on the functional/structural properties of A1. A tryptophan residue (TrpA697/TrpB677) is π-stacked with PhQ. The C4=O group of PhQ is hydrogen bonded (H-bonded) to the backbone of a leucine residue (LeuA722/LeuB706). The C1=O carbonyl group of PhQ is free from H-bonding, so H-bonding to PhQ in the A1 binding site is decidedly asymmetrical.
Figure 1b, c indicates clusters of water molecules near PhQ in the A1 binding site. A series of water molecules located near LeuA722/LeuB706 form a network of H-bonds [water molecule numbering is A5018, A5030, A5031, A5055 for A1A, B5014, B5015, B5037, B5055 for A1B]. These clusters of water molecules have been postulated to have an impact on the redox properties of A1, and also to contribute to increasing the strength of the asymmetric H-bonding to the C4=O group of PhQ, at least for PhQ in the A1A binding site (Karyagina et al. 2007; Rohani et al. 2019). Figure 1 also indicates a cluster of water molecules between the aromatic ring of PhQ and FX [water molecules numbered A5007, A5015, A5022, A5043, A5049 for A1A, B5018, B5019, B5030, B5055, B5056, B5058 for A1B].
Figure 1 indicates a high degree of structural symmetry between the pigments on the A- and B-branch ET chains. However, TrpB673 breaks this symmetry. The A-branch counterpart of TrpB673 is GlyA693. While TrpB673 is on the PsaB protein subunit, it is spatially located between A1A, A1B, and FX (Fig. 1b, c). Given the unique location of TrpB673 in the ET chain it has been proposed that TrpB673 participates as an intermediate acceptor in ET between A1 and FX (Ivashin and Larsson 2003). Although this hypothesis has since been shown improbable from the perspective of ET theory (Moser and Dutton 2006), the role of TrpB673 in ET in PSI is not well understood. An EPR study on PSI complexes from Chlamydomonas (C.) reinhardtii in which Trp was replaced with Gly shows that the mutation alters primarily ET on the B-branch, indicating only an indirect involvement of Trp with the forward ET (Ali et al. 2006).
Here time-resolved step-scan (TRSS) FTIR difference spectroscopy (DS) at 77 K was used to study a PSI mutant from S6803 in which TrpB673 was changed to Phe. In spectra for this mutant bands associated with both P700+A1– and 3P700 are found. Adjusting the PSI samples’ exposure to actinic illumination at RT, prior to freezing, resulted in different ratio of P700+A1– and 3P700 states formed. Furthermore, 3P700 state formation was quenched in actinic light exposed PSI mutant samples incubated in the presence of PhQ. This study indicates that the TrpB673 mutation likely disturbs the group of water molecules in the environment near the aromatic ring of PhQ, leading to light-induced phylloquinol (PhQH2) formation, presumably through a double-protonation event induced by the disturbed water molecules. Added PhQ in the buffer is able to displace PhQH2 in the A1 binding site.
Materials and methods
Construction/growth of W673F PSI
The recipient strains were generated by deletion of a portion of the psaB gene as described previously (Xu et al. 2003) with some modifications. The S6803 psaB knockout (ΔpsaB) recipient strain was obtained by the recombinant DNA sequence that was cloned from the 608 bp to 1014 bp on the psaB open reading frame, and from 9 to 751 bp after psaB with an introduced EcoR I restriction site between these two flanking DNA fragments. Then the 1.3 kbp kanamycin resistance cassette gene was added at the EcoR I restriction site, which was served for the homologous recombination. After 9 generations of segregation on BG-11 containing plates, with 20 µg/mL kanamycin antibiotic under very low light intensity, the stable ΔpsaB strain was obtained and confirmed by PCR analysis. For mutagenesis in the C-terminal region of the PsaB protein, the generated plasmid pBC contains the C-terminal region of the psaB gene with resistance genes for ampicillin and chloramphenicol antibiotic and 760 bp region downstream of the psaB gene (Xu et al. 2003), which served as the templates for PCR based site-directed mutagenesis. The site-directed mutated DNA was used to transform the recipient strain ΔpsaB. The transformation and transformant selection were performed under weak light and heterotrophic growth as described previously (Xu et al. 2003). The chloramphenicol resistant colonies were selected and segregated for four generations. The fragment containing the mutated site was amplified by PCR and it was sequenced to ensure the mutation.
The PsaB-W673F mutant, and ΔpsaB recipient strain were grown in BG-11 medium supplemented with 5 mM glucose with their designed antibiotics. PsaB-W673F was cultured at 30 °C, under low light intensity (< 5 µE m−2 s−1) while the ΔpsaB recipient strain is maintained in the dark but supplied with 40 µE m−2 s−1 light for 10 min every day. Cells were collected during the late exponential growth phase, washed and resuspended in SMN buffer (0.4 M sucrose, 10 mM NaCl, and 10 mM Mops-NaOH, pH 7.0) for further use (Xu et al. 2003).
Isolation of PSI particles
Thylakoid membranes were prepared from cells in late exponential growth phase as described before (Xu et al. 2003). To purify PSI complexes from the thylakoid membranes, the concentration of the thylakoids were adjusted to 0.5 mg/mL Chl in 10 mM Mops-NaOH, pH 7.0 buffer, and solubilized for 2 h at 4 °C by adding n-dodecyl-d-maltopyranoside (DM) to a concentration of 1% (w/v). The solution was centrifuged for 20 min at 15,000 g to remove insoluble debris, and the supernatant was loaded onto a 10–30% (w/v) sucrose gradient prepared in 10 mM Mops-NaOH pH 7.0 buffer containing 0.05% (w/v) DM. The gradients were centrifuged for 16–18 h at 150,000×g at 4 °C, and the lower, dark green band containing PSI trimers was collected. The PSI trimers were concentrated using Pierce concentrators (20,000 molecular weight cut-off membranes (Thermo Scientific)) to a final concentration of ~ 1 µg/µl Chl and stored at − 80 °C until further use.
Preparation of PSI samples for FTIR DS
20 µL of WB673F PSI particles at Chl-a concentration of 1.66 mg/mL were suspended in 980 µL Tris buffer (pH 8.0) with 0.04% DM. The solution was ultra-centrifuged at 408,000×g for 3 h to produce a soft pellet. 0.1 µL each of 20 mM sodium ascorbate and 10 µM phenazine methosulfate were added to the soft pellet to act as exogenous electron donor to P700+. The pellet was squeezed between two CaF2 windows until the optical density at ~ 1654 cm–1 corresponding to amide I absorption was below 1.0. Actinic flashes (~ 1 mJ energy and 5 ns duration) at 532 nm were from a Minilite Nd:YAG laser (Continuum, San Jose, CA) operating at 10 Hz. In some experiments samples were exposed to repetitive flashes at RT, prior to freezing to 77 K. Samples were exposed to actinic illumination for 1, 4, or 16 h. For measurement at 77 K, samples were mounted in an Optistat DN-V liquid nitrogen cryostat (Oxford Instrument, United Kingdom). All samples were frozen in the dark regardless of the pre-exposure to actinic flashes at RT.
PhQ was reincorporated into W673F mutant PSI complexes by using the same method as outlined previously to incorporate quinones into PSI that lacked a functional quinone in the A1 binding site (Makita and Hastings 2016b). Briefly, 500 × molar excess PhQ, dissolved in ethanol, was added to suspension of PSI particles so that the volume ratio of ethanol in the mixture is below 2%. The mixture was stirred at 277 K for 12 h in the dark, before ultra-centrifugation to form a soft pellet.
FTIR difference spectroscopy (DS)
All FTIR DS were collected using a Bruker Vertex80 FTIR spectrometer (Bruker Optics, Billerica, MA). (P700+–P700) FTIR DS at 77 K were obtained using CW light from a 10 mW helium neon laser, as described previously (Wang et al. 2004). Time-resolved step-scan (TRSS) FTIR DS were collected also as described previously (Makita and Hastings 2018). Briefly, time-resolved spectra were collected with 6 µs temporal resolution, over a 3.5 ms time window, in the 1950–1100 cm−1 spectral range, at 4 cm−1 spectral resolution. 2000–1000 cm−1 bandpass filters were placed both before and after the sample. The CaF2 sample compartment windows, and the cryostat shroud windows, also blocked IR light below ~ 1100 cm−1 from reaching the detector. Laser flash of 5 ns duration at 532 nm were from a Minilite Nd:YAG laser (Continuum, San Jose, CA) operating at 10 Hz. At each interferometer step (in step-scan measurements), data from 20 laser flashes were coadded (averaged). Each full step-scan acquisition was repeated ~ 40 times and all data were averaged to improve the signal-to-noise ratio.
The time-resolved spectra obtained at 77 K were globally analyzed using Glotaran (Snellenburg et al. 2012), and fitted to a sum of exponential components. The dominant component/phase has a lifetime of ~ 390 μs and the spectra associated with this lifetime are called decay associated spectra (DAS). Global analysis was primarily used to separate spectral features associated with a heating artifact caused by the actinic laser flashes used to initiate photochemistry (Makita and Hastings 2019).
Results
Characteristics of the W673F mutant PSI
WB673F and WB673A mutant cells require glucose for growth. We failed to isolate PSI complexes from the WB673A mutant cells, however, preventing spectroscopic characterization of WB673A mutant PSI. The WB673F mutation is a more conservative replacement than WB673A, and the WB673F mutant cells grew well (on glucose under low light), and we were able to prepare PSI trimers from the WB673F mutant strain. Interestingly, the WB673F mutant strain cannot grow photoautotrophically. Further details of the growth rate and other physiological characterizations will be described elsewhere.
In this study, three types of FTIR DS are considered: (1) Photoaccumulated (P700+–P700) FTIR DS; (2) Time-resolved (P700+A1––P700A1) FTIR DAS; (3) (3P700–P700) FTIR DAS. In these FTIR DS and DAS, positive peaks correspond to P700+, 3P700 and the A1– states, and negative peaks correspond to the P700 and A1 ground (or neutral) states. All spectra except for (3P700–P700) DS are normalized to the 1717(+)/1697(−) cm−1 difference band appearing in both the photoaccumulated (static) and the time-resolved spectra.
Photoaccumulated (P700+–P700) DS at 77 K
Figure 2 shows (P700+–P700) FTIR DS collected at 77 K, for PSI from WT and PSI from the WB673F mutant. Two sets of spectra are shown for WB673F PSI (Fig. 2b). One sample was exposed to 10 Hz actinic laser flashes for 16 h at RT prior to freezing (Fig. 2b, red). The other sample was not subjected to actinic illumination at RT, and was frozen in the dark (Fig. 2b, blue). The samples that were exposed to actinic illumination at RT prior to freezing will be referred to as pre-flashed samples, while samples kept in the dark prior to and during freezing will be referred to as non-flashed samples.

Photoaccumulated (P700+–P700) FTIR DS for a WT PSI and b WB673F mutant PSI at 77 K. For WB673F mutant PSI, FTIR DS were collected for non-flashed (blue) and pre-flashed (532 nm laser flashes for 16 h at 10 Hz) (red) samples. The spectra are normalized using the 1717( +)/1697(–) cm–1 FTIR difference band. c (WB673F–WT) double difference spectrum (DDS), obtained by subtracting the spectrum in A from the average of the spectra in b
In the (P700+–P700) FTIR DS for WT PSI (Fig. 2a), positive bands are observed at 1754, 1741, 1717, 1688, 1675, 1655, 1622, 1601, 1590, 1567, 1550, 1524, and 1431 cm–1, while negative bands are observed at 1748, 1733, 1697, 1681, 1668, 1637, 1610, 1543, 1535, and 1423 cm–1. These difference features in (P700+–P700) FTIR DS for cyanobacterial PSI have been discussed in detail before (Breton 2006b; Hastings et al. 2019; Sivakumar et al. 2005).
No significant changes in the (P700+–P700) FTIR DS are observed for pre-flashed and non-flashed WB673F PSI samples (Fig. 2b). However, small changes are observed in the (P700+–P700) DS for WT and WB673F PSI samples. These small changes are visualized more easily in the (WB673F–WT) double difference spectrum (DDS) (Fig. 2c). The most intense features in the DDS in Fig. 2c are near ~ 1650 and ~ 1550 cm−1. Features near these frequencies are usually associated with amide I and II protein vibrations, respectively. So most likely there is some mutation-induced alteration in the protein environment, presumably near A1/FX given that the mutation is near A1 and FX. Most likely, the charge on the iron-sulfur clusters have an electrostatic impact on the protein amide vibrations near A1 and FX, and these amide vibrations are modified by the mutation and appear as features in the DDS in Fig. 2c.
Time-resolved FTIR DS at 77 K
Figure 3a shows (3P700–P700) FTIR DAS obtained at 77 K. Time-resolved (P700+A1––P700A1) FTIR DAS obtained using WT and non-flashed mutant PSI at 77 K are shown in Fig. 3b and c, respectively. Time-resolved (P700+A1––P700A1) FTIR DAS obtained using mutant PSI that has been pre-flashed repetitively at RT with 532 nm laser flashes at 10 Hz, for 1, 4, and 16 h are shown in Fig. 3d–f, respectively.

a (3P700–P700) FTIR DS at 77 K, from (Makita and Hastings 2018). b Time-resolved FTIR DAS for WT PSI at 77 K. Time-resolved FTIR DAS for non-flashed (c), 1 h pre-flashed (d), 4 h pre-flashed (e) and 16 h pre-flashed (f) WB673F mutant PSI at 77 K. Vertical dotted lines are shown at 1635 and 1594 cm–1. Time-resolved FTIR DAS (b–f) represent the ~ 390 µs phase
Low temperature flash-induced absorption changes at 1697 cm–1 for the differently illuminated mutant PSI complexes are outlined in Fig. S1. At 1697 cm–1, an intense negative band is present in (P700+–P700) and (P700+A1––P700A1) FTIR DS, but not in (3P700–P700) FTIR DS. The transient absorption changes at 1697 cm–1 (at 77 K) is, therefore, associated with P700+A1– charge recombination. Specifically, the 1697 cm–1 band in Fig. 2a and b is well known to be associated with the 131-keto C=O group of the PB pigment of P700 (Breton 2006a; Hastings 2015). By fitting the data at 1697 cm–1 to two stretched exponential functions and a constant, the lifetime of the major decay phase is calculated to be 389 µs for all off the different PSI complexes. The parameters obtained from fitting the kinetics at 1697 cm–1 are listed in Table S1. A similar time constant is observed for WT PSI (Sivakumar et al. 2005).
The time-resolved (P700+A1––P700A1) FTIR DAS for WT (Fig. 3b) and non-flashed mutant (Fig. 3c) PSI are similar. In addition to the difference signals associated with P700+/P700 discussed above (Fig. 2), the (P700+A1––P700A1) FTIR DS displays positive bands associated with A1– at 1495 and 1415 cm–1 due to the C1=O and C4=O stretching vibrations of PhQ, respectively (Makita et al. 2017; Rohani et al. 2019). The C4=O stretching vibration is downshifted relative to the C1=O vibration due to H-bonding. The characteristic bands of A1– at 1495 and 1415 cm–1 is found also in spectra obtained using pre-flashed WB673F PSI (Fig. 3d, e).
The time-resolved (P700+A1––P700A1) FTIR DS obtained using WT and non-flashed WB673F PSI are very similar (Fig. 3b and c) indicating that the mutation does not greatly impact the environment of PhQ in the A1 binding site, in either the neutral or the anion state. The time-resolved spectra for pre-flashed WB673F PSI (Fig. 3d–e and f), however, exhibit clear differences compared to the WT and non-flashed WB673F PSI spectra, most notably near 1635 and 1594 cm–1. For WB673F PSI that was pre-flashed for 1 h (Fig. 3d), a positive band is starting to appear at 1594 cm–1 with a similar amplitude to the band at 1601 cm–1. In addition, the relative intensity of the negative band at 1635 cm–1 increases with respect to the 1697 cm–1 band (compare intensity ratios in the spectra in Fig. 3b and c). When mutant PSI samples are pre-flashed for 4 h (Fig. 3e), these changes at 1635 and 1594 cm−1 grow, and after 16 h of pre-flashing the 1635(−) and 1594( +) cm−1 bands are now the most prominent bands in the spectrum. These spectral features that grow with the increasing number of flashes at RT prior to cooling are characteristic of features found in the (3P700–P700) FTIR DS (Fig. 3a), where the bleaching at 1635 cm−1 is well-known to correspond to the 131-keto C=O of the PA pigment of P700, which downshifts to 1594 cm−1 upon 3P700 formation (Breton et al. 1999). The time-resolved FTIR DAS for pre-flashed WB673F PSI therefore result from a combination of signals found in (P700+A1––P700A1) and (3P700–P700) FTIR DS, with the contribution from (3P700–P700) signals increasing as the number of pre-flashes at RT increases. Note that the P700+A1– and 3P700 states decay with similar lifetimes (~ 360 µs and ~ 200 to 300 µs, respectively) that cannot easily be separated in the global analysis, so the DAS contains contributions from both states with similar lifetime.
The spectra in Fig. 3 indicate 3P700 formation in a fraction of WB673F PSI samples only when the samples are exposed to actinic illumination at RT prior to cooling, and the relative fraction depends on the number of pre-flashes at RT. From the relative peak intensities of the 1697 and 1594 cm−1 bands (see SI for details), we estimate the relative fraction of WB673F PSI undergoing 3P700 formation is 25, 38, and 58% for 1, 4, and 16 h pre-flashed samples, respectively (Table S2). Note that in the time-resolved measurements at 77 K the frozen sample is exposed to repetitive actinic laser flashes at 10 Hz for 5–20 h. However, there is no indication for the appearance of a 1635(−)/1594( +) cm–1 difference band during the course of measurements at 77 K.
It is well known that 3P700 formation can occur upon P700+A0− charge recombination (Golbeck and Bryant 1991), which is common when the A0– → A1 forward ET process is inhibited, either because the A1 binding site is empty (Sieckmann et al. 1993), or the quinone in the binding site is no longer functional in ET (Breton et al. 1999; Makita and Hastings 2018; Schlodder et al. 2001). PhQ is clearly present in the non-flashed mutant PSI samples (anion bands are observed), and it is likely that flash illumination at RT caused some sort of alteration of the PhQ in the A1 binding site. Given the large number of flashes required this alteration occurs with low yield. Given the location of the mutation, the forward ET from A1– to FX is likely inhibited because A1 is no longer a functional intermediate in ET. One likely possibility is the light-induced double protonation of PhQ in the mutant PSI samples.
PhQ is clearly observed to be a functional electron acceptor in non-flashed WB673F PSI. Therefore, it is unlikely that the mutation results in an empty A1 binding pocket. The 3P700 state is observed at 77 K only in the mutant and only under exposure to many hours of actinic laser flashes at 10 Hz, at RT. Therefore, it is likely that the mutation opens up a pathway for movement of water molecules that can lead to protonation of PhQ in the A1 binding site. Obviously, this is a rare event, as the effects are observed only after thousands of laser flashes.
One might predict that a protonated PhQ in the A1 binding site will have a lower affinity for the binding site than the native PhQ. So, by incubating pre-flashed mutant PSI samples in the presence of PhQ one might find that PhQ could replace the protonated species in the A1 binding site, restoring regular ET through A1, and diminishing triplet state formation. To test this prediction we incubated pre-flashed WB673F PSI samples in the presence of a large molar excess of PhQ. After washing the sample, we undertook time-resolved FTIR experiments on the pre-flashed WB673F PSI samples that had been incubated in the presence of PhQ.
Figure 4 shows time-resolved FTIR DAS obtained using two samples from WB673F mutant PSI where PhQ has been re-introduced (samples were incubated in the presence of a large molar excess of PhQ). Before re-introducing PhQ, both samples were exposed to actinic flashes at RT for ~ 4 h to disable A1, so both PSI samples are equivalent to that in Fig. 3e. After incubation in the presence of PhQ in the dark for ~ 12 h, one sample (Fig. 4a) was frozen to 77 K in the dark. The other (Fig. 4b) was exposed to another series of ~ 4 h actinic laser flashes at RT before cooling to 77 K. In Fig. 4a, the relative amplitude of the 1635 cm−1 band and lack of intense band at 1594 cm−1 indicates that the 3P700 state is not present. The spectrum in Fig. 4a indicates that the ET function of PhQ in the A1 binding site is recovered simply by adding exogenous PhQ to the PSI samples. On the other hand, Fig. 4b shows characteristic features of the (3P700–P700) FTIR DS, much like in the FTIR DS for pre-flashed WB673F PSI (Fig. 3f). Therefore, even after the A1 function is restored by exogenous PhQ, exposure to repetitive actinic flashes at RT is capable of re-deactivating PhQ in the A1 binding site. The FTIR DAS in Fig. 4 show that the effect of pre-flashing WB673F PSI is reversible. That is, PhQ in the A1 binding site in the mutant can be disabled through exposure to actinic flashes, and re-enabled when PhQ is re-incorporated by displacing the non-functional quinone in the binding site, and then re-disabled through additional exposure to repetitive laser flash illumination. The likely cause of 3P700 formation in pre-flashed WB673F mutant PSI is a mutant-induced structural modification of the binding site, which allows PhQ protonation to occur, albeit with low yield.

Time-resolved (P700+A1––P700A1) FTIR DAS at 77 K for WB673F mutant PSI with exogenous PhQ added. PhQ was added to WB673F mutant PSI that had been first pre-flashed for 4 h at RT. a WB673F mutant PSI + PhQ, where PSI was cooled in the dark prior to measurement. b Same samples as in a except the WB673F + PhQ PSI was pre-flashed again for 4 h at RT prior to freezing. Dotted guidelines are shown at 1635 and 1594 cm–1
Discussion
The rate of forward ET from A1− to FX on the B-branch is roughly an order of magnitude larger than for the corresponding ET on the A-branch. Given the similarity in edge-to-edge distances between PhQ and FX on either branch, the order of magnitude difference in the ET rate is likely due to a difference in midpoint potentials (Em) between the PhQ’s on each branch. Based on kinetic simulations, this difference is approximately ~ 50 mV (Makita and Hastings 2016a; Santabarbara and Casazza 2019; Santabarbara et al. 2019). Such a difference in Em likely results because of a difference in the PhQ’s interaction with the surrounding local environment (Kawashima and Ishikita 2017). Electrostatic calculations have identified structural factors (such as side-chain orientation and protonation states) that could lead to differences in the Em of PhQ on the A and B branches (Ishikita and Knapp 2003; Kawashima and Ishikita 2017). However, how pigment–protein interactions impact the functionality of PhQ’s as ET cofactors in the A1 binding sites are still poorly understood. The work presented here suggests that TrpB673, a residue with indole side chain lying at a nexus between A1A, A1B, and FX, could be important in impeding protonation of PhQ (on perhaps both branches) in PSI, by blocking access of water molecules to PhQ in the A1 binding site.
Direct impact of WB673F on P700 and A1
The photoaccumulated (P700+–P700) FTIR DS and the time-resolved (P700+A1––P700A1) FTIR DS suggests that the replacement of TrpB673 with Phe does not directly impact P700 or PhQ in the A1 site as the vibrational frequencies associated with these pigments are unaffected by the mutation. The (P700+–P700) FTIR DS exhibits weak mutation-induced changes, but these changes are focused mostly in the regions where amide I and amide II protein absorption is expected. Therefore, these weak spectral changes are likely due to mutation-induced modifications in the protein environment surrounding FX. Since TrpB673 is in the vicinity of A1A, A1B, and FX cofactors, it is unlikely that P700 will be affected by the mutation. The A1 pigments on the other hand, are within 4.0–6.5 Å of the indole side chain of TrpB673 (Fig. 1a). However, no significant changes are observed in the (P700+A1––P700A1) FTIR DS either. In particular, the C1=O and C4=O modes of A1A– are found at 1495 and 1415 cm–1, respectively for both WT and mutant PSI. The transient absorption kinetics at 1697 cm–1 for PSI at 77 K indicate that P700+A1A– charge recombination in mutant PSI has a lifetime of ~ 390 µs (Fig. S1), compared to ~ 360 µs for WT PSI (Makita and Hastings 2016b). Within the noise level of the experiment, the time constants for WT and mutant PSI are the same, suggesting that the midpoint potentials of A1A has not been affected by the mutation.
At 77 K, time-resolved measurements on cyanobacterial PSI from S6803 probe the A-branch ET process only. As such the measurement does not provide insight to mutation-induced alterations associated with the PhQ on the B-branch.
Quinol formation in WB673F PSI
The time-resolved FTIR DS for pre-flashed WB673F PSI show that the exposure to actinic flashes at RT leads to formation of 3P700 at 77 K, with the relative amplitude of the 3P700 signals increasing as the time associated with pre-flashing increases. Since the ability of PSI samples to form 3P700 can be reversed by addition of exogenous PhQ, and re-created with additional exposure to repetitive flashes at RT, 3P700 formation is likely triggered by the conversion of PhQ in the A1 binding site to the doubly protonated quinol form (PhQH2).
PhQH2 formation requires two electrons and two protons to be donated to a neutral state quinone (Gunner et al. 2008). Quinols will not accept any additional electrons (or protons) and can no longer function as ET cofactors. Quinol formation is an essential part of ET in type II RCs, such as PSII and purple bacterial RCs, in which a quinol formation in the QB binding site leads to dissociation of the pigment from the binding site, which can then be replaced by a quinone from the membrane pool (Joliot and Joliot 2006; Okamura et al. 2000; Saito et al. 2013). However, quinones in the A1 binding site in PSI (and in the QA binding site in type II RCs) function purely as 1-electron intermediaries in ET, and quinol formation would be detrimental to this function. The PSI RC, therefore, is designed to avoid or inhibit quinol formation in the A1 binding site, even in the presence of nearby clusters of water molecules.
While double-reduction may be achievable through multiple rapid turnovers, double-protonation in PSI is a highly unlikely process, as PhQ in the A1 binding site lacks a potential proton donor, or a proton transfer pathway to the carbonyl groups of PhQ. PhQH2 formation can be induced in WT PSI, but usually requires intense illumination in combination with harsh reducing agents while cooling (Breton et al. 1999; Frank et al. 1979; Gast et al. 1983; Rutherford and Sétif 1990; Schlodder et al. 2001).
Quinol formation in PSI can, however, be induced without harsh chemical pretreatments, when PhQ in the A1 binding site is exchanged for a benzoquinone (BQ) (Lefebvre-Legendre et al. 2007; Makita and Hastings 2018; McConnell et al. 2011). In a process termed photo-inactivation (McConnell et al. 2011), BQ in the A1 binding site can be converted to benzoquinol (BQH2) simply be subjecting PSI to a series of 10 Hz actinic laser flashes for 10–30 min (McConnell et al. 2011). In this process, water molecules between A1 and FX serve as proton donors (Makita and Hastings 2018). These water molecules cannot donate protons when PhQ occupies the A1 binding site, and it is the lack of an aromatic ring in the BQ species that opens a pathway for these water molecules to access and protonate BQ in the A1 binding site. While this mechanism allows for the double-reduction/double-protonation of a BQ, BQH2 formation in the A1 site is highly inefficient, requiring thousands of actinic laser flashes at 10 Hz for complete photo-inactivation (Makita and Hastings 2018; McConnell et al. 2011). It is important to note that the results from these studies of PSI with BQ incorporated, and other previous studies on PSI, indicate that the midpoint potential of the incorporated quinone is not the major factor that enables the double-reduction in A1. The higher midpoint potentials of BQs, compared to PhQ, make the double-reduction process more thermodynamically favorable. However, previous studies on PSI with higher potential naphthoquinones (NQs) incorporated showed no sign of double-reduction of the NQ in the A1 binding site (Makita and Hastings 2015). Through these observations, it was suggested that the double-reduction is triggered not by the midpoint potential, but by the structural modification that opens a protonation pathway.
The mechanism underlying PhQH2 formation in WB673F PSI is likely similar to the photo-inactivation process observed for PSI with BQ’s incorporated. The lifetime of the P700+A1– state is unaffected (Fig. S1, Table S1), which suggests that the midpoint potential of A1 is relatively unaffected and therefore not the direct cause of double-reduction. A source of protons for the PhQH2 formation is likely the same set of water molecules that are involved in the BQH2 formation process, given that TrpB673 is located within close proximity to these water molecules (Fig. 1B, C). While a similar photo-inactivation mechanism is expected, the yield of the photo-inactivation process is much less efficient for WB673F mutant PSI. For PSI with BQ incorporated, 10–30 min of exposure to 10 Hz actinic laser flashes was sufficient to achieve near-complete photo-inactivation. For WB673F mutant PSI, however, 16 h of exposure to actinic laser flash with similar repetition rate and energy only resulted in partial photo-inactivation (Fig. 3). Photo-inactivation in the WB673F mutant is considerably more inefficient than the already inefficient process in PSI with BQ incorporated. It seems likely therefore that the aromatic ring of PhQ can still restrict access of the water molecules to protonating the PhQ carbonyl groups. The WB673F mutation likely disrupts the environment of the water network between A1A, A1B, and FX, such that these water molecules now have some increased capability of protonating PhQ in the A1 binding site. Since forward ET proceeds predominantly (~ 95%) down the A-branch at 77 K (Makita and Hastings 2015), the water molecules are capable of protonating the A1A PhQ. The very inefficient photo-inactivation process observed here might be partly due to the fact that it is PhQ in the A1A binding site that is being probed, and this PhQ is somewhat distant from TrpB673, making any mutant-induced structural alteration of the A1A binding site smaller than for the A1B binding site. If this is the case, we would predict B-branch photo-inactivation with much fewer actinic laser flashes than is used here.
While we favor the mechanism described above for 3P700 formation, several other scenarios can be hypothesized. It is possible that the WB673F mutation causes an alteration of the branch utilization ratio, and the “disabled” PSI population corresponds to modified B-branch activity. Modification of the A/B-branching ratio in PSI due to specific experimental conditions such as mutation or removal of ET cofactors, has been reported previously (Badshah et al. 2018; Poluektov et al. 2019). It is possible that the WB673F mutation increases B-branch utilization, and that it is A1B that is being doubly reduced and protonated through pre-flashing. For this hypothesis to explain the experimental data, however, it must be assumed that the mutation induced rerouting of ET has also modified A1B such that the P700+A1B– recombination kinetics and (P700+A1B––P700A1B) FTIR DS are now nearly identical to that obtained for A1A in native PSI. While the recombination kinetics and FTIR DS have not been reported for A1B in native PSI at 77 K, the predicted difference in the midpoint potential (Santabarbara et al. 2019) and previous EPR results for A1B (Berthold et al. 2012; Poluektov et al. 2019; Poluektov and Utschig 2015) suggest that the kinetic and spectral properties are likely different from those of A1A. The P700+A1– kinetic and spectral properties identified for WB673F PSI in this study, however, indicate that the features observed are characteristic of A1A.
An interquinone ET process, where an electron is transferred from A1B → FX → A1A (Santabarbara et al. 2010), might also help explain mutation induced 3P700 formation. In this case one would also have to consider the case where the mutation modifies the midpoint potential of A1B to be more positive than A1A, and then also consider A1A → FX → A1B ET. Regardless of the ET direction, we will assume that interquinone ET is the mechanism underlying double reduction. In the first case, where ET proceeds from A1B to A1A, accumulation of doubly-reduced A1A should also result in P700+A1B– recombination. In the second case, where ET proceeds from A1A to A1B, P700+A1B– recombination is expected in a large fraction of the non-flashed samples. So, in both cases, P700+A1B– recombination should be observed in either non-flashed or pre-flashed PSI samples. Such a conclusion is not compatible with the data presented here, where the observed kinetic or spectral features can only be assigned to A1A.
Lastly, it is also possible that the mutation induces heterogeneous binding of PhQ in the A1 binding site, creating populations with slightly different conformations. The “disabled” fraction, then, is the conformation that allows the double-reduction, while a fraction with active A1 even after prolonged illumination is the conformation that does not allow the double-reduction. This scenario would explain why the fraction of disabled PSI remains relatively low, even after 16 h of pre-flashing. One limitation in this hypothesis is that while it assumes conformational heterogeneity at room temperature, there is no indication of such a heterogeneity at 77 K. If PhQ is bound in several different conformations in the A1 binding site, then the semiquinone C = O modes (at 1495 and 1415 cm–1) might be expected to shift in frequency for the different conformations, leading to multiple different bands, or at least some broadening of the main C = O bands. Such features are not observed in the FTIR DS for WB673F PSI (Fig. 3).
Confirmation of the double reduction/protonation of PhQ, and the exact mechanism of the process, requires further investigation. One possible approach to confirm the a PhQ protonation process is to repeat the experiments at different pH, as accumulation of PhQH2 via protonation is expected to be accelerated at lower pH. Such a pH induced acceleration was observed in previous work where BQ in the A1 binding site is doubly protonated (McConnell et al. 2011).
Involvement of B-branch ET, or mutation modified branch utilization, could possibly be investigated using transient absorption spectroscopy at room temperature, with time resolution preferably below 1–2 ns.
The role of WB673 in PSI
Despite the interesting location of TrpB673 at the intersection of the A and B ET branches, the role of TrpB673 in PSI ET is not well understood. It has been suggested that the indole side chain of TrpB673 is involved in ET between A1 and FX (Ivashin and Larsson 2003). This notion is unlikely, however, as the A1–TrpB673–FX distances would suggest very rapid ET, much more rapid than the observed A1– to FX ET rates of ~ 15 and 300 ns (Moser and Dutton 2006).
Previously TrpB673 has been replaced by Gly and it was shown that this mutation mainly affected B-branch ET (Ali et al. 2006). So TrpB673 could impact the redox properties of A1B (or of both A1B and FX), and could be involved in modulating the forward ET between A1B and FX. However, recent calculations have found that TrpB673 does not directly impact on the midpoint potential of A1B (Ishikita and Knapp 2003). It was suggested, however, that the presence of a bulky side chain introduced a twist in the protein backbone near the A1B site, compared to the A1A site where GlyA693 is found instead of Trp. (Kawashima and Ishikita 2017). For both the A1B and A1A sites, the amino acid adjacent to TrpB673 and GlyA693 is Ser (SerB672 and SerA692, respectively). In the A1A site, SerA692 is oriented to form an H-bond with a nearby cluster of water molecules. In the A1B site, however, because of the different twist in the backbone due to the steric effect of the bulky indole side chain of TrpB673, SerB672 has no partner to form an H-bond with. It is suggested that these differences in the local environment induced by TrpB673 cause a difference in the midpoint potentials between A1B and A1A (Kawashima and Ishikita 2017).
Previous computational studies also highlight how the water clusters between A1A, A1B, and FX contribute to the midpoint potentials of the A1 pigments. The cluster of water molecules, surrounding TrpB673, are in H-bonding distance to AspB575. While no proton transfer pathway exists beyond AspB575, the interaction may result in protonation of AspB575, which in turn impacts the midpoint potentials of A1 significantly (Kawashima and Ishikita 2017). If AspB575 remains protonated, the midpoint potentials of A1A and A1B are increased by 106 and 76 mV, respectively, compared to when AspB575 remains ionized (Kawashima and Ishikita 2017).
The study presented here shows that the replacement of TrpB673 with Phe results in double-protonation of PhQ in the A1A binding site. The only likely proton donors near PhQ are the clusters of water molecules indicated in Fig. 1b, c. A cluster of 5 water molecules [A5007, A5015, A5022, A5043, A5049] surround the A1A site and 6 water molecules [B5018, B5019, B5030, B5055, B5056, B5058] for the A1B site. TrpB673 bisects these water clusters. Although a Phe side-chain is likely bulky enough to sustain the original backbone twist, the observed PhQ protonation suggests mutation-induced alteration of these water clusters, which then allows PhQ protonation.
By combining the observations from this study, and the previous study on WB664G mutant PSI (Ali et al. 2006), and taking into account the computational studies on TrpB673 in PSI (Ishikita and Knapp 2003; Kawashima and Ishikita 2017), the role of TrpB673 in PSI ET can be summarized. The bulky indole side chain of Trp is required to create a backbone twist in the A1B site, that is absent in the A1A site, which contributes to a decrease in the A1B midpoint potential (Kawashima and Ishikita 2017). The twist also allows for TrpB673 to face the direction of the groups of water molecules, and to be inserted between the A1A and A1B water clusters. The inserted TrpB673 stabilizes the water molecules to form fixed H-bond patterns for both the A1A and A1B sites. The presence of these water networks is important in two ways. AspB575 near A1A is within H-bonding distance to one of the water molecules. The interaction of AspB575 with the water is important in controlling the protonation state of AspB575, which alters the midpoint potentials of A1 by ~ 100 mV (Kawashima and Ishikita 2017). The fixed H-bonded network of water molecules is also important in preventing the water from accessing PhQ in the A1 binding site and serving as proton donors. Availability of proton donors enable double-protonation of PhQ to form PhQH2 and effectively disables A1 as ET cofactor.
Conclusions
Time-resolved FTIR DS at 77 K has been used to study native and WB673F mutant PSI. The mutation does not directly impact the environment near PhQ in the A1 binding site (or P700). However, prolonged exposure to actinic laser flashes at RT results in PhQ protonation in the mutant but not in the WT. PhQ protonation is probed in PSI via 3P700 formation at 77 K. Protonated PhQ (PhQH2) in the mutant A1 binding site can be reversed by incubating samples in the presence regular PhQ (at RT). Presumably PhQ has a greater affinity for the A1 binding site compared to the protonated species. We suggest that water clusters near A1A, A1B and FX are disturbed in the mutant allowing protons access to the PhQ carbonyl groups. TrpB673 plays a structural role in blocking proton access to the A1 binding site. However, TrpB673 also has an electrostatic impact that could also modulate ET from A1− to FX.
Abbreviations
- BQ:
-
Benzoquinone
- C = O:
-
Carbonyl
- Chl a :
-
Chlorophyll a
- DAS:
-
Decay associated spectrum
- DFT:
-
Density functional theory
- DS:
-
Difference spectra/spectrum/spectroscopy
- ET:
-
Electron transfer
- FTIR:
-
Fourier transform infrared
- H-bond:
-
Hydrogen bond
- NQ:
-
Naphthoquinone
- PhQ:
-
Phylloquinone (2-methyl-3-phytyl-1,4-naphthoqiuinone)
- PSI:
-
Photosystem I
- PSII:
-
Photosystem II
- RC:
-
Reaction center
- S6803:
-
Synechocystis Sp. PCC 6803
- T.:
-
Thermosynechococcus
- TRSS:
-
Time-resolved step-scan
- WT:
-
Wild type
References
Agalarov R, Brettel K (2003) Temperature dependence of biphasic forward electron transfer from the phylloquinone(s) A1 in photosystem I: only the slower phase is activated. Biochim Biophys Acta 1604:7–12
Ali K, Santabarbara S, Heathcote P, Evans MCW, Purton S (2006) Bidirectional electron transfer in photosystem I: Replacement of the symmetry-breaking tryptophan close to the PsaB-bound phylloquinone (A1B) with a glycine residue alters the redox properties of A1B and blocks forward electron transfer at cryogenic temperatures. Biochim Biophys Acta 1757:1623–1633
Antoshvili M, Caspy I, Hippler M, Nelson N (2018) Structure and function of photosystem I in Cyanidioschyzon merolae. Photosynth Res 139(1–3):499–508
Badshah SL et al (2018) Mutations in algal and cyanobacterial Photosystem I that independently affect the yield of initial charge separation in the two electron transfer cofactor branches. Biochim Biophys Acta BBA 1859:42–55
Berthold T et al (2012) Exploring the electron transfer pathways in photosystem I by high-time-resolution electron paramagnetic resonance: observation of the B-side radical pair P700+A1B– in whole cells of the deuterated green alga Chlamydomonas reinhardtii at cryogenic temperatures. J Am Chem Soc 134:5563–5576
Breton J (2006a) FTIR studies of the primary electron donor. In: Golbeck J (ed) Photosystem I: the light driven plastocyanin: ferredoxin oxidoreductase. Advances in photosynthesis and respiration. Springer, Dordrecht, vol 24, pp 271–28924
Breton J (2006b) FTIR Studies of the Primary Electron Donor, P700. In: Golbeck J (ed) Photosystem I: Advances in photosynthesis and respiration, vol 24. Springer, Dordrecht, pp 271–289
Breton J, Nabedryk E, Leibl W (1999) FTIR study of the primary electron donor of photosystem I (P700) revealing delocalization of the charge in P700+ and localization of the triplet character in 3P700. Biochemistry 38:11585–11592
Brettel K (1997) Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim Biophys Acta 1318:322–373
Byrdin M, Santabarbara S, Gu F, Fairclough WV, Heathcote P, Redding K, Rappaport F (2006) Assignment of a kinetic component to electron transfer between iron–sulfur clusters FX and FA/B of Photosystem I. Biochim Biophys Acta 1757:1529–1538
Cherepanov DA, Milanovsky GE, Gopta OA, Balasubramanian R, Bryant DA, Semenov AY, Golbeck JH (2018) Electron-phonon coupling in cyanobacterial photosystem I. J Phys Chem B 122:7943–7955
Frank HA, McLean MB, Sauer K (1979) Triplet states in photosystem I of spinach chloroplasts and subchloroplast particles. Proc Natl Acad Sci USA 76:5124
Fromme P, Jordan P, Krauss N (2001) Structure of photosystem I. Biochim Biophys Acta 1507:5–31
Gast P, Swarthoff T, Ebskamp FCR, Hoff AJ (1983) Evidence for a new early acceptor in Photosystem I of plants. An ESR investigation of reaction center triplet yield and of the reduced intermediary acceptors. Biochim Biophys Acta 722:163–175
Golbeck JH, Bryant DA (1991) Photosystem I. Curr Top Genet 16:83–177
Guergova-Kuras M, Boudreaux B, Joliot A, Joliot P, Redding K (2001) Evidence for two active branches for electron transfer in photosystem I. Proc Natl Acad Sci USA 98:4437–4442
Gunner MR, Madeo J, Zhu Z (2008) Modification of quinone electrochemistry by the proteins in the biological electron transfer chains: examples from photosynthetic reaction centers. J Bioenergy Biomembr 40:509
Hastings G (2015) Vibrational spectroscopy of photosystem I. Biochim Biophys Acta 1847:55–68
Hastings G, Hoshina S, Webber AN, Blankenship RE (1995) Universality of energy and electron transfer processes in photosystem I. Biochemistry 34:15512–15522
Hastings G, Kleinherenbrink FAM, Lin S, McHugh TJ, Blankenship RE (1994) Observation of the reduction and reoxidation of the primary electron acceptor in photosystem I. Biochemistry 33:3193–3200
Hastings G, Makita H, Agarwala N, Rohani L, Shen G, Bryant DA (2019) Fourier transform visible and infrared difference spectroscopy for the study of P700 in photosystem I from Fischerella thermalis PCC 7521 cells grown under white light and far-red light: Evidence that the A–1 cofactor is chlorophyll f. Biochim Biophys Acta 1860:452–460
Ishikita H, Knapp E-W (2003) Redox potential of quinones in both electron transfer branches of photosystem I. J Biol Chem 278:52002–52011
Ivashin N, Larsson S (2003) Electron transfer pathways in photosystem I reaction centers. Chem Phys Lett 375:383–387
Joliot P, Joliot A (1999) In vivo analysis of the electron transfer within photosystem I: are the two phylloquinones involved? Biochemistry 38:11130–11136
Joliot P, Joliot A (2006) Cyclic electron flow in C3 plants. Biochim Biophys Acta 1757:362–368
Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krausz N (2001) Three-dimensional structure of cyanobacterial photosystem I at 2.5A resolution. Nature 411:909–917
Jordan R, Nessau U, Schlodder E (1998) Charge recombination between the reduced iron-sulphur clusters and P700+. In: Garab G (ed) Photosynthesis: mechanisms and effects. Springer, Dordrecht, pp 663–666
Karyagina I et al (2007) Contributions of the protein environment to the midpoint potentials of the A1 phylloquinones and the FX iron−sulfur cluster in photosystem I. Biochemistry 46:10804–10816
Kawashima K, Ishikita H (2017) Structural factors that alter the redox potential of quinones in cyanobacterial and plant photosystem I. Biochemistry 56:3019–3028
Lefebvre-Legendre L, Rappaport F, Finazzi G, Ceol M, Grivet C, Hopfgartner G, Rochaix J-D (2007) Loss of phylloquinone in chlamydomonas affects plastoquinone pool size and photosystem II synthesis. J Biol Chem 282:13250–13263
Lüneberg J, Fromme P, Jekow P, Schlodder E (1994) Spectroscopic characterization of PS I core complexes from thermophilic Synechococcus sp: Identical reoxidation kinetics of A−1 before and after removal of theiron-sulfur-clusters FA and FB. FEBS Lett 338:197–202
Makita H, Hastings G (2015) Directionality of electron transfer in cyanobacterial photosystem I at 298 and 77 K. Febs Lett 589:1412–1417
Makita H, Hastings G (2016a) Modeling electron transfer in photosystem I. Biochim Biophys Acta BBA 1857:723–733
Makita H, Hastings G (2016b) Time-resolved visible and infrared absorption spectroscopy data obtained using photosystem I particles with non-native quinones incorporated into the A1 binding site. Data Brief 7:1463–1468
Makita H, Hastings G (2017) Inverted-region electron transfer as a mechanism for enhancing photosynthetic solar energy conversion efficiency. Proc Natl Acad Sci USA 114:9267–9272
Makita H, Hastings G (2018) Photosystem I with benzoquinone analogues incorporated into the A1 binding site. Photosynth Res 137:85–93
Makita H, Hastings G (2019) Time-Resolved Step-Scan FTIR Difference Spectroscopy for the Study of the P700 Triplet State in Photosystem I. Frontiers in Science, Technology, Engineering and Mathematics 3
Makita H, Rohani L, Zhao N, Hastings G (2017) Quinones in the A1 binding site in photosystem I studied using time-resolved FTIR difference spectroscopy. Biochim Biophys Acta 1858:804–813
Makita H, Zhao N, Hastings G (2015) Time-resolved visible and infrared difference spectroscopy for the study of photosystem I with different quinones incorporated into the A1 binding site. Biochim Biophys Acta BBA 1847:343–354
Malavath T, Caspy I, Netzer-El SY, Klaiman D, Nelson N (2018) Structure and function of wild-type and subunit-depleted photosystem I in Synechocystis. Biochim Biophys Acta 1859:645–654
Mazor Y, Borovikova A, Caspy I, Nelson N (2017) Structure of the plant photosystem I supercomplex at 2.6 Å resolution. Nat Plants 3:17014
McConnell MD, Cowgill JB, Baker PL, Rappaport F, Redding KE (2011) Double reduction of plastoquinone to plastoquinol in photosystem 1. Biochemistry 50:11034–11046
Milanovsky GE, Petrova AA, Cherepanov DA, Semenov AY (2017) Kinetic modeling of electron transfer reactions in photosystem I complexes of various structures with substituted quinone acceptors. Photosynth Res 133:185–199
Moser C, Dutton PL (2006) Application of marcus theory to photosystem I electron transfer. In: Golbeck J (ed) Photosystem I advances in photosynthesis and respiration, vol 24. Springer, Dordrecht, pp 583–594
Okamura MY, Paddock ML, Graige MS, Feher G (2000) Proton and electron transfer in bacterial reaction centers. Biochim Biophys Acta 1458:148–163
Ozawa S, Kosugi M, Kashino Y, Sugimura T, Takahashi Y (2012) 5′-Monohydroxyphylloquinone is the dominant naphthoquinone of PSI in the green alga Chlamydomonas reinhardtii. Plant Cell Physiol 53:237–243
Poluektov OG, Niklas J, Utschig LM (2019) Spin-correlated radical pairs as quantum sensors of bidirectional ET mechanisms in photosystem I. J Phys Chem B 123:7536–7544
Poluektov OG, Utschig LM (2015) Directionality of electron transfer in type I reaction center proteins: high-frequency EPR study of PS I with removed iron-sulfur centers. J Phys Chem B 119:13771–13776
Qin X et al (2019) Structure of a green algal photosystem I in complex with a large number of light-harvesting complex I subunits. Nat Plants 5:263–272
Rohani L, Makita H, Levitz A, Henary M, Hastings G (2019) Calculated vibrational properties of semiquinones in the A1 binding site in photosystem I. Biochim Biophys Acta 1860:699–707
Rutherford AW, Sétif P (1990) Orientation of P700, the primary electron donor of Photosystem I. Biochim Biophys Acta 1019:128–132
Saito K, Rutherford AW, Ishikita H (2013) Mechanism of proton-coupled quinone reduction in Photosystem II. Proc Natl Acad Sci USA 110:954–959
Santabarbara S, Casazza AP (2019) Kinetics and energetics of phylloquinone reduction in photosystem I: insight from modeling of the site directed mutants. Front Plant Sci 10:852
Santabarbara S, Casazza AP, Hastings G (2019) Modelling electron transfer in photosystem I: limits and perspectives. Physiol Plant 166:73–87
Santabarbara S, Heathcote P, Evans MCW (2005) Modelling of the electron transfer reactions in Photosystem I by electron tunnelling theory: the phylloquinones bound to the PsaA and the PsaB reaction centre subunits of PS I are almost isoenergetic to the iron–sulfur cluster FX. Biochim Biophys Acta 1708:283–310
Santabarbara S et al (2010) Interquinone electron transfer in photosystem I as evidenced by altering the hydrogen bond strength to the phylloquinone(s). J Phys Chem B 114:9300–9312
Schlodder E, Falkenberg K, Gergeleit M, Brettel K (1998) Temperature dependence of forward and reverse electron transfer from A1-, the reduced secondary electron acceptor in photosystem I. Biochemistry 37:9466–9476
Schlodder E, Paul A, Cetin M (2001) Triplet states in photosystem I complexes from Synechococcus elongatus. In: 12th International Congress on Photosynthesis, Brisbane, Australia, August 18–23. Photosynthesis Research
Setif P, Brettel K (1993) Forward electron transfer from phylloquinone A1 to iron-sulfur centers in spinach photosystem I. Biochemistry 32:7846–7854
Shen G et al (2002) Assembly of photosystem I. II. Rubredoxin is required for the in vivo assembly of FX in Synechococcus sp PCC 7002 as shown by optical and EPR spectroscopy. J Biol Chem 277(23):20355–20366
Shinkarev VP, Zybailov B, Vassiliev IR, Golbeck JH (2002) Modeling of the P700+ charge recombination kinetics with phylloquinone and plastoquinone-9 in the A1 site of photosystem I. Biophys J 83:2885–2897
Sieckmann I, Brettel K, Bock C, van der Est A, Stehlik D (1993) Transient electron paramagnetic resonance of the triplet state of P700 in photosystem I: evidence for triplet delocalization at room temperature. Biochemistry 32:4842–4847
Sivakumar V, Wang R, Hastings G (2005) A1 reduction in intact cyanobacterial photosystem I particles studied by time-resolved step-scan Fourier transform infrared difference spectroscopy and isotope labeling. Biochemistry 44:1880–1893
Snellenburg JJ, Laptenok SP, Seger R, Mullen KM, van Stokkum IHM (2012) Glotaran: a Java-based Graphical User Interface For The R-package TIMP. J Stat Softw 49:1–22
Srinivasan N, Golbeck JH (2009) Protein–cofactor interactions in bioenergetic complexes: the role of the A1A and A1B phylloquinones in Photosystem I. Biochim Biophys Acta 1787:1057–1088
Suga M, Ozawa S-I, Yoshida-Motomura K, Akita F, Miyazaki N, Takahashi Y (2019) Structure of the green algal photosystem I supercomplex with a decameric light-harvesting complex I. Nat Plants 5:626–636
Walker D (1993) Energy, plants and man, 2nd edn. University Science Books, Brighton
Wang R, Sivakumar V, Johnson TW, Hastings G (2004) FTIR difference spectroscopy in combination with isotope labeling for identification of the carbonyl modes of P700 and P700+ in photosystem I. Biophys J 86:1061–1073
Webber AN, Lubitz W (2001) P700: the primary electron donor of photosystem I. Biochim Biophys Acta 1507:61–79
Xu W et al (2003) Electron transfer in cyanobacterial photosystem I - I. Physiological and spectroscopic characterization of site-directed mutants in a putative electron transfer pathway from A(0) through A(1) to F-x. J Biol Chem 278:27864–27875
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC-0017937 to GH. WX acknowledges support from the National Science Foundation’s EPSCoR Program and Louisiana RCS Program (NSF(2010)-PFUND-217 and LEQSF(2013-16)-RD-A15). The statements made herein are solely the responsibility of the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Agarwala, N., Makita, H., Luo, L. et al. Reversible inhibition and reactivation of electron transfer in photosystem I. Photosynth Res 145, 97–109 (2020). https://doi.org/10.1007/s11120-020-00760-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-020-00760-9