
Abstract
In a society where women often want successful careers and equal opportunities to men, the early nature of ovarian aging often forces women to make difficult life choices between career and family development. Fertility in women begins to decline after the age of 37 years and it is rare for pregnancies to occur after 45. This reproductive decline in women is inevitable and culminates in menopause, which is a major driver of age-related diseases. In a world where biomedical advances are leading to modifiable biological outcomes, it is time to focus on mitigating female reproductive senescence to maintain fertility and preserve age-related hormonal functions, with the goal of providing increased life choices and enhancing healthspan. To date, reproductive longevity research remains an understudied field. More needs to be done to unravel the biology of the ovarian follicles, which are the functional units of reproductive lifespan and are comprised of cell types including the oocyte (female gamete) and a group of specialized supporting somatic cells. Biological attempts to maintain the quality and quantity of follicles in animal models through manipulating pathways involved in aging can potentially prolong female reproductive lifespan and healthspan. Here, we summarize the molecular events driving ovarian aging and menopause and the interventional strategies to offset these events. Developing solutions to female reproductive senescence will open doors to discover ways to enhance true healthy longevity for both men and women.
Similar content being viewed by others
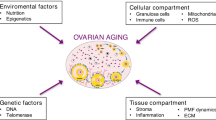

Introduction
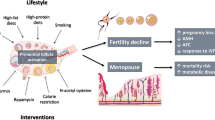
In women, the inevitability of reproductive decline is accepted as a natural phenomenon, with consequences on the life choices that are made. Fertility begins to significantly decline after approximately the age of 37 years old (although considerable variation exists) and it is rare for pregnancy to occur after 45 years old. Moreover, menopause is a driver of age-related diseases. In a society where women often want to (and deserve to) have successful careers and equal opportunities to men, prevailing biology often forces choices between sub-optimal outcomes. In a world where biomedical advances are increasingly leading to modifiable biological outcomes, it is time to focus on female reproductive decline as a modifiable event, where maintenance of fertility and preservation of age-related hormonal functions are emphasized with the goal of providing increased life choices and healthspan.
Notably, women live longer than men in most parts of the world — an average of 4–7 years longer in developed countries.1,2 Yet, the female gonad, the ovary, ages exceptionally early and rapidly before any other parts of the female body system.3,4,5,6 Each woman is born with about 1–2 million oocytes in the form of primordial follicles in her ovaries. After birth, this pool of follicles declines gradually and continuously. This depletion process is accelerated and coupled with a decrease in oocyte quality after about 31 years of age,7 leading to a gradual loss of fecundity. At the same time, the production of the main ovarian gonadal steroid family, estrogens, declines with the depletion of ovarian follicles. Ultimately, when the number of ovarian follicles drops below 1000,7 the woman reaches natural sterility, also known as menopause, which occurs at an approximate age of 50 years.3,4,5,6,8,9
With increased longevity, women spend on average, nearly 40% of their lives in menopause, which is a clinical phenomenon whereby the number of ovarian follicles is so low that there are insufficient levels of estrogens to bring about further ovarian activity and stimulation of the womb lining to result in cyclical menstrual bleeding. This results in the cessation of periods. If a woman aged over 45 years has no spontaneous periods consecutively (and not pregnant) for more than 12 months, she is deemed to have reached clinical menopause. Unfortunately, 1% of all women suffer from a condition known as premature ovarian insufficiency (POI),10 a state whereby the end of reproductive lifespan occurs before they are 40 years old due to a premature and irreversible loss of ovarian follicles. Importantly, several age-related chronic diseases are disproportionately affected by the onset of menopause.11,12 For instance, there are heightened risks of neurocognitive decline,13 cardiovascular diseases (CVDs),14 metabolic dysfunction,15 sarcopenia,16 insulin resistance, osteoporosis11 and sexual dysfunction. In addition to the permanent loss of fertility, women are at increased susceptibility to cardio-metabolic diseases, leading to premature mortality.14,17
The ovarian follicle as the basic functional unit of reproductive lifespan and healthspan
Mammalian ovaries, with the ovarian follicles serving as the functional units, work in sync with the mature hypothalamic-pituitary system, known as the hypothalamic-pituitary-ovarian axis, regulate menstrual cycles and govern the reproductive lifespan and healthspan of a woman (Fig. 1). During the menstrual cycle, the ovarian follicles support the maturation of oocytes, which are the female gametes carrying all genetic information and storing nutrients essential for embryo development upon successful fertilization. An enhanced ovarian follicle pool and better follicular quality thus indicate a longer reproductive lifespan.18

Along the hypothalamic-pituitary-ovarian axis, the hypothalamus secretes gonadotropin-releasing hormone (GnRH) which travels down to stimulate the pituitary gland which in turn secretes follicle stimulating hormone (FSH) and luteinizing hormone (LH). FSH and LH reach the ovaries in the bloodstream to signal the development of ovarian follicles to produce estrogen and progesterone during the follicular and luteal phase of the menstrual cycle. Estrogen rises steadily via a positive feedback loop to result in LH surge from the anterior pituitary gland, leading to ovulation. In a “young ovary”, in each cycle, several resting primordial follicles with immature ova are activated. They develop during folliculogenesis and normally only one ovarian follicle will be “selected” as the dominant follicle and eventually releases the mature ovum (ovulation). The ruptured follicle then transforms into the corpus luteum and degenerates to form the corpus albicans if no implantation occurred. Follicular supporting cells such as the granulosa and theca cells, and the corpus luteum provide endocrine support necessary for ovulation, preparation for implantation and pregnancy, with the release of estrogen and progesterone (symbolized as blue dots). As a woman ages, the finite pool of primordial follicles depletes during each ovulatory cycle and along with constant follicular atresia, results in the degeneration and loss of ovarian follicles and their oocytes, becoming the “aged ovary”. The “aged ovary” shrinks due to age-related fibrosis and releases little estrogen and progesterone due to the extremely low number of viable ovarian follicles.
Ovarian follicles are comprised of somatic cells, known as the granulosa and thecal cells, surround the oocytes and support the growth and development of oocytes. They are the energy hub to produce essential reproductive hormones, progesterone and androgens produced by theca cells and estrogens produced by granulosa cells (Fig. 1).19 These reproductive hormones, especially estrogens, maintain female reproductive health and provide systemic beneficial effects on other biologic systems. Estrogen is the paramount hormone in regulating the female reproductive system and maintaining women’s health, with roles in the cardiovascular system, cognitive function, skin homeostasis and bone metabolism, as reviewed previously.20,21,22,23 Similarly, progesterone, a hormone that is released by the corpus luteum following ovulation and plays a role in maintaining pregnancy, exerts its effects on the reproductive system, but also supports neuroregeneration and neuroprotection.24
The number of ovarian follicles is finite at birth in a baby girl, comprising the entire ovarian reserve for her lifetime.25 As the woman ages, the follicles are constantly recruited for maturation: they either reach ovulation (~400) or (the majority) undergo atresia.26 Along with this decline in follicle number, follicle quality also decreases with age. This has been proposed to be caused by abnormalities in meiotic spindle assembly and chromosomal distribution in oocytes, as well as mitochondrial dysfunction in both oocytes and the surrounding follicular cells. These biological changes are associated with advancing maternal age and accompanied by declining ovarian steroidogenesis functions due to dysfunctional ovarian folliculogenesis.27,28,29,30,31,32,33 Notably, even in young women with diminished ovarian reserve, the chances of achieving high-quality embryos and successful pregnancy in in vitro fertilization (IVF) have been shown to be much greater compared to older women, despite obtaining similar numbers of eggs.34 Thus, both quality and quantity of ovarian follicles are important, and their irrevocable decline will result in the end of a woman’s reproductive lifespan and, ultimately, a decline in overall health. Therefore, if a woman is born with a very low number of ovarian follicles, or experiences faster depletion of healthy follicles, she will reach menopause earlier. This contrasts with men, who continue to produce spermatozoa (for fertility) and androgens in their testes throughout their lives, albeit with lower quantity and quality as they age.
Menopause: an evolutionary vestigial inheritance?
Menopause appears to decouple reproductive from somatic lifespan in women, leading to reproductive senescence primarily due to a sudden deprivation of serum estrogens.35 Furthermore, menopause is extremely rare and only known to be present in 2–3 species of mammals; humans being (Homo sapiens) is the only terrestrial mammal with menopause (Fig. 2).36 Interestingly, these species are associated with a longer lifespan as compared to most without a long post-reproductive lifespan.

The human female is one of a few selected species of mammals with a significant post-reproductive lifespan. The significance of menopause is extensively being debated on whether it is a vestigial evolutionary inheritance or has crucial implication in conferring evolutionary survival of the fittest to our early days’ ancestors as been reproduced from Ellis et al.36 (Open access license: https://creativecommons.org/licenses/by/4.0/. Image legend has been shifted to the top left of the image from the original image).
Beyond the permanent loss of fertility, the hypoestrogenic environment due to menopause is a profound accelerator of aging in women. Importantly, the age of menopause is potentially hereditary, with genome-wide association studies demonstrating the presence of genetic variants linked to menopause that are involved in DNA repair and maintenance. One genetic variant is also linked to systemic aging.10 Interestingly, the human ovary, is thought to be the first organ to decline in function with age. The evolutionary history of menopause is thought to be attributed to the “Grandmother Hypothesis”,37,38 whereby energy expended for reproduction is ceased and redirected to tending to their young and future generations to sustain survival of the larger group, as studied in the menopausal female killer whale (Orcinus orca).39 As modern humans have gained longevity due to the shift from prehistoric ancestor roles of hunters-gatherers to current times of technological advancement and enhanced lifespan, menopause may be seen as a vestigial inheritance from our ancestors. Menopause itself, was only coined in the 1820’s, by French physician Charles-Pierre-Louis de Gardanne.40 At that time, the average female lifespan was around 40–50 years of age, which is congruent to what is currently known as age of menopause in women which is ~49–50 years of age.41
Until relatively recent times, unhygienic sanitation and lifestyle coupled with the lack of access of quality healthcare, including treatment of infectious diseases, and a range of other factors contributed to early mortality in a non-gender selective manner.42,43,44,45 The relative differences between reproductive lifespan and overall lifespan were likely to be small on an evolutionary timeframe during that period; however, with societal and medical improvements these may have result in large differences between reproductive lifespan and healthspan in women from an evolutionary perspective.
There are selected mammals that have extended reproductive lifespan46 such as the fin whale (Balaenoptera physalus) and elephant (Loxodonta) (Fig. 2), with longevity closely linked to the homogametic sex, as compared to the heterogametic sex. Interestingly, however, as compared to humans’ closest relatives such as chimpanzees (Pan troglodytes),47 it seems that the duration of reproductive lifespan is somewhat conserved. Yet the lifespan of chimpanzee regardless in the wild or captivity is far shorter.48 In contrast, prolonged human (Homo sapiens) reproductive age is linked to longevity.49 The conservation of energy due to lower resting metabolic rate during menopause,50 may have contributed to the longevity in humans, as compared to our closer vertebrate species, while this may not necessarily have translated to healthspan in women with increasing health problems associated with earlier menopause and after the onset of menopause.41
Pregnancies in women of advanced maternal age are linked to increased risks of obstetrical complications in the mother, such as gestational diabetes, pre-eclampsia, hypertension, and increased risks of fetal problems such as aneuploidy, e.g., Down’s Syndrome and other congenital malformations.51,52,53,54,55 This is associated with the decline in ovarian follicle quality, which results in oocytes of poorer quality during maternal aging. Therefore, reproductive longevity research would have to address both the extension of reproductive lifespan and the improvement of reproductive healthspan, such that women of advanced maternal age can achieve healthy pregnancies and healthy babies.
Reproductive senescence, hypoestrogenism and inflammation as the driver of aging
Indeed, humans are just a handful of species with an evolutionary divergence of somatic senescence from reproductive senescence, with reaching menopause as the significant driver of aging in women. As discussed above, the deprivation of estrogens from the ovarian follicles dictates this reproductive inevitability. Estrogen confers cardio-protective effects, through improving circulation and vascular health, maintaining oxidative balance, while reducing fibrosis and arterial stiffness in the female vasculature (Fig. 3).56,57,58,59,60,61 In preclinical models, estrogen acts as an anti-inflammatory agent, retarding CVD onset and progression, and this appears to be both sexually dimorphic and age sensitive, which explains the lower risk of CVD in premenopausal women compared to men and postmenopausal women.62 Epidemiological evidence correlating chronic diseases to the onset of menopause suggests that women in the reproductive age group are less prone to these comorbidities when compared to men.63,64,65,66 Consequently, with the postmenopausal decline in estrogen levels, women become predisposed to chronic inflammatory conditions such as atherosclerosis and type 2 diabetes.64,65,66,67,68,69 Estrogen directly binds to the estrogen receptors (ERs), ERα or ERβ,70 leading to their translocation into the nucleus to target genes driven by promoters containing estrogen responsive elements (EREs) and disrupting the inflammatory cascade by preventing NF-κB transcriptional activation.71 Disruption of NF-κB has a profound effect on the synthesis and secretion of pro-inflammatory cytokines such as TNFα, IL6, and IL1β. Furthermore, estrogen (exogenous in vivo replacement of 17β-Estradiol, but not progesterone) modulates the cell surface expression of Toll-like receptor 4 (TLR4) in macrophages, which is critical to the pro-inflammatory polarization (M1) phenotype and accompanying cytokine secretion.72,73

However, the biological significance of the post-reproductive lifespan in women remains unknown.
Reproductive aging and inflammation on musculoskeletal health
Expression of ERs occurs in nearly all musculoskeletal and connective tissues, such as ligament and tendons.74,75 As the expression of ERs is ubiquitous in satellite cells (adult muscle stem cells) of muscle fibers, estrogen stimulates the activation and proliferation of these quiescent stem cells (satellite cells) during muscle repair or injury.76 Although both ERα and ERβ are present, ERα contributes to the critical signaling role of muscle stem cell maintenance and muscle regeneration in women.77 Systematically, chronic inflammation also affects the rejuvenation capability of satellite cells.78 In contrast, while estrogen is able to improve muscle mass and function, female athletes are known to experience significantly higher incidences of anterior cruciate ligament ruptures (due to increased laxity) during the different phases of the menstrual cycle due to the high estrogen levels, when compared to their male counterparts,78 whereas, menopause is linked to an elevated risk of musculoskeletal trauma and sarcopenia. This implies that, while estrogen is beneficial directly towards muscle health, it could reduce the stiffness of connective tissues, thereby reducing function and promoting injury.74
Bone mass integrity is maintained through the balance between bone resorption by osteoclasts and bone formation by osteoblasts.79 Osteoporosis is preceded by the micro-architectural loss of bone mass, which gradually reduces the ability of the bone to support body weight. Postmenopausal women are at higher risk of developing osteoporosis, and they form the largest number of osteoporotic cases.80,81 Serum estrogen levels are directly related to bone mineral density and inversely linked to bone fracture risks.82 Fundamentally, a hypoestrogenic environment tilts the balance of bone towards resorption, as estrogen exerts profound effects on the development and survivability of both osteoblasts and osteoclasts.83 Estrogen stimulates the maturation of osteoblasts through upregulation of the TGFβ signaling pathway, which is an apoptotic inducer through Fas ligand in osteoclasts.79,80,84,85 Furthermore, ERs are highly expressed in osteoblastic lineage cells, with ERα being the more prominent receptor. In ERα-knockout female mice, significant bone loss was observed, a phenotype similar to ovariectomized rodents.86,87,88,89 ERα also confers selective suppression of the receptor activator of NF-κβ (RANKL), a critical cytokine in bone resorption that is secreted by hematopoietic and mesenchymal lineages.88
In estrogen deficient states, RANKL upregulation is seen in B lymphocytes.90 Conditional suppression of RANKL in B lymphocytes but not T lymphocytes, ablated the signs of bone loss in mice.90 Nonetheless, ERα expression is also found in T lymphocytes and RANKL is secreted from T lymphocytes as well. Although deletion of RANKL from T lymphocytes did little to protect from bone loss, estrogens enhance T cell activation and proliferation.91 T lymphocytes in return stimulate osteoclast differentiation and bone resorption, while secreting inflammatory cytokines such as TNFα and IL6, which further exacerbate bone loss.91,92,93 T lymphocytes therefore could elicit a RANKL-independent, yet synergistic, pathway in osteolysis through the secretion of TNFα.92 In fact, TNFα is equipotent to RANKL in mediating osteoclastogenesis, further exerting synergistic effects in the presence of RANKL.92 Collectively, evidence from preclinical studies suggest that estrogen deficiency-induced postmenopausal osteoporosis has a strong immunomodulatory component intertwined with sex steroid actions. Therefore, an immunomodulatory effect from the precursors of T and B cells is likely to be implicated in postmenopausal osteoporosis with RANKL being suggested to be the major contributory factor to hypoestrogenism-induced osteoporosis in bone lining cells.88
Calcium makes up the major component in bone that provides strength and structure,94 and the absorption of calcium from the gut is very much reliant on the availability of vitamin D in the body.95 Aging is related to an increase in the production of vitamin D96 but malabsorption of calcium, which has a compounding effect on the onset of osteoporosis.97 Postmenopausal women suffer a negative calcium balance that slowly stabilizes over the years, but remains tilted towards calcium loss.98 As natural aging reduces the efficacy of vitamin D production in the liver and skin, it further aggravates the onset of androgenic-induced osteoporosis in women. Therefore, dietary supplementation of vitamin D and calcium, not either or, is only a temporary solution for postmenopausal women at high risk of osteoporosis. Similarly, data from Women’s Health Initiative (WHI) studies demonstrate that menopausal hormone replacement therapy (HRT)99,100 causes a significant reduction in the incidence of bone fractures in postmenopausal women.99,101 However, the use of menopausal HRT, including transient and lowest dose usage, in mediating benefits on musculoskeletal health needs to be weighed with increased risks of breast cancer and stroke. Several reviews have discussed this issue in more detail.100,102
Reproductive aging and inflammation on cardio-metabolic health
Menopause presents a void in sex hormone-mediated homeostasis of glucose and lipid metabolism, which has profound implications in obesity and glucose impairment. Sex hormones are known to regulate the distribution of visceral fat in humans.103,104,105 Fat distribution is altered in pre- and postmenopausal women, where subcutaneous fat is progressively being displaced by abdominal fat. Interestingly, adipocytes express both ERα and ERβ, yet only ERα is found to be in brown adipocytes.106,107 Brown adipose tissue maintains energy expenditure by enhancing lipid and glucose metabolism.108 It is also part of the endocrine system that secretes adipokines, which regulate inflammatory responses and confer cardio-protection.109,110 Hence, alteration of ERα in animal models leads to central obesity and diabetes, and in humans this change in fat deposition is also associated with the development of CVD risk factors.111,112
During menopause, changes in metabolism include lower energy expenditure, predisposition to central adiposity and an impairment of insulin sensitivity. Clinical evidence is mixed on whether HRT confers CVD protection in postmenopausal women. The Nurse’s Health Study (NHS), a prospective longitudinal study spanning 20 years recruited 121,000 female nurses, with 70,533 being postmenopausal, suggested that HRT significantly reduced the risks of CVD, as compared to non-users.105 In contrast, the Heart and Estrogen/Progesterone Replacement Study (HERS), a randomized controlled trial demonstrated that administering conjugated equine estrogens (CEE) and medroxyprogesterone acetate confers no beneficial effects.102 Likewise, two subsequent randomized controlled trials of the WHI involving either estrogen + progesterone (WHI E + P) (16,608 postmenopausal women were assessed)100 or estrogen alone (WHI E)99 (10,739 postmenopausal women who had undergone hysterectomy were followed) were stopped abruptly following an elevated risk of coronary heart disease in the former trial and an increase in incidence of stroke in the latter. Successive smaller studies, the Danish Osteoporosis Prevention Study (DOPS)113 and Early versus Late Intervention Trial with Estradiol (ELITE),114 were more promising in demonstrating the protective effects of menopausal hormone therapy in reversing the risks of CVD and atherosclerosis. DOPS followed the study population of postmenopausal women for 10 years, and showed that estrogen treatment given to women, significantly suppressed their risks of mortality, CVD, and heart failure. Notably, in DOPS there was no significant elevation of cancer risks.
The optimal period of estrogen rescue is likely to be at the early postmenopausal stage, as demonstrated in ELITE, where there were reduced atherosclerotic events noted at early, but not late stage of postmenopausal intervention. In the Kronos Early Estrogen Prevention Study (KEEPS),115 only relief of vasomotor symptoms was observed, with a beneficial trend in CVD. In DOPS, ELITE and KEEPS, no obvious adverse effects were observed in the use of estrogens. Findings in ELITE reinforced the “Timing Hypothesis”, in which estrogen’s Jekyll and Hyde role is dependent on intervention prior to the onset of erosion of coronary plaque, which can be worsened by hormonal therapy.116 Impairment in glucose metabolism represents a more significant phenotype of menopause. Despite the poor outcome of the HERS and WHI trials in de-escalating the onset of CVD risks, these studies highlighted the significant role that estrogens have in reducing the onset of type-2 diabetes,99,100,102 a significant risk factor for CVD. Additionally, women who underwent estrogen replacement therapy were observed to have lower hemoglobin A1c (HbA1c), a risk factor for diabetes.117,118
Interventions with HRT are directly beneficial to women who suffer from premature POI, while the beneficial effects in women who attain menopause at the expected age of about 50 years likely depend on treatment inception soon after menopause. Potential health benefits by these hormones can be maintained, such as protective effects against CVDs,119,120 brain aging121,122 and osteoporosis,123 thus enhancing the women’s healthspan, which is especially true for women who suffer from POI (i.e., menopause before they are 40 years old). It seems that in older women who were more than 5–10 years post menopause and above 60 years of age, HRT can be detrimental and cause increased risks of breast cancer and stroke with long-term use, as demonstrated in the WHI studies, leading to controversies about its use.124,125,126,127 At the same time, several systematic reviews have failed to demonstrate protective effects of HRT against age-related health risks such as CVD events128 and cognitive decline129 in postmenopausal women, as HRT does not reverse processes that have already aged. However, it remains essential for women with premature and early ovarian insufficiency who require HRT for maintenance of their healthspan before the age of natural menopause (49–50 years).
The estimated life expectancy has been steadily improved to an average of 72.6 years (and 79.4 years in developed regions) for human beings.1 However, there has not been an equivalent improvement in healthspan, as the older individuals suffer from frailty and other aging-related medical conditions that affect multiple organs in the body.130,131,132 This is further accentuated by ovarian or reproductive senescence. This partial uncoupling of healthspan from lifespan has raised the idea that for people beyond a certain age, i.e., 65 years old, life extension should not be the primary goal.133,134 Thus, a major need exists to define the critical period for intervention in women to enhance reproductive healthspan and to reduce all-cause morbidity. This likely involves mimicking the beneficial effects of estrogen postmenopausally, while avoiding the associated side effects. This will be one ultimate goal of reproductive longevity research, which will not only attempt to tackle the issue of reproductive lifespan, but also enhance female healthspan and lifespan.
Strategies to delay or even reverse ovarian aging as means to maintain reproductive lifespan and healthspan
Assisted reproductive technology (ART), established in the late 20th century, has become more popular and accessible now11 to circumvent infertility problems due to the loss of oocyte quantity and/or quality to difficulty in fertilization and embryogenesis. Adjunct strategies, such as oocyte and ovarian tissue cryopreservation, have also matured, leading to rising success rates and utility.135,136 However, these strategies do have their limitations as they circumvent, instead of directly targeting, the root cause of fertility decline — ovarian aging. Thus, while allowing a woman to try to conceive, these strategies are unable to guarantee reproductive success as oocyte quality constantly decreases with age and advanced maternal age has become the most common factor for IVF failure.137,138 Furthermore, ART does not restore or maintain the levels and protective effects of ovarian gonadal steroids, such as estrogens.
Given that ovarian follicles are the functional units of ovaries, studies had attempted to enhance ovarian health and prevent reproductive aging through employment of various interventions studied in the context of aging and reported to enhance lifespan in animal models.139 These studies report molecules/compounds that target specific signaling pathways, spanning from antioxidants, precursors of oxidative stress pathways, mitochondrial chain precursors, isoflavones, polyphenol derivatives, and plant-based compounds, to commonly used medications like metformin and, recently, the mammalian target of rapamycin (mTOR) inhibitors (Table 1). Interestingly, most of this exploratory work was performed in rodent models, and results may differ between these models and humans. Rodent models provide key insights into possible human interventions, but follow-up human studies are needed. This is in part because female rodents and women share progressively increasing irregularity in ovulatory cycles (known as estrous cycle in rodents) and increasing fetal aneuploidy, as decline of oocyte quality and quantity becomes imminent with age, although female mice do not experience menopause like women.140 Primate models can be employed to study reproductive senescence, although they are expensive and time-consuming.
Utilizing agents studied in the context of general aging for correction of reproductive aging
Referring to Table 1, antioxidants represent the biggest category of compounds studied for reproductive aging, as the free radical theory has been the classical aging theory that attributes aging phenomena to accumulated cellular oxidative stress.141 According to this theory, the accumulation of reactive oxygen species (ROS) leads to ovarian oxidative stress and changes in ovarian microenvironment, resulting in cellular senescence and a decrease in oocyte quality and quantity.142,143,144,145 Indeed, oxidative stress has been shown to be associated with aging-related oocyte deterioration.146,147,148 In addition, previous studies found an increase in ROS and a decrease in levels of antioxidants in oocytes of older women receiving IVF, which are also associated with ART failure.149,150,151 Antioxidants such as N-acetyl-L-cysteine (NAC),152,153,154,155 flavonoids,156 vitamins C and E157 and coenzyme Q10 (CoQ10) have been tested in rodent models, although many of these agents had other roles in addition to being ROS scavengers (Table 1). Pro-longevity effects of antioxidants in the female reproductive system include maintenance of ovarian reserve, varying improvements of primordial and healthy ovarian follicle counts, a decreased proportion of atretic ovarian follicles and improvement in litter size and estrous cycle regularity. However, the use of antioxidants has also been associated with side effects, including those related to female reproduction, such as long-term disruption of ovarian and uterine functions with pharmacological doses of vitamins C and E.158 In fact, antioxidants such as vitamins A and E were shown to increase mortality in clinical trials.159 This may be explained by potential beneficial functions of oxidative stress in some contexts, for example, reduction of insulin resistance triggered by exercise-induced ROS.160 Additionally, several systematic reviews failed to demonstrate any positive outcome of antioxidant supplementation in the context of general aging159 and other age-related diseases, such as cataract, dementia and CVD.161,162,163,164 In the context of ovarian aging, the in vivo studies on antioxidants were almost exclusively done in relatively young or middle-aged animals and failed to assess the effects in more aged ovaries (from older animals), potentially limiting the translatability to older or even postmenopausal women. Furthermore, only a few antioxidants have been studied in women with age-related ART failure and the results remain highly inconsistent.165,166 The oral supplementation of some antioxidants such as resveratrol was even associated with decreased pregnancy rates, the converse of what was expected.167 Reservatrol also has many aging-associated actions that are independent of its antioxidative effects,168 further complicating the interpretation of these findings.
Manipulation of glucose metabolism to mimic the effects of caloric restriction (CR) has been a focus in longevity studies. CR, the chronic reduction in total calorie intake without malnutrition, robustly improves both healthspan and lifespan in many organisms.169 As CR is not easy to be implemented in real life, genes and pathways involved in longevity mechanisms of CR are being identified and there are multiple candidate compounds that can potentially mimic the longevity effects of CR. Metformin, an approved anti-diabetic drug, achieves a reduction in blood glucose by enhancing peripheral insulin sensitivity and suppressing gluconeogenesis in the liver,170,171 which resemble some metabolic effects of CR.172 Indeed, metformin has demonstrated promising results in enhancing healthspan and lifespan in animal models172,173 and clinical trials.174 In the context of ovarian aging, multiple studies revealed that CR preserved oocyte quality, fertility and/or ovarian reserve in aged female rodents.6,175,176 Similarly, a recent study reported that six months of metformin treatment increased serum estrogen level and follicle quantity and resulted in more regular estrous cycles in normally aged mice.177 Such effects are consistent with another study showing an association between fasting-induced lower blood glucose level and reduced primordial follicular activation in mice.178 However, this observation was not corroborated by another rodent study,179 which found comparable follicle counts between metformin-fed rats and control rats. As there were differences in duration of treatment and rodent species, further investigations are needed to assess the effects of metformin in mammalian ovaries. Importantly, older animals need to be used to examine the true biological effects, as the previous studies were conducted on young rodents. These limitations apply to another study investigating 2-deoxyglucose (2-DG), where young mice and a short treatment duration were employed.180 2-DG is a synthetic glucose analog that competitively inhibits glycolysis, reduces insulin levels and decreases body temperature in rats, and thus has also been considered as a candidate CR mimetic.181 Interestingly, this study180 revealed inhibition of primordial follicle activation by 2-DG, indicating its potential in preservation of primordial ovarian follicles — hence protecting the ovarian reserve. However, the chronic treatment of 2-DG has been shown to cause cardiotoxicity and increased mortality in male rodents.182 Although it remains unknown whether this is also the case in females, such toxicity has decelerated the transition of 2-DG to clinical use.
Mitochondrial dysfunction has been identified as a hallmark for aging and implicated in ovarian aging and infertility, as oocytes are uniquely enriched with mitochondria.183 Aging-related changes in mitochondria, including accumulation of mitochondrial DNA mutations, altered membrane potential and impaired metabolism, undermine mitochondrial functions and are proposed to link to ovarian aging phenotypes.184 Besides the aforementioned antioxidant therapies, such as CoQ10, a combination of vitamins C and E, and flavonoids, aimed to increase nicotinamide adenine dinucleotide (NAD+), an essential cofactor and enzyme substrate in several crucial redox reactions and metabolic pathways which declines with age,185 has been shown to alleviate ovarian aging by improving mitochondrial function. NAD+ supplementation was tested in two recent studies in young and middle-aged mice.186,187 These studies demonstrated that the oocytes were rejuvenated, with enhanced fertility attributable to a reduction in levels of ROS and improvement in ovarian mitochondrial metabolism.186 However, knowledge about mitochondrial boosters including NAD+ precursors remain limited as they were only tested in rodents and in ages up to 14 months in mice. Therefore, mitochondrial boosters are likely to gain more attention in the future, especially given the new findings linking mitochondrial dysfunction to female reproductive aging via impaired NADH/NAD+ redox functions.188 Notably, the sirtuins, a family of NAD+-dependent deacylases and key regulators of aging, have both mitochondrial and non-mitochondrial functions such as DNA repair and inflammatory response.189 Thus, the mechanism of NAD+ supplementation in ovarian longevity may be pleiotropic.
mTOR is a serine/threonine protein complex that is sensitive to rapamycin and mTOR suppression has been shown to extend lifespan in several species.190,191,192,193,194,195,196,197,198,199 mTOR also coordinates several key cellular signaling and metabolic pathways implicated in follicular development and ovarian aging.200 Follicular mTOR signaling stimulates primordial follicle activation, which is the start of post-puberty follicular development and directly determines the follicular reserve and reproductive lifespan. It was demonstrated in vivo that ovarian mTOR overactivation triggers premature follicular activation and early follicle depletion,201,202,203,204,205 together with more atretic follicles and degenerated oocytes.205 At the same time, AKT-mediated mTOR signaling was found to regulate granulosa cell autophagy in folliculogenesis and its inhibition was found to induce follicular atresia through promoting granulosa cell autophagy.206 Studies in rodent models4,207,208,209,210 have demonstrated that inhibition of mTOR signaling can improve ovarian reserve, as indicated by the increase in primordial follicle counts and extension of reproductive lifespan. There has also been some evidence indicating a decrease in the absolute atretic follicle count,210,211 probably due to overall suppression of follicular activation. Unfortunately, rapamycin was also found to cause disruption of estrous cycles and loss of fertility, due to a cessation of follicle activation after prolonged use of more than 4 months. This was expected because mTOR signaling plays a strong role in follicular growth as discussed. Nevertheless, this disruption seems to be reversible, as a recent study4 devised a 2-week transient rapamycin treatment that successfully restored follicular development and estrus cycles in post-treatment mice, observing an improvement in the treated mice’s reproductive capacity and ovarian lifespan, regardless of their ages at treatment up to 16 months of age. This approach may be consistent with the longevity effects of rapamycin, where a transient treatment even in relatively late age is sufficient to extend healthspan and lifespan.211 mTOR inhibitors could potentially be useful in the treatment of reproductive aging, but further studies are required due to considerations on utilizing mTOR inhibitors as strategic short-term treatment modality to achieve long-term protective effects against ovarian aging in women. Apart from inhibition, as mTOR plays a crucial role in reproduction, its chronic activation in reproductive aging should be extensively examined with small molecules such as MHY1485212 and 3BDO,213 which are potent mTOR activators.
Other options such as dehydroepiandrosterone (DHEA), a precursor for estrogen in peripheral tissues, and melatonin, a sleep-promoting hormone with antioxidant properties, have shown some success in extending reproductive lifespan and/or improving ovarian responses in both animal and clinical studies. The concentration of DHEA decreases progressively with age.214 As an essential prohormone in ovarian steroidogenesis, DHEA was suggested to promote gonadotropin action and ovarian functions, as indicated by the improvement in anti-Mullerian hormone (AMH) level and IVF outcomes in women with diminished ovarian reserve.215,216 This is further supported by an ovine study that also showed an increase in follicular AMH expression and granulosa cell proliferation after DHEA treatment.217 Yet, the same study also found an accelerated primordial follicle activation and an increase in antral follicle count, which raises the concern of pre-mature follicle depletion especially upon long-term administration. In contrast, melatonin, also an endogenous hormone in the body, has more extensive evidence in delaying ovarian aging. In rodents, it was found to improve oocyte quality and quantity and maintain follicular reserve.217,218,219,220,221,222,223,224 A recent study223 demonstrated the endogenous role of melatonin in suppressing follicular overactivation and atresia and delaying ovarian aging. It was proposed that melatonin acts through its antioxidant capacity and the MT1/AMPK pathway,220,221,223 though further mechanistic study is needed. However, these studies varied greatly in age of subjects, end-point measurements and duration of treatment. For instance, supplementation of melatonin was conducted mostly on very young mice daily for half to one year,218,220,221,222 which makes the translation of this treatment in clinical settings impractical due to the widespread metabolic and physiological activities that melatonin exerts on the body, raising concerns on any unanticipated adverse effects which can occur due to prolonged administration. Additionally, several clinical trials investigating oral melatonin administration in women with infertility or IVF failures have demonstrated conflicting results, which are summarized in Table 1.224,225,226,227,228,229
A list of compounds tested in this context are listed in Table 1. However, many of the studies are primarily observational and although many compounds are purported to “extend reproductive lifespan”, results were often derived from findings in relatively young animals treated for varying durations. Furthermore, the exact mechanisms of actions for these interventions still lack clarity, and the therapeutic targets as well as their sites of action are yet to be elucidated. There remains a lot to be studied in understanding the underlying biological mechanisms governing reproductive aging at the ovarian follicle level and in the surrounding ovarian environment.
Future directions
There is a compelling need to develop new strategies to offer women to:
-
(1)
Choose when she can fulfill her hopes of childbearing, once the mysteries behind ovarian senescence are better understood.
-
(2)
Protect and enhance her reproductive lifespan and healthspan using innovative solutions that are safe, with minimal or no side effects, to overcome the negative consequences of reproductive aging.
-
(3)
Advocate for reproductive longevity and equality in women, and for science to assist in tackling the inevitability of reproductive aging for future generations.
New intervention strategies should target the root cause of the reproductive aging process, ideally at a specific organ-cell-molecular target level, and be relatively short-term and have minimal side effects.
Conclusion
Current knowledge on the mechanisms of ovarian or reproductive aging remains limited. Clinical management of fertility issues is always confounded by maternal age with reproductive outcomes limited by the age of women, i.e., older age and reduced ovarian reserves result in higher risks of reproductive failure.230 Few options are available for the management of women with POI and natural menopause because we still do not fully understand the biology of ovarian senescence in women. The balance between reproductive lifespan (fertility) and healthspan (general good health) will be the long-term goal and imperative as part of the movement for gender equality. A woman should not be limited to her reproductive lifespan and “accept” what biology has imposed on her reproductive choices and life choices, in addition to her decline of healthspan later in life. Women represent half of the world population, and they also carry the future of the world. Reproductive longevity in women also determines healthy longevity in their male relatives,231 which emphasizes the importance of solving female reproductive senescence and will open doors to discover ways to enhance true healthy longevity for both men and women.
References
United Nations Department of Economic and Social Affairs. World Mortality 2019 Data Booklet (United Nations Department of Economic and Social Affairs, 2019).
Ginter, E. & Simko, V. Women live longer than men. Bratisl. Lek. Listy 114, 45–49 (2013).
Li, Q. et al. Current understanding of ovarian aging. Sci. China Life Sci. 55, 659–669 (2012).
Dou, X. et al. Short-term rapamycin treatment increases ovarian lifespan in young and middle-aged female mice. Aging Cell 16, 825–836 (2017).
Zhang, J. et al. Can ovarian aging be delayed by pharmacological strategies? Aging 11, 817–832 (2019).
Garcia, D. N. et al. Effect of caloric restriction and rapamycin on ovarian aging in mice. Geroscience 41, 395–408 (2019).
Faddy, M. J., Gosden, R. G., Gougeon, A., Richardson, S. J. & Nelson, J. F. Accelerated disappearance of ovarian follicles in mid-life: implications for forecasting menopause. Hum. Reprod. 7, 1342–1346 (1992).
Broekmans, F. J., Soules, M. R. & Fauser, B. C. Ovarian aging: mechanisms and clinical consequences. Endocr. Rev. 30, 465–493 (2009).
Santoro, N. The menopausal transition. Am. J. Med. 118(Suppl 12B), 8–13 (2005).
Laven, J. S. Genetics of early and normal menopause. Semin. Reprod. Med. 33, 377–383 (2015).
Zhu, D., Li, X., Macrae, V. E., Simoncini, T. & Fu, X. Extragonadal effects of follicle-stimulating hormone on osteoporosis and cardiovascular disease in women during menopausal transition. Trends Endocrinol. Metab. 29, 571–580 (2018).
Lizcano, F. & Guzman, G. Estrogen deficiency and the origin of obesity during menopause. Biomed. Res. Int. 2014, 757461 (2014).
Song, X. et al. Reproductive and hormonal factors and risk of cognitive impairment among Singapore Chinese women. Am. J. Obstet. Gynecol. 223, 410.e411–410.e423 (2020).
Muka, T. et al. Association of age at onset of menopause and time since onset of menopause with cardiovascular outcomes, intermediate vascular traits, and all-cause mortality: a systematic review and meta-analysis. JAMA Cardiol. 1, 767–776 (2016).
Thong, E. P., Codner, E., Laven, J. S. E. & Teede, H. Diabetes: a metabolic and reproductive disorder in women. Lancet Diabetes Endocrinol. 8, 134–149 (2020).
Geraci, A. et al. Sarcopenia and menopause: the role of estradiol. Front. Endocrinol. 12, 682012 (2021).
Zhu, D. et al. Age at natural menopause and risk of incident cardiovascular disease: a pooled analysis of individual patient data. Lancet Public Health 4, e553–e564 (2019).
Scheffer, G. J. et al. The number of antral follicles in normal women with proven fertility is the best reflection of reproductive age. Hum. Reprod. 18, 700–706 (2003).
Drummond, A. E. The role of steroids in follicular growth. Reprod. Biol. Endocrinol. 4, 16–16 (2006).
Khosla, S., Oursler, M. J. & Monroe, D. G. Estrogen and the skeleton. Trends Endocrinol. Metab. 23, 576–581 (2012).
Russell, J. K., Jones, C. K. & Newhouse, P. A. The role of estrogen in brain and cognitive aging. Neurotherapeutics 16, 649–665 (2019).
Wilkinson, H. N. & Hardman, M. J. The role of estrogen in cutaneous ageing and repair. Maturitas 103, 60–64 (2017).
Knowlton, A. A. & Lee, A. R. Estrogen and the cardiovascular system. Pharmacol. Ther. 135, 54–70 (2012).
Sitruk-Ware, R. & El-Etr, M. Progesterone and related progestins: potential new health benefits. Climacteric 16, 69–78 (2013).
Gleicher, N., Weghofer, A. & Barad, D. H. Defining ovarian reserve to better understand ovarian aging. Reprod. Biol. Endocrinol. 9, 23 (2011).
Mesiano, S., Jones, E. E. Chapter 55: The female reproductive system. In: Medical physiology 3rd ed. (eds Boron, W., Boulpaep, E.) (Philadelphia, Elsevier, 2017).
Pellestor, F., Anahory, T. & Hamamah, S. Effect of maternal age on the frequency of cytogenetic abnormalities in human oocytes. Cytogenet. Genome Res. 111, 206–212 (2005).
Kuliev, A., Cieslak, J. & Verlinsky, Y. Frequency and distribution of chromosome abnormalities in human oocytes. Cytogenet. Genome Res. 111, 193–198 (2005).
Battaglia, D. E., Goodwin, P., Klein, N. A. & Soules, M. R. Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum. Reprod. 11, 2217–2222 (1996).
Liu, Y. et al. Age-related changes in the mitochondria of human mural granulosa cells. Hum. Reprod. 32, 2465–2473 (2017).
Seifer, D. B., DeJesus, V. & Hubbard, K. Mitochondrial deletions in luteinized granulosa cells as a function of age in women undergoing in vitro fertilization. Fertil. Steril. 78, 1046–1048 (2002).
Sreerangaraja Urs, D. B. et al. Mitochondrial function in modulating human granulosa cell steroidogenesis and female fertility. Int. J. Mol. Sci. 21, 3592 (2020).
Tatone, C. et al. Evidence that carbonyl stress by methylglyoxal exposure induces DNA damage and spindle aberrations, affects mitochondrial integrity in mammalian oocytes and contributes to oocyte ageing. Hum. Reprod. 26, 1843–1859 (2011).
Chang, Y., Li, J., Li, X., Liu, H. & Liang, X. Egg quality and pregnancy outcome in young infertile women with diminished ovarian reserve. Med. Sci. Monit. 24, 7279–7284 (2018).
Pollycove, R., Naftolin, F. & Simon, J. A. The evolutionary origin and significance of menopause. Menopause 18, 336–342 (2011).
Ellis, S. et al. Postreproductive lifespans are rare in mammals. Ecol. Evol. 8, 2482–2494 (2018).
Hawkes, K. Human longevity: the grandmother effect. Nature 428, 128–129 (2004).
Hawkes, K., O’Connell, J. F., Jones, N. G., Alvarez, H. & Charnov, E. L. Grandmothering, menopause, and the evolution of human life histories. Proc. Natl. Acad. Sci. USA 95, 1336–1339 (1998).
Whitehead, H. Life history evolution: what does a menopausal killer whale do? Curr. Biol. 25, R225–R227 (2015).
Singh, A., Kaur, S. & Walia, I. A historical perspective on menopause and menopausal age. Bull. Indian Inst. Hist. Med. Hyderabad 32, 121–135 (2002).
Velez, M. P. et al. Age at natural menopause and physical functioning in postmenopausal women: the Canadian Longitudinal Study on Aging. Menopause 26, 958–965 (2019).
Vasold, M. [Mortality in Nuremberg in the 19th century (about 1800 to 1913)]. Wurzbg Medizinhist Mitt 25, 241–338 (2006).
Anderson, R. N. Deaths: leading causes for 2000. Natl. Vital Stat. Rep. 50, 1–85 (2002).
Shaw-Taylor, L. An introduction to the history of infectious diseases, epidemics and the early phases of the long-run decline in mortality. Econ. Hist. Rev. 73, E1–E19 (2020).
Oeppen, J. & Vaupel, J. W. Demography. Broken limits to life expectancy. Science 296, 1029–1031 (2002).
Xirocostas, Z. A., Everingham, S. E. & Moles, A. T. The sex with the reduced sex chromosome dies earlier: a comparison across the tree of life. Biol. Lett. 16, 20190867 (2020).
Hawkes, K. & Smith, K. R. Do women stop early? Similarities in fertility decline in humans and chimpanzees. Ann. N. Y. Acad. Sci. 1204, 43–53 (2010).
Wood, B. M., Watts, D. P., Mitani, J. C. & Langergraber, K. E. Favorable ecological circumstances promote life expectancy in chimpanzees similar to that of human hunter-gatherers. J. Hum. Evol. 105, 41–56 (2017).
Shadyab, A. H. et al. Ages at menarche and menopause and reproductive lifespan as predictors of exceptional longevity in women: the Women’s Health Initiative. Menopause 24, 35–44 (2017).
Hodson, L. et al. Lower resting and total energy expenditure in postmenopausal compared with premenopausal women matched for abdominal obesity. J. Nutr. Sci. 3, e3 (2014).
Vincent-Rohfritsch, A., Le Ray, C., Anselem, O., Cabrol, D. & Goffinet, F. Pregnancy in women aged 43 years or older: maternal and perinatal risks. J. Gynecol. Obstet. Biol. Reprod. 41, 468–475 (2012).
Yogev, Y. et al. Pregnancy outcome at extremely advanced maternal age. Am. J. Obstet. Gynecol. 203, 558.e1–558.e7 (2010).
Sheen, J. J. et al. Maternal age and risk for adverse outcomes. Am. J. Obstet. Gynecol. 219, 390.e1–390.e15 (2018).
Londero, A. P., Rossetti, E., Pittini, C., Cagnacci, A. & Driul, L. Maternal age and the risk of adverse pregnancy outcomes: a retrospective cohort study. BMC Pregnancy Childbirth 19, 261 (2019).
Mikwar, M., MacFarlane, A. J. & Marchetti, F. Mechanisms of oocyte aneuploidy associated with advanced maternal age. Mutat. Res. 785, 108320 (2020).
Babiker, F. A. et al. 17beta-estradiol antagonizes cardiomyocyte hypertrophy by autocrine/paracrine stimulation of a guanylyl cyclase A receptor-cyclic guanosine monophosphate-dependent protein kinase pathway. Circulation 109, 269–276 (2004).
Lagranha, C. J., Deschamps, A., Aponte, A., Steenbergen, C. & Murphy, E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 106, 1681–1691 (2010).
Iorga, A. et al. Rescue of pressure overload-induced heart failure by estrogen therapy. J. Am. Heart Assoc. 5, e002482 (2016).
Pedram, A., Razandi, M., Narayanan, R. & Levin, E. R. Estrogen receptor beta signals to inhibition of cardiac fibrosis. Mol. Cell Endocrinol. 434, 57–68 (2016).
Adams, M. R. et al. Inhibition of coronary artery atherosclerosis by 17-beta estradiol in ovariectomized monkeys. Lack of an effect of added progesterone. Arteriosclerosis 10, 1051–1057 (1990).
Christ, J. P. et al. Estrogen deprivation and cardiovascular disease risk in primary ovarian insufficiency. Fertil. Steril. 109, 594–600.e1 (2018).
Giordano, S. et al. Estrogen and cardiovascular disease: is timing everything? Am. J. Med. Sci. 350, 27–35 (2015).
Spence, R. D. & Voskuhl, R. R. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Front. Neuroendocrinol. 33, 105–115 (2012).
Cooper, G. S. & Stroehla, B. C. The epidemiology of autoimmune diseases. Autoimmun. Rev. 2, 119–125 (2003).
DECODE Study Group. Age- and sex-specific prevalences of diabetes and impaired glucose regulation in 13 European cohorts. Diabetes Care 26, 61–69 (2003).
Qiao, Q. et al. Age- and sex-specific prevalence of diabetes and impaired glucose regulation in 11 Asian cohorts. Diabetes Care 26, 1770–1780 (2003).
Gubbels Bupp, M. R. Sex, the aging immune system, and chronic disease. Cell Immunol. 294, 102–110 (2015).
Vitale, C., Mendelsohn, M. E. & Rosano, G. M. C. Gender differences in the cardiovascular effect of sex hormones. Nat. Rev. Cardiol. 6, 532–542 (2009).
Lerner, D. J. & Kannel, W. B. Patterns of coronary heart disease morbidity and mortality in the sexes: a 26-year follow-up of the Framingham population. Am. Heart J. 111, 383–390 (1986).
Pennell, L. M., Galligan, C. L. & Fish, E. N. Sex affects immunity. J. Autoimmun. 38, J282–J291 (2012).
Ghisletti, S., Meda, C., Maggi, A. & Vegeto, E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol. Cell Biol. 25, 2957–2968 (2005).
Rettew, J. A., Huet, Y. M. & Marriott, I. Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology 150, 3877–3884 (2009).
Rettew, J. A., McCall, S. H. T. & Marriott, I. GPR30/GPER-1 mediates rapid decreases in TLR4 expression on murine macrophages. Mol. Cell Endocrinol. 328, 87–92 (2010).
Chidi-Ogbolu, N. & Baar, K. Effect of estrogen on musculoskeletal performance and injury risk. Front. Physiol. 9, 1834 (2018).
Luo, T. & Kim, J. K. The role of estrogen and estrogen receptors on cardiomyocytes: an overview. Can. J. Cardiol. 32, 1017–1025 (2016).
Keefe, A. C. et al. Muscle stem cells contribute to myofibres in sedentary adult mice. Nat. Commun. 6, 7087 (2015).
Collins, B. C. et al. Estrogen regulates the satellite cell compartment in females. Cell Rep. 28, 368–381.e6 (2019).
Shultz, S. J., Sander, T. C., Kirk, S. E. & Perrin, D. H. Sex differences in knee joint laxity change across the female menstrual cycle. J. Sports Med. Phys. Fitness 45, 594–603 (2005).
Fischer, V. & Haffner-Luntzer, M. Interaction between bone and immune cells: implications for postmenopausal osteoporosis. Semin. Cell Dev. Biol. 123, 14–21 (2022).
Krum, S. A. & Brown, M. Unraveling estrogen action in osteoporosis. Cell Cycle 7, 1348–1352 (2008).
Mehta, J., Kling, J. M. & Manson, J. E. Risks, benefits, and treatment modalities of menopausal hormone therapy: current concepts. Front. Endocrinol. 12, 564781 (2021).
Cauley, J. A. Estrogen and bone health in men and women. Steroids 99, 11–15 (2015).
Borjesson, A. E., Lagerquist, M. K., Windahl, S. H. & Ohlsson, C. The role of estrogen receptor alpha in the regulation of bone and growth plate cartilage. Cell Mol. Life Sci. 70, 4023–4037 (2013).
Krum, S. A. et al. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 27, 535–545 (2008).
Hughes, D. E. et al. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat. Med. 2, 1132–1136 (1996).
Almeida, M. et al. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J. Clin. Invest. 123, 394–404 (2013).
Raisz, L. G. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J. Clin. Invest. 115, 3318–3325 (2005).
Streicher, C. et al. Estrogen regulates bone turnover by targeting RANKL expression in bone lining cells. Sci. Rep. 7, 6460 (2017).
Yousefzadeh, N., Kashfi, K., Jeddi, S. & Ghasemi, A. Ovariectomized rat model of osteoporosis: a practical guide. EXCLI J. 19, 89–107 (2020).
Onal, M. et al. Receptor activator of nuclear factor kappaB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J. Biol. Chem. 287, 29851–29860 (2012).
Mohammad, I. et al. Estrogen receptor alpha contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal. 11, eaap9415 (2018).
Fuller, K., Murphy, C., Kirstein, B., Fox, S. W. & Chambers, T. J. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology 143, 1108–1118 (2002).
Weitzmann, M. N. & Ofotokun, I. Physiological and pathophysiological bone turnover — role of the immune system. Nat. Rev. Endocrinol. 12, 518–532 (2016).
Vannucci, L. et al. Calcium intake in bone health: a focus on calcium-rich mineral waters. Nutrients 10, 1930 (2018).
Christakos, S., Dhawan, P., Porta, A., Mady, L. J. & Seth, T. Vitamin D and intestinal calcium absorption. Mol. Cell Endocrinol. 347, 25–29 (2011).
Gallagher, J. C. Vitamin D and aging. Endocrinol. Metab. Clin. North Am. 42, 319–332 (2013).
Veldurthy, V. et al. Vitamin D, calcium homeostasis and aging. Bone Res. 4, 16041 (2016).
Tella, S. H. & Gallagher, J. C. Prevention and treatment of postmenopausal osteoporosis. J. Steroid Biochem. Mol. Biol. 142, 155–170 (2014).
Anderson, G. L. et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 291, 1701–1712 (2004).
Rossouw, J. E. et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288, 321–333 (2002).
Levin, V. A., Jiang, X. & Kagan, R. Estrogen therapy for osteoporosis in the modern era. Osteoporos Int. 29, 1049–1055 (2018).
Hulley, S. et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 280, 605–613 (1998).
Heine, P. A., Taylor, J. A., Iwamoto, G. A., Lubahn, D. B. & Cooke, P. S. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc. Natl. Acad. Sci. USA 97, 12729–12734 (2000).
Murata, Y., Robertson, K. M., Jones, M. E. & Simpson, E. R. Effect of estrogen deficiency in the male: the ArKO mouse model. Mol. Cell Endocrinol. 193, 7–12 (2002).
Grodstein, F. et al. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann. Intern. Med. 133, 933–941 (2000).
Miller, W. L. & Auchus, R. J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 (2011).
Rodriguez-Cuenca, S. et al. Sex steroid receptor expression profile in brown adipose tissue. Effects of hormonal status. Cell Physiol. Biochem. 20, 877–886 (2007).
Chen, H. J., Meng, T., Gao, P. J. & Ruan, C. C. The role of brown adipose tissue dysfunction in the development of cardiovascular disease. Front. Endocrinol. 12, 652246 (2021).
Planavila, A. et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat. Commun. 4, 2019 (2013).
Hondares, E. et al. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J. Biol. Chem. 286, 12983–12990 (2011).
Park, C. J. et al. Genetic rescue of nonclassical ERalpha signaling normalizes energy balance in obese Eralpha-null mutant mice. J. Clin. Invest. 121, 604–612 (2011).
Casazza, K., Page, G. P. & Fernandez, J. R. The association between the rs2234693 and rs9340799 estrogen receptor alpha gene polymorphisms and risk factors for cardiovascular disease: a review. Biol. Res. Nurs. 12, 84–97 (2010).
Schierbeck, L. L. et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ 345, e6409 (2012).
Hodis, H. N. et al. Methods and baseline cardiovascular data from the Early versus Late Intervention Trial with Estradiol testing the menopausal hormone timing hypothesis. Menopause 22, 391–401 (2015).
Harman, S. M. et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women: a randomized trial. Ann. Intern. Med. 161, 249–260 (2014).
Phillips, L. S. & Langer, R. D. Postmenopausal hormone therapy: critical reappraisal and a unified hypothesis. Fertil. Steril. 83, 558–566 (2005).
Cavero-Redondo, I., Peleteiro, B., Alvarez-Bueno, C., Rodriguez-Artalejo, F. & Martinez-Vizcaino, V. Glycated haemoglobin A1c as a risk factor of cardiovascular outcomes and all-cause mortality in diabetic and non-diabetic populations: a systematic review and meta-analysis. BMJ Open 7, e015949 (2017).
Zhong, G. C., Ye, M. X., Cheng, J. H., Zhao, Y. & Gong, J. P. HbA1c and risks of all-cause and cause-specific death in subjects without known diabetes: a dose-response meta-analysis of prospective cohort studies. Sci. Rep. 6, 24071 (2016).
Iorga, A. et al. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex Differ. 8, 33 (2017).
Yang, X. P. & Reckelhoff, J. F. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 20, 133–138 (2011).
Zárate, S., Stevnsner, T. & Gredilla, R. Role of estrogen and other sex hormones in brain aging. neuroprotection and DNA repair. Front. Aging Neurosci. 9, 430–430 (2017).
Siddiqui, A. N. et al. Neuroprotective role of steroidal sex hormones: an overview. CNS Neurosci. Ther. 22, 342–350 (2016).
Gambacciani, M. & Levancini, M. Hormone replacement therapy and the prevention of postmenopausal osteoporosis. Prz Menopauzalny 13, 213–220 (2014).
Fait, T. Menopause hormone therapy: latest developments and clinical practice. Drugs Context 8, 212551–212551 (2019).
Vinogradova, Y., Coupland, C. & Hippisley-Cox, J. Use of hormone replacement therapy and risk of breast cancer: nested case-control studies using the QResearch and CPRD databases. BMJ 371, m3873 (2020).
Lobo, R. A. Hormone-replacement therapy: current thinking. Nat. Rev. Endocrinol. 13, 220–231 (2017).
D’Alonzo, M., Bounous, V. E., Villa, M. & Biglia, N. Current evidence of the oncological benefit-risk profile of hormone replacement therapy. Medicina 55, 573 (2019).
Boardman, H. M. P. et al. Hormone therapy for preventing cardiovascular disease in post‐menopausal women. Cochrane Database Syst. Rev. 2015, CD002229 (2015).
Lethaby, A., Hogervorst, E., Richards, M., Yesufu, A. & Yaffe, K. Hormone replacement therapy for cognitive function in postmenopausal women. Cochrane Database Syst. Rev. 2008, CD003122 (2018).
Clegg, A., Young, J., Iliffe, S., Rikkert, M. O. & Rockwood, K. Frailty in elderly people. Lancet 381, 752–762 (2013).
Jaul, E. & Barron, J. Age-related diseases and clinical and public health implications for the 85 years old and over population. Front. Public Health 5, 335–335 (2017).
Cheng, X. et al. Population ageing and mortality during 1990–2017: a global decomposition analysis. PLoS Med. 17, e1003138 (2020).
Olshansky, S. J. From lifespan to healthspan. JAMA 320, 1323–1324 (2018).
Crimmins, E. M. Lifespan and healthspan: past, present, and promise. Gerontologist 55, 901–911 (2015).
Argyle, C. E., Harper, J. C. & Davies, M. C. Oocyte cryopreservation: where are we now? Hum. Reprod. Update 22, 440–449 (2016).
Iussig, B. et al. A brief history of oocyte cryopreservation: arguments and facts. Acta Obstet. Gynecol. Scand. 98, 550–558 (2019).
Bhattacharya, S., Maheshwari, A. & Mollison, J. Factors associated with failed treatment: an analysis of 121,744 women embarking on their first IVF cycles. PLoS One 8, e82249 (2013).
Centers for Disease Control and Prevention. 2019 Assisted Reproductive Technology Fertility Clinic and National Summary Report (US Dept of Health and Human Services, 2021).
Llarena, N. & Hine, C. Reproductive longevity and aging: geroscience approaches to maintain long-term ovarian fitness. J. Gerontol. A Biol. Sci. Med. Sci. 76, 1551–1560 (2021).
Finch, C. E. The menopause and aging, a comparative perspective. J. Steroid Biochem. Mol. Biol. 142, 132–141 (2014).
Harman, D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300 (1956).
Liochev, S. I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 60, 1–4 (2013).
Sohal, R. S. & Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 273, 59–63 (1996).
Finkel, T. & Holbrook, N. J. Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247 (2000).
Sasaki, H. et al. Impact of oxidative stress on age-associated decline in oocyte developmental competence. Front. Endocrinol. 10, 811–811 (2019).
Becatti, M. et al. A biochemical approach to detect oxidative stress in infertile women undergoing assisted reproductive technology procedures. Int. J. Mol. Sci. 19, 592 (2018).
Wiener-Megnazi, Z. et al. Oxidative stress indices in follicular fluid as measured by the thermochemiluminescence assay correlate with outcome parameters in in vitro fertilization. Fertil. Steril. 82, 1171–1176 (2004).
Appasamy, M. et al. Evaluation of the relationship between follicular fluid oxidative stress, ovarian hormones, and response to gonadotropin stimulation. Fertil. Steril. 89, 912–921 (2008).
Oyawoye, O. et al. Antioxidants and reactive oxygen species in follicular fluid of women undergoing IVF: relationship to outcome. Hum. Reprod. 18, 2270–2274 (2003).
Das, S. et al. Reactive oxygen species level in follicular fluid — embryo quality marker in IVF? Hum. Reprod. 21, 2403–2407 (2006).
Terao, H. et al. Role of oxidative stress in follicular fluid on embryos of patients undergoing assisted reproductive technology treatment. J. Obstet. Gynaecol. Res. 45, 1884–1891 (2019).
Liu, L., Trimarchi, J. R., Navarro, P., Blasco, M. A. & Keefe, D. L. Oxidative stress contributes to arsenic-induced telomere attrition, chromosome instability, and apoptosis. J. Biol. Chem. 278, 31998–32004 (2003).
Navarro, P. A., Liu, L., Ferriani, R. A. & Keefe, D. L. Arsenite induces aberrations in meiosis that can be prevented by coadministration of N-acetylcysteine in mice. Fertil. Steril. 85(Suppl 1), 1187–1194 (2006).
Huang, J., Okuka, M., McLean, M., Keefe, D. L. & Liu, L. Telomere susceptibility to cigarette smoke-induced oxidative damage and chromosomal instability of mouse embryos in vitro. Free Radic. Biol. Med. 48, 1663–1676 (2010).
Liu, J. et al. Delay in oocyte aging in mice by the antioxidant N-acetyl-L-cysteine (NAC). Hum. Reprod. 27, 1411–1420 (2012).
Chen, Z. G. et al. Effects of plant polyphenols on ovarian follicular reserve in aging rats. Biochem. Cell Biol. 88, 737–745 (2010).
Tarín, J. J., Pérez-Albalá, S. & Cano, A. Oral antioxidants counteract the negative effects of female aging on oocyte quantity and quality in the mouse. Mol. Reprod. Dev. 61, 385–397 (2002).
Tarı́n, J. J., Pérez-Albalá, S., Pertusa, J. F. & Cano, A. Oral administration of pharmacological doses of Vitamins C and E reduces reproductive fitness and impairs the ovarian and uterine functions of female mice. Theriogenology 57, 1539–1550 (2002).
Bjelakovic, G., Nikolova, D., Gluud, L. L., Simonetti, R. G. & Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, CD007176 (2012).
Ristow, M. et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 106, 8665–8670 (2009).
Evans, J. R. & Lawrenson, J. G. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst. Rev. 7, CD000254 (2017).
Mathew, M. C., Ervin, A. M., Tao, J. & Davis, R. M. Antioxidant vitamin supplementation for preventing and slowing the progression of age-related cataract. Cochrane Database Syst. Rev. 6, CD004567 (2012).
Rutjes, A. W. S. et al. Vitamin and mineral supplementation for maintaining cognitive function in cognitively healthy people in mid and late life. Cochrane Database Syst. Rev. 12, CD011906 (2018).
Al-Khudairy, L. et al. Vitamin C supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 3, CD011114 (2017).
Agarwal, A., Durairajanayagam, D. & du Plessis, S. S. Utility of antioxidants during assisted reproductive techniques: an evidence based review. Reprod. Biol. Endocrinol. 12, 112–112 (2014).
Tesarik, J. Towards personalized antioxidant use in female infertility: need for more molecular and clinical studies. Biomedicines 9, 1933 (2021).
Ochiai, A. et al. Influence of resveratrol supplementation on IVF-embryo transfer cycle outcomes. Reprod. Biomed. Online 39, 205–210 (2019).
Zhang, L. X. et al. Resveratrol (RV): a pharmacological review and call for further research. Biomed. Pharmacother. 143, 112164 (2021).
Heilbronn, L. K. & Ravussin, E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am. J. Clin. Nutr. 78, 361–369 (2003).
Féry, F., Plat, L. & Balasse, E. O. Effects of metformin on the pathways of glucose utilization after oral glucose in non-insulin-dependent diabetes mellitus patients. Metabolism 46, 227–233 (1997).
Hundal, R. S. et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49, 2063–2069 (2000).
Onken, B. & Driscoll, M. Metformin induces a dietary restriction–like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS One 5, e8758 (2010).
Martin-Montalvo, A. et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 4, 2192 (2013).
Campbell, J. M., Bellman, S. M., Stephenson, M. D. & Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res. Rev. 40, 31–44 (2017).
Isola, J. V. V. et al. Mild calorie restriction, but not 17α-estradiol, extends ovarian reserve and fertility in female mice. Exp. Gerontol. 159, 111669 (2022).
Selesniemi, K., Lee, H.-J. & Tilly, J. L. Moderate caloric restriction initiated in rodents during adulthood sustains function of the female reproductive axis into advanced chronological age. Aging Cell 7, 622–629 (2008).
Qin, X. et al. Metformin prevents murine ovarian aging. Aging 11, 3785–3794 (2019).
Xu, S. et al. Glucose activates the primordial follicle through the AMPK/mTOR signaling pathway. Clin. Transl. Med. 10, e122 (2020).
Oner, G., Ozcelik, B., Ozgun, M. T. & Ozturk, F. The effects of metformin and letrozole on endometrium and ovary in a rat model. Gynecol. Endocrinol. 27, 1084–1086 (2011).
Barilovits, S. J. et al. Characterization of a mechanism to inhibit ovarian follicle activation. Fertil. Steril. 101, 1450–1457 (2014).
Lane, M. A., Ingram, D. K. & Roth, G. S. 2-Deoxy-D-glucose feeding in rats mimics physiologic effects of calorie restriction. J. Anti Aging Med. 1, 327–337 (1998).
Minor, R. K. et al. Chronic ingestion of 2-deoxy-D-glucose induces cardiac vacuolization and increases mortality in rats. Toxicol. Appl. Pharmacol. 243, 332–339 (2010).
Chiang, J. L. et al. Mitochondria in ovarian aging and reproductive longevity. Ageing Res. Rev. 63, 101168 (2020).
Wang, T., Zhang, M., Jiang, Z., Seli, E. Mitochondrial dysfunction and ovarian aging. Am. J. Reprod. Immunol. 77, bqaa001 (2017).
Camacho-Pereira, J. et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23, 1127–1139 (2016).
Yang, Q. et al. Increasing ovarian NAD(+) levels improve mitochondrial functions and reverse ovarian aging. Free Radic. Biol. Med. 156, 1–10 (2020).
Bertoldo, M. J. et al. NAD+ repletion rescues female fertility during reproductive aging. Cell Rep. 30, 1670–1681.e7 (2020).
Yang, L. et al. Mitochondrial DNA mutation exacerbates female reproductive aging via impairment of the NADH/NAD(+) redox. Aging Cell 19, e13206 (2020).
Bonkowski, M. S. & Sinclair, D. A. Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 17, 679–690 (2016).
Jia, K. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897–3906 (2004).
Vellai, T. et al. Genetics influence of TOR kinase on lifespan in C. elegans. Nature 426, 620–620 (2003).
Kapahi, P. et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14, 885–890 (2004).
Lamming, D. W. et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643 (2012).
Wu, J. J. et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 4, 913–920 (2013).
Miller, R. A. et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13, 468–477 (2014).
Neff, F. et al. Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Invest. 123, 3272–3291 (2013).
Chen, C., Liu, Y., Liu, Y. & Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2, ra75 (2009).
Miller, R. A. et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 66, 191–201 (2011).
Harrison, D. E. et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009).
Guo, Z. & Yu, Q. Role of mTOR signaling in female reproduction. Front. Endocrinol. 10, 692–692 (2019).
Adhikari, D. et al. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum. Mol. Genet. 19, 397–410 (2010).
Adhikari, D. et al. Disruption of Tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol. Hum. Reprod. 15, 765–770 (2009).
Zhang, H. et al. Somatic cells initiate primordial follicle activation and govern the development of dormant oocytes in mice. Curr. Biol. 24, 2501–2508 (2014).
Lu, X. et al. Stimulation of ovarian follicle growth after AMPK inhibition. Reproduction 153, 683–694 (2017).
Tanaka, Y. et al. Deletion of tuberous sclerosis 1 in somatic cells of the murine reproductive tract causes female infertility. Endocrinology 153, 404–416 (2012).
Choi, J., Jo, M., Lee, E. & Choi, D. AKT is involved in granulosa cell autophagy regulation via mTOR signaling during rat follicular development and atresia. Reproduction 147, 73–80 (2014).
Zhang, X. M. et al. Rapamycin preserves the follicle pool reserve and prolongs the ovarian lifespan of female rats via modulating mTOR activation and sirtuin expression. Gene 523, 82–87 (2013).
Tong, Y. et al. Rapamycin-sensitive mTORC1 signaling is involved in physiological primordial follicle activation in mouse ovary. Mol. Reprod. Dev. 80, 1018–1034 (2013).
Adhikari, D. et al. Pharmacological inhibition of mTORC1 prevents over-activation of the primordial follicle pool in response to elevated PI3K signaling. PLoS One 8, e53810 (2013).
Luo, L. L., Xu, J. J. & Fu, Y. C. Rapamycin prolongs female reproductive lifespan. Cell Cycle 12, 3353–3354 (2013).
Bitto, A. et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 5, e16351 (2016).
Wang, Y. et al. The essential role of PRAK in tumor metastasis and its therapeutic potential. Nat. Commun. 12, 1736 (2021).
Woo, Y. et al. Rapamycin promotes ROS-mediated cell death via functional inhibition of xCT expression in melanoma under gamma-irradiation. Front. Oncol. 11, 665420 (2021).
Orentreich, N., Brind, J. L., Rizer, R. L. & Vogelman, J. H. Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J. Clin. Endocrinol. Metab. 59, 551–555 (1984).
Zhang, J. et al. Dehydroepiandrosterone improves the ovarian reserve of women with diminished ovarian reserve and is a potential regulator of the immune response in the ovaries. BioScience Trends 9, 350–359 (2015).
Gleicher, N., Weghofer, A. & Barad, D. H. Improvement in diminished ovarian reserve after dehydroepiandrosterone supplementation. Reprod. Biomed. Online 21, 360–365 (2010).
Narkwichean, A. et al. Effects of dehydroepiandrosterone on in vivo ovine follicular development. Hum. Reprod. 29, 146–154 (2013).
Meredith, S., Jackson, K., Dudenhoeffer, G., Graham, L. & Epple, J. Long-term supplementation with melatonin delays reproductive senescence in rats, without an effect on number of primordial follicles. Exp. Gerontol. 35, 343–352 (2000).
Fernández, B. E., Díaz, E., Fernández, C., Núñez, P. & Díaz, B. Ovarian aging: melatonin regulation of the cytometric and endocrine evolutive pattern. Curr. Aging Sci. 6, 1–7 (2013).
Tamura, H. et al. Long-term melatonin treatment delays ovarian aging. J. Pineal. Res. 62, (2017).
Song, C. et al. Melatonin improves age-induced fertility decline and attenuates ovarian mitochondrial oxidative stress in mice. Sci. Rep. 6, 35165 (2016).
Zhang, L. et al. Melatonin regulates the activities of ovary and delays the fertility decline in female animals via MT1/AMPK pathway. J. Pineal. Res. 66, e12550 (2019).
Yang, C. et al. Melatonin delays ovarian aging in mice by slowing down the exhaustion of ovarian reserve. Commun. Biol. 4, 534 (2021).
Bellipanni, G., Bianchi, P., Pierpaoli, W., Bulian, D. & Ilyia, E. Effects of melatonin in perimenopausal and menopausal women: a randomized and placebo controlled study. Exp. Gerontol. 36, 297–310 (2001).
Takasaki, A., Nakamura, Y., Tamura, H., Shimamura, K. & Morioka, H. Melatonin as a new drug for improving oocyte quality. Reprod. Med. Biol. 2, 139–144 (2003).
Tamura, H. et al. Oxidative stress impairs oocyte quality and melatonin protects oocytes from free radical damage and improves fertilization rate. J. Pineal. Res. 44, 280–287 (2008).
Batioglu, A. S., Sahin, U., Gurlek, B., Ozturk, N. & Unsal, E. The efficacy of melatonin administration on oocyte quality. Gynecol. Endocrinol. 28, 91–93 (2012).
Fernando, S. et al. Melatonin in assisted reproductive technology: a pilot double-blind randomized placebo-controlled clinical trial. Front. Endocrinol. 9, 545 (2018).
Espino, J. et al. Impact of melatonin supplementation in women with unexplained infertility undergoing fertility treatment. Antioxidants 8, 338 (2019).
George, K. & Kamath, M. S. Fertility and age. J. Hum. Reprod. Sci. 3, 121–123 (2010).
Smith, K. R. et al. Familial aggregation of survival and late female reproduction. J. Gerontol. A Biol. Sci. Med. Sci. 64, 740–744 (2009).
Abdollahifar, M. A. et al. Vitamin C restores ovarian follicular reservation in a mouse model of aging. Anat. Cell Biol. 52, 196–203 (2019).
Zhuang, X. L. et al. Effects of genistein on ovarian follicular development and ovarian life span in rats. Fitoterapia 81, 998–1002 (2010).
Appt, S. E. et al. The effect of diet and cardiovascular risk on ovarian aging in cynomolgus monkeys (Macaca fascicularis). Menopause 17, 741–748 (2010).
Liu, M. et al. Resveratrol protects against age-associated infertility in mice. Hum. Reprod. 28, 707–717 (2013).
Ben-Meir, A. et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 14, 887–895 (2015).
Timoteo-Ferreira, F. et al. Apocynin dietary supplementation delays mouse ovarian ageing. Oxid. Med. Cell Longev. 2019, 5316984 (2019).
Silva, E. et al. Antioxidant supplementation modulates age-related placental bed morphology and reproductive outcome in mice. Biol. Reprod. 93, 56 (2015).
Xian, Y. et al. Antioxidants retard the ageing of mouse oocytes. Mol. Med. Rep. 18, 1981–1986 (2018).
Akino, N. et al. Activation of Nrf2/Keap1 pathway by oral Dimethylfumarate administration alleviates oxidative stress and age-associated infertility might be delayed in the mouse ovary. Reprod. Biol. Endocrinol. 17, 23 (2019).
Wei, M. et al. Ovarian failure-resistant effects of catalpol in aged female rats. Biol. Pharm. Bull. 37, 1444–1449 (2014).
Jinno, M. et al. Low-dose metformin improves pregnancy rate in IVF repeaters without polycystic ovary syndrome: its indication and mechanism. Fertil. Steril. 94, S29–S29 (2010).
Zhang, Z. et al. alpha-ketoglutarate delays age-related fertility decline in mammals. Aging Cell 20, e13291 (2021).
Yu, J., Yaba, A., Kasiman, C., Thomson, T. & Johnson, J. mTOR controls ovarian follicle growth by regulating granulosa cell proliferation. PLoS One 6, e21415 (2011).
Acknowledgements
L.D. is funded for her PhD studies by National University of Singapore. The authors acknowledge the generous contribution of the Bia-Echo Foundation to Yong Loo Lin School of Medicine, National University of Singapore in the set-up of NUS Bia-Echo Asia Centre of Reproductive Longevity and Equality to change the science and narrative of female reproductive longevity.
Author information
Authors and Affiliations
Contributions
Z.H. and B.K.K. conceptualized and led the review. D.B.L.T. and L.D. performed the literature review, summarized data, constructed the figures and wrote the first version of the manuscript. B.K.K. and Z.H. revised and critically reviewed the subsequent draft revisions. All authors read, edited, and approved the final article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dong, L., Teh, D.B.L., Kennedy, B.K. et al. Unraveling female reproductive senescence to enhance healthy longevity. Cell Res 33, 11–29 (2023). https://doi.org/10.1038/s41422-022-00718-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-022-00718-7
- Springer Nature Singapore Pte Ltd.
This article is cited by
-
Identification and validation of senescence-related genes in polycystic ovary syndrome
Journal of Ovarian Research (2024)
-
Reactive oxygen species-scavenging nanomaterials for the prevention and treatment of age-related diseases
Journal of Nanobiotechnology (2024)
-
Nur77 improves ovarian function in reproductive aging mice by activating mitophagy and inhibiting apoptosis
Reproductive Biology and Endocrinology (2024)
-
SenNet recommendations for detecting senescent cells in different tissues
Nature Reviews Molecular Cell Biology (2024)
-
The stromal microenvironment and ovarian aging: mechanisms and therapeutic opportunities
Journal of Ovarian Research (2023)