
Abstract
Plant growth-limiting factors, such as low nutrient availability and weak pathogen resistance, may hinder the production of several crops. Plant growth-promoting bacteria (PGPB) used in agriculture, which stimulate plant growth and development, can serve as a potential tool to mitigate or even circumvent these limitations. The present study evaluated the feasibility of using bacteria isolated from the maize rhizosphere as PGPB for the cultivation of this crop. A total of 282 isolates were collected and clustered into 57 groups based on their genetic similarity using BOX-PCR. A representative isolate from each group was selected and identified at the genus level with 16S rRNA sequencing. The identified genera included Bacillus (61.5% of the isolates), Lysinibacillus (30.52%), Pseudomonas (3.15%), Stenotrophomonas (2.91%), Paenibacillus (1.22%), Enterobacter (0.25%), Rhizobium (0.25%), and Atlantibacter (0.25%). Eleven isolates with the highest performance were selected for analyzing the possible pathways underlying plant growth promotion using biochemical and molecular techniques. Of the selected isolates, 90.9% were positive for indolepyruvate/phenylpyruvate decarboxylase, 54.4% for pyrroloquinoline quinine synthase, 36.4% for nitrogenase reductase, and 27.3% for nitrite reductase. Based on biochemical characterization, 9.1% isolates could fix nitrogen, 36.6% could solubilize phosphate, 54.5% could produce siderophores, and 90.9% could produce indole acetic acid. Enzymatic profiling revealed that the isolates could degrade starch (90.1%), cellulose (72.7%), pectin (81.8%), protein (90.9%), chitin (18.2%), urea (54.5%), and esters (45.4%). Based on the data obtained, we identified three Bacillus spp. (LGMB12, LGMB273, and LGMB426), one Stenotrophomonas sp. (LGMB417), and one Pseudomonas sp. (LGMB456) with the potential to serve as PGPB for maize. Further research is warranted to evaluate the biotechnological potential of these isolates as biofertilizers under field conditions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Brazil, the USA, and China, are the largest producers of maize (Zea mays L.) [1], a major cereal crop worldwide [2]. The intensification of maize production, high degree of nutrient removal from agricultural areas, lack of adequate nutritional management of soil, and adoption of monoculture demand continuous replacement of nutrients for crops [3]. To solve this problem, fertilizers, particularly chemicals, must be used, which incur high production costs and cause environmental pollution. Given the risks associated with the indiscriminate use of these inputs, such as the eutrophication of soil and groundwater or emission of greenhouse gases (GHGs) [4], novel tools to promote plant growth are required. An alternative to overcome this challenge is the use of rhizobacteria that can fix atmospheric nitrogen and promote plant growth through other microbiological processes [5].
Many genera of plant growth-promoting bacteria (PGPB), including Pseudomonas, Burkholderia, Bacillus, Bradyrhizobium, Rhizobium, Gluconacetobacter, Herbaspirillum, and Azospirillum, among others, have been identified [6,7,8,9,10,11]. However, some PGPB may be pathogenic to plants, humans, and other non-human animals, presenting the risk of environmental spread [12]. PGPB may improve root development and nutrient absorption, thereby lowering production costs, reducing fertilizer use, and mitigating environmental impacts [13].
Nitrogen is the major limiting factor for maize biomass production [14, 15]. The use of synthetic nitrogen fertilizers is limited because of high nitrogen loss to the environment through microbial immobilization, leaching, and volatilization [16]. Therefore, bioproducts derived from PGPB [17] have been used as commercial inoculants worldwide to improve productivity [18].
Currently, several PGPB-based products are available on the market for different crops, such as Azospirillum brasilense for maize [6, 19], Rhizobium tropici for common bean (Phaseolus vulgaris L.) [20], and Bradyrhizobium japonicum for soybean (Glycine max (L.) Merr.) [21]. Of note, there is certain affinity between bacterial strains and cultivars [22].
Some key pathways are required to enable the benefits of using PGPB, and specific pathways can be identified via detection of marker genes, such as nitrogenase reductase (nifH), which is involved in nitrogen fixation, or phloroglucinol (phlD) and pyrroloquinoline quinine synthesis (pqqC), which are involved in phosphate solubilization [23], among others. The detection of such genes is the first indication of a candidate PGPB [24]. Moreover, molecular approaches, such as the detection of genes involved in plant growth-promoting mechanisms, have certain advantages, including the greater ease of execution; higher sensitivity, specificity, and reproducibility; and shorter execution time even for a large number of isolates; however, qualitative data must still be evaluated via biochemical approaches [25,26,27]. In addition, some microorganisms may promote plant growth through other mechanisms, such as the production of enzymes, particularly hydrolytic enzymes (amylase and cellulase, among others), which may interfere with pathogen control [28, 29].
Rhizobacteria possess several properties [30], justifying the global interest in them as PGPB and biocontrol agents, particularly for maize [31]. However, limited studies have been conducted relative to the global area under maize cultivation, and additional research is paramount considering the wide variability in results with many factors, such as climate, natural microbiota, available nutrients, and crop characteristics. Therefore, the use of PGPB should be optimized to specific agroecosystems [32].
To this end, the present study explored the biotechnological potential of bacterial isolates collected from the maize rhizosphere as PGPB using in vitro and in vivo evaluations. Indole acetic acid (IAA) synthesis and pathways relevant to both plant growth promotion and pathogen resistance (through biocontrol or induction of systemic resistance) were also assessed. BOX-PCR was used to evaluate the genetic variability of the collection, and 16S rRNA sequencing was used to identify isolates at the genus level.
Material and methods
Biological material
The bacteria (n = 282) used in the present study were previously isolated from the maize rhizosphere by our research group [33]; however, Ikeda et al. [33] included only 217 selected bacteria from over 500 isolates in their study. Therefore, in the present study, we expanded the number of isolates and performed analyses. The isolates were deposited at the Microorganism Genetics Laboratory (LabGeM), Department of Genetics, UFPR, Curitiba, State of Paraná, Brazil.
Bacteria were isolated from the rhizosphere of different maize genotypes cultivated in field trials [33]. Ikeda et al. (2013) cultivated maize in the southern Brazilian region of Campo Largo, State of Paraná. Maize roots were submerged in distilled water for 1 min, followed by immersion in 70% ethanol (v/v) for 1 min, 3% sodium hypochlorite (v/v) for 3 min, and 70% ethanol (v/v) for 30 s, and then washed three times with sterile distilled water for 1 min. Following surface sterilization, the samples were fragmented into five pieces of 8 mm and aseptically transferred to Petri plates containing a nitrogen-free solid culture medium [33]. Growth was assessed daily.
Characterization of isolates
Isolated fingerprinting with BOX-PCR
For the extraction of genomic DNA, the isolates were grown in 3 mL of Luria Bertani (LB) liquid medium for 18 h at 30 °C with agitation at 200 rpm, and the DNA was extracted as described by Szilagyi-Zecchin et al. [34]. DNA quality was accessed by agarose electrophoresis. DNA quantity was determined using a NanoDrop devise (Thermo Fisher Scientific, USA) and standardized to 50 ng.
The initial characterization of the isolates was performed by BOX-PCR using the A1R primer (5′-CTACGGCAAGGCGACGCTGACG-3′, Invitrogen™) [35]. The reaction was conducted under the conditions described by Kaschuk et al. [36] with required modifications of extension temperatures and time. The reaction mixture (25 μL) contained 200 ng DNA, 10 × PCR buffer, 0.5 U·μL−1 Taq DNA polymerase, 0.2 μM primer, 0.2 mM dNTPs, and 3 mM MgCl2. The amplification conditions were as follows: initial denaturation at 95 °C for 7 min; 35 cycles of 94 °C for 1 min, 53 °C for 1 min, and 72 °C for 3 min; and final extension at 72 °C for 4 min.
The amplified fragments were separated by 0.7% agarose gel (25 cm × 20 cm, pH 8.0) electrophoresis at 140 V for 120 min. The gels were stained with ethidium bromide, visualized under UV light, and photographed. The band pattern was manually defined, comparing the presence and absence (transformed into binary matrices) of bands among the isolates. The dissimilarity of BOX-PCR fingerprints was calculated using the Jaccard (J) coefficient [37] with the vegan package in R [38], and clustering was performed using the bootstrapped (104 generations) unweighted pair group method with arithmetic mean (UPGMA) algorithm with the pvclust package in R [39]. A dendrogram was constructed using the ggtree package in R [40, 41].
Genus identification
The isolates were identified by amplifying the partial sequences of the 16S rRNA gene using the fD1 (5′-AGAGTTTGATCCTGGCTCAG-3′) and rD1 (5′-AAGGAGGTGATCCAGCC-3′) primers [42]. The reaction was conducted under the conditions described by Menna et al. [43], with required modifications of amplification time and temperature. The reaction mixture (25 μL) contained 200 ng DNA, 10 × PCR buffer, 0.5 U·μL−1 Taq DNA polymerase, 0.2 μM primers, 0.2 mM dNTPs, and 3 mM MgCl2. The amplification conditions were as follows: initial denaturation at 95 °C for 5 min; 30 cycles of 94 °C for 45 s, 55 °C for 45 s, and 72 °C for 2 min; and final extension at 72 °C for 10 min. The obtained fragments were sequenced using Big Dye with the ABI3500 DNA Sequencer, as described by Kimoto et al. [44].
The obtained sequences were compared those available in the List of Prokaryotic Names with Standing in Nomenclature (LPSN) (http://www.bacterio.net) [45,46,47] using the BLAST tool [48], aligned using Muscle software [49], and edited using BioEdit 7.2.5 [50] and MEGA 7 [51]. Bayesian phylogenetic trees were generated using MrBayes 3.2.7a [52], incorporating the evolutionary model indicated after testing with jModel Test [53, 54] and performing simultaneous runs for random trees for 107 generations to reach an LnL deviation of 0.01 or below. The trees were visualized using FigTree 1.4.
Evaluation of plant growth-promoting characteristics
The plant growth-promoting potential of 57 representative isolates from each group of the BOX-PCR profile was evaluated. The experiment was performed in a climatic chamber (Walk-In Chamber). The experimental design was completely randomized, and the experiment was performed in triplicate.
Seeds of a commercial hybrid maize cultivar (AG 8780, Agroceres®) were manually treated with an insecticide (250 mL 100 kg−1 of seeds), containing carboxin and thiram (belonging to the carboxanilide and dimethyldithiocarbamate chemical groups, respectively, Vitavax-Thiram®) as active ingredients, and a fungicide (1.5 L 100 kg−1 of seeds) containing imidacloprid and thiodicarb (belonging to the neonicotinoid and oxime methylcarbamate chemical groups, respectively, Cropstar®) as active ingredients, using a seed treater 36 h before sowing. As the positive controls, seeds were inoculated with a commercial formulation containing Azospirillum brasilense (AzoTotal®, Total Biotecnologia; AbV5 and AbV6 strains; 2 × 108 CFU mL−1; 100 mL 20 kg−1 of seeds). As the negative controls, seeds without inoculation were used. The toxicity of the fungicide and insecticide against the bacteria was evaluated by the inoculation of bacteria on solid LB medium containing the same concentrations of the fungicide and insecticide used in the experiment, and no toxicity was observed (data not shown).
For cultivation, the seeds were disinfected with 70% alcohol for 1 min and 3% sodium hypochlorite for 3 min, washed six times with distilled water, inoculated with 1 mL of the bacterial suspension in LB medium (1 × 108 cells mL−1), and sown in autoclaved vermiculite, two per pot (volume, 1.7 L). The plants were maintained at a mean temperature of 28 °C under a 12 h photoperiod daily. Irrigation was applied at 50 mL per pot daily, alternating between sterile distilled water and sterile Hoagland and Arnon nutrient solution [55].
At 13 days after sowing, the plants were thinned to maintain only one plant per pot. After 30 days, the plants were collected, and root growth was measured using a root scanner (WinRhizo® device). For the whole plant analysis, the plants were first stored in paper bags, and the wet mass was measured. The samples were then dried in an oven at 60 °C for 72 h to determine the dry weight.
Homogeneity of variances was determined using the Bartlett test, and the normality of data was assessed using the Kolmogorov–Smirnov test, with Lilliefors correction. Nonparametric data were submitted to the Kruskal–Wallis test [nonparametric single-factor analysis of variance (ANOVA)], and parametric data were subjected to ANOVA. Means were compared using Tukey’s test (p ≤ 0.05). The data were plotted using Sigma®Plot 12.0 (https://systatsoftware.com/), and all statistical analyses were performed using Assistat 7.6 (http://www.assistat.com/indexp.html).
Molecular characterization of genes related to plant growth promotion
To investigate the candidate genes related to plant growth-promoting pathways in the best isolates identified in the root growth assay, DNA was amplified with specific primers (Table 1).
The reaction mixture (25 μL) contained 100 ng DNA, 10 × PCR buffer, 0.5 U·μL−1 Taq DNA polymerase, 0.2 μM primers, 0.2 mM dNTPs, and 3 mM MgCl2. The amplification conditions for nitrogenase reductase (nifH) [56] were as follows: initial denaturation at 97 °C for 4 min; 1 cycle of denaturation at 96 °C for 20 s, annealing at 65 °C for 30 s, and elongation at 72 °C for 30 s; 2 cycles of denaturation at 96 °C for 20 s, annealing at 62 °C for 30 s, and elongation at 72 °C for 35 s; 3 cycles of denaturation at 96 °C for 20 s, annealing at 59 °C for 30 s, and elongation at 72 °C for 42 s; 4 cycles of denaturation at 96 °C for 20 s, annealing at 56 °C for 30 s, and elongation at 72 °C for 45 s; 5 cycles of denaturation at 96 °C for 20 s, annealing at 53 °C for 30 s, and elongation at 72 °C for 50 s; 25 cycles of denaturation at 94 °C for 20 s, annealing at 50 °C for 45 s, and elongation at 72 °C for 60 s; and final extension at 72 °C for 10 min. For phloroglucinol synthesis (phlD) [57], the amplification followed an initial denaturation at 95 °C for 3 min, 35 cycles of 60 s at 94 °C, 60 s at 60 °C, and 60 s at 72 °C, and final extension at 72 °C for 5 min.
The amplification conditions for phosphate solubilization (pqqC) [58] were as follows: initial denaturation at 96 °C for 10 min; 30 cycles of 30 s at 96 °C, 30 s at 63 °C, and 1 min at 72 °C; and a final extension of 10 min at 72 °C. Indole acetic acid synthesis (ipdC) [59] was amplified using an initial denaturation step at 94 °C for 5 min; 35 cycles of 30 s at 95 °C, 30 s at 60 °C, and 30 s at 72 °C; and final extension at 72 °C for 7 min. The amplification conditions for nitrite reductase (nirK) [60] were as follows: initial denaturation at 95 °C for 5 min; 1 cycle of denaturation at 95 °C for 30 s, annealing at 45 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 44.5 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 44 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 43.5 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 43 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 42.5 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 42 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 41.5 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 41 °C for 40 s, and elongation at 72 °C for 40 s; 1 cycle of denaturation at 95 °C for 30 s, annealing at 40 °C for 40 s, and elongation at 72 °C for 40 s; 20 cycles of 30 s at 95 °C; 40 s at 43 °C; and 40 s at 72 °C; and final extension of 7 min at 72 °C. The amplification conditions for ethylene degradation (acdS) [61] were as follows: initial denaturation at 95 °C for 5 min; 35 cycles of 30 s at 95 °C, 30 s at 50 °C, and 30 s at 72 °C; and final extension at 72 °C for 7 min.
Biochemical evaluation of plant growth-promoting parameters
Siderophore production was analyzed as described by Schwyn and Neilands [62] using solid DYGS medium with CAS solution, carefully mixed into 72.9 mg of hexadecyltrimethylammonium (HDTMA) and dissolved in 40 mL of distilled water. The results were considered positive when the color of the medium changed from blue to yellow.
Phosphate solubilization was evaluated as described Sylvester-Bradley et al. [63] using the GL culture medium supplemented with 0.25 g L−1 K2HPO4 and 1 g L−1 CaCl2. The results were considered positive when a halo was formed around the colony.
Biological nitrogen fixation was evaluated as described by Araújo et al. [64] using a nitrogen-free semi-solid JNFb medium. First, the isolates were grown in nitrogen-free solid JNFb medium at 32 °C. After 4 days, the isolates were transferred to ampoules with 5 mL of nitrogen-free semi-solid medium and incubated at the same temperature (120 rpm) for 7 days; the culture was repeated twice. The isolates were successively reinoculated to prevent pellicle formation due to the nitrogen reserves of the accumulated bacteria cells [33]. Bacterial growth was assessed based on the formation of a pellicle on the medium surface.
IAA synthesis was evaluated using the methodology described by Kuss et al. [65], modified with the use of King B medium. Salkowski solution was added to reveal the results. Absorbance was measured at 520 nm using spectrophotometry. The final values were multiplied by the molecular weight of commercial auxin (C10H9NO2 = 175.19 g mol−1) and expressed in micrograms per milliliter. The data were transformed using √x + ½.
Amylase production was tested in MM9 medium [66] [200 mL of salt solution (64 g Na2HPO4.2H2O, 15 g KH2PO4, 2.5 g NaCl, and 5 g NH4Cl q.s.p. 1,000 mL distilled water), 2 mL of 1 M MgSO4, 10 g glucose, 0.1 mL of 1 M CaCl2, and 15 g agar, q.s.p. 1,000 mL distilled water; pH 7.0] containing 0.5% yeast extract and 1% soluble starch [67]. The result was considered positive when a halo was formed around the bacterial colony following iodine addition.
Pectinase and chitinase production were also evaluated in MM9 medium supplemented with 1% pectin [67] and 0.08% colloidal chitin [68, 69], respectively. For pectin, the result was considered positive when a halo was formed around the bacterial colony following lugol addition. For chitin, the result was considered positive when a halo appeared around the colony following chitin degradation.
Cellulase test were performed as described by Renwick et al. [64]. Cellulase production was revealed by the addition of Congo Red to the mineral culture medium [70] (0.02 g CaCO3, 0.01 g FeSO4.7H2O, 1.71 g KCl, 0.05 g MgSO4.7H2O, 4.11 g Na2HPO4.12H2O, and 15 g agar; q.s.p. 1,000 mL distilled water) containing 0.5% carboxymethylcellulose.
Esterase production was assayed in a solid esterase/lipase culture medium supplemented with 1% Tween 80 [71]. Protease production was evaluated in skimmed milk and agar medium [72]. For both esterase and protease, the results were considered positive when a halo was formed around the colonies.
Urease production was evaluated in a urease culture medium as described by Dye [73]. The result was considered positive when the culture medium turned blue.
All tests were performed in triplicate. The bacterial culture temperature for all biochemical tests was 32 °C. For IAA analysis, the homogeneity of variances was tested using Bartlett test, and the normality of data was assessed using the Kolmogorov–Smirnov test, with Lilliefors correction. All data were subjected to ANOVA and Tukey’s test (p ≤ 0.05). Statistical analyses were performed using Assistat v. 7.6 (http://www.assistat.com/indexp.html).
Nucleotide sequence accession numbers
The 16S rRNA sequences of the isolates have been deposited in NCBI GenBank (https://www.ncbi.nlm.nih.gov/) under accession numbers MT780814–MT780870.
Results
Characterization of isolates
The band pattern of the 282 isolates was obtained using BOX-PCR. The amplified products were scored in terms of the presence (1) or absence (0) of band at a position in each of the isolates, resulting in 57 different genetic profiles [Fig. 1; values to the left of the nodes represent significant bootstrap values (> 50%)]. The BOX-PCR profiles were considered different when the level of dissimilarity was equal to or higher than 80%.

Dendrogram obtained using BOX-PCR amplification products from maize isolates. Clusters were formed based on 57 distinct genetic profiles. Cluster analysis was performed using the UPGMA algorithm and Jaccard’s coefficient. Numbers in parenthesis represent the number of isolates with the same genetic profile and the selected representative. Numbers at nodes represent the bootstrap support of 104 generations of clustering (only values exceeding 50% are shown). Genera were identified based on the partial sequence of the 16S rRNA gene using Bayesian analysis
Dendrogram analysis revealed that the isolates differed in terms of the 57 profiles, separated into three main clusters. The first cluster was formed by LGMB456 (representing one isolate) and LGMB459 (representing three isolates) (Pseudomonas spp.), the second by LGMB33 (representing one isolate) (Bacillus sp.), and the third by two main subgroups, each subdivided into several branches, representing the remaining 54 profiles (of the remaining 277 isolates).
Based on these results, a single representative isolate was selected at random from each profile, resulting in 57 isolates representing the respective genetic groups.
Identification of isolates at the genus level
One representative from each of the 57 BOX-PCR profiles was selected for 16S rRNA sequencing. Based on phylogenetic analysis (Table 2, Fig. S1–S8), the isolates were identified as belonging to the genera Bacillus (35 profiles), Lysinibacillus (12 profiles), Pseudomonas (4 profiles), Stenotrophomonas (2 profiles), Enterobacter (1 profile), Paenibacillus (1 profile), Rhizobium (1 profile), and Atlantibacter (1 profile).
Selection of high-performance isolates using an in vivo assay
Dry weight of the whole plants (in g) (p = 0.0001) was significantly higher for LGMB324 (Lysinibacillus sp.) than that for 38 other isolates belonging to the genera Stenotrophomonas sp., Pseudomonas spp., Lysinibacillus spp., Rhizobium sp., Bacillus spp., Enterobacter sp., and Atlantibacter sp. as well as the negative and positive controls but was comparable to that for the 20 other isolates belonging to the genera Lysinibacillus spp., Bacillus spp., Pseudomonas sp., Stenotrophomonas sp., and Paenibacillus sp. Total root length (in cm) (p = 0.01063) was significantly higher for LGMB12 (Bacillus sp.) than that for 28 other isolates belonging to genera Pseudomonas spp., Bacillus spp., Lysinibacillus spp., and Enterobacter sp. but was comparable to that for 30 other isolates belonging to the genera Lysinibacillus spp., Rhizobium sp., Bacillus spp., Atlantibacter sp., Stenotrophomonas spp., Paenibacillus sp., and Pseudomonas sp. as well as the negative and positive controls. The length of roots ranging in diameter from 0 to 0.5 mm (in cm) (p = 0.00736) was significantly higher for LGMB12 (Bacillus sp.) than that for 43 other isolates belonging to the genera Lysinibacillus spp., Stenotrophomonas sp., Pseudomonas spp., Bacillus spp., Enterobacter sp., Rhizobium sp., and Paenibacillus sp. as well as the positive controls but was comparable to that for 15 other isolates belonging to the genera Lysinibacillus spp., Bacillus spp., Atlantibacter sp., Stenotrophomonas sp., and Pseudomonas sp. as well as the negative controls. The wet mass of the whole plant (in g) (p = 0.1662), total root volume (in cm3) (p = 0.1412), and mean root diameter (mm) (p = 0.2523) were comparable across all treatments (Tukey’s test, at 5% significance) (Fig. 2).

Evaluation of plant growth-promoting bacteria for maize development after 30 days of inoculation of seeds and growth in a climatic chamber. Dry weight of the whole plants (in g, black dots), total root length (in cm, blue bars), and total length of roots ranging in diameter from 0 to 0.5 mm (in cm, red bars) were evaluated. The data were analyzed with Tukey’s test, and a p < 0.05 was considered significant. The Y axis on the left indicates the total root length and total length of roots ranging in diameter from 0 to 0.5 mm, expressed in centimeters, and the Y axis on the right indicates the dry weight of the whole plants, expressed in grams. The controls are represented by ( −) negative (no bacterial inoculated) and ( +) positive seeds (inoculated with Azospirillum brasilense AbV5 and AbV6). The error bars with SD are represented by vertical lines. Each column and dot on the graph represent triplicate analyses of plants whose seeds were inoculated with the LGMB marked on the X axis, comprising 177 samples. The asterisks (*) next to the isolate name indicate the number of parameters that were significantly higher: *Significant for one parameter; **significant for two parameters; and ***significant for three parameters
Based on the analysis of plant growth, 11 isolates stood out, presenting results statistically equal or superior to the other isolates tested for dry weight, total root length, and total length of roots ranging in diameter from 0 to 0.5 mm. LGMB12, LGMB273, LGMB444 (Bacillus spp.), LGMB23, LGMB45, LGMB324 (Lysinibacillus spp.), and LGMB417 (Stenotrophomonas sp.) were significant in terms of all characteristics, whereas LGMB319 and LGMB426 (Bacillus spp.), LGMB429 (Paenibacillus sp.), and LGMB456 (Pseudomonas sp.) were significant in terms of two of the three characteristics (dry weight and total length of roots ranging in diameter from 0 to 0.5 mm; although not significant, total root length exceeded 800 cm).
Genetic and biochemical characteristics involved in plant growth promotion
The 11 isolates selected in the above evaluations, based on the higher root growth of maize plants, were further analyzed to determine the possible pathways involved in plant growth promotion.
Molecular characterization of genes revealed that the isolates likely possess specific plant growth-promoting mechanisms. Therefore, the results of gene expression analyses were confirmed using biochemical tests for phosphate solubilization, IAA production, and biological nitrogen fixation.
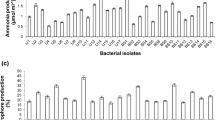
Molecular assays detected (Table 3) the marker gene for phosphate solubilization (pqqC) in 54.5% of the isolates, and all these isolates could solubilize phosphate in the biochemical test [LGMB12, LGMB273, LGMB426 (Bacillus spp.), LGMB23 (Lysinibacillus sp.), LGMB417 (Stenotrophomonas sp.), and LGMB456 (Pseudomonas sp.)]. Moreover, the marker gene for biological nitrogen fixation (nifH) was detected in 36.4% of the isolates [LGMB12 and LGMB426 (Bacillus spp.), LGMB417 (Stenotrophomonas sp.), and LGMB429 (Paenibacillus sp.)]. However, in the biochemical test, amplification with the nifH primers was successful in only one isolate [LGMB417 (Stenotrophomonas sp.)] (Table 3). The ipdC gene, which encodes a precursor of IAA, was detected in 90.9% isolates, except in LGMB444 (Bacillus sp.). In biochemical test, LGMB45 (Lysinibacillus sp.) showed higher IAA production, albeit not significantly different from LGMB23 and LGMB324 (Lysinibacillus spp.) or LGMB456 (Pseudomonas sp.) (Table 3).
Furthermore, biochemical tests revealed that six isolates produced siderophores [LGMB12, LGMB273, LGMB319, LGMB426, LGMB444 (Bacillus spp.), and LGMB324 (Lysinibacillus sp.)] (Table 3). In addition, the nirK gene, which is involved in denitrification, was detected in 27.3% isolates [LGMB45 (Lysinibacillus sp.), LGMB426 (Bacillus sp.), and LGMB429 (Paenibacillus sp.)]. The evaluated isolates did not possess phlD and acdS genes, which are responsible for the synthesis of phloroglucinol and degradation of plant ethylene precursor, respectively.
In addition, LGMB426 (Bacillus sp.) presented the highest number of evaluated characteristics, being able to solubilize phosphate, synthesize IAA, and produce siderophores.
Production of enzymes with biotechnological applications in agriculture
The enzymatic profiles (Table 4) showed that 90.1% isolates produced amylase, 54.5% produced urease, 72.7% produced cellulase, 18.2% produced chitinase, 81.8% produced pectinase, 90.9% produced protease, and 45.4% produced esterase. LGMB444 (Bacillus sp.) exhibited the highest diversity of enzymes (Table 4), being able to produce all enzymes tested, including amylase, cellulase, pectinase, chitinase, urease, esterase, and protease.
Discussion
Microbial inoculants of elite strains may help improve crop productivity through diverse mechanisms, including nitrogen fixation, phosphate solubilization, siderophore synthesis, phytohormone production, and hydrolytic enzyme synthesis (chitinase, cellulase, and protease, among others), which enhance soil fertility and agricultural yield as well as favor plant growth [74,75,76].
In the present study, high intraspecific diversity was detected among 282 isolates collected from maize rhizosphere based on the BOX-PCR profiles. BOX-PCR fingerprinting is a precise discriminatory technique to determine genetic relatedness and diversity, particularly for the genus Bacillus, which shows multiple distinct band patterns [77]. This modality is of great importance in studies comprising a large number of samples [78,79,80] to identify at the strain level, which is not possible based solely on their morphology [81].
Diversity of maize root bacterial isolates is well-known, and bacteria of the genera Bacillus, Pseudomonas, Paenibacillus, and Enterobacter are known to possess the potential to promote plant growth [9, 10, 34, 82, 83]. In the present study, we identified several isolates belonging to these genera as potential PGPB (Bacillus, Fig. S1; Pseudomonas, Fig. S2; Paenibacillus, Fig. S3; and Enterobacter, Fig. S4), in addition to bacteria belonging to other genera, including Rhizobium (Fig. S5), Lysinibacillus (Fig. S6), Stenotrophomonas (Fig. S7), and Atlantibacter (Fig. S8). These genera are agriculturally important and predominant colonizers of the rhizospheres of various crops, and they possess a broad spectrum of antagonistic activities [84], which is an advantage for bacteria intended for use as inoculants. However, the isolates must be identified to the species level for use as inoculants to exclude the ones pathogenic to plants, humans, and other non-human animals [12].
For use as a candidate biofertilizer, a bacterium must possess the potential to promote crop growth and increase yield, being active mainly in the rhizosphere [85]. As such, enhanced root growth promotes nutrient absorption, and root length is an indicator of the nutrient uptake and acquisition efficiency of plants [86].
In the present study, the 11 isolates that significantly increased root length were selected to assess the possible mechanisms underlying plant growth promotion. Specifically, inoculation of bacteria that solubilize phosphate can serve as an alternative for the application of fertilizers, since majority of these phosphate solubilizers are present in the soil [87, 88]. A considerable proportion of bacteria tested in the present study could promote plant nutrient uptake by making phosphorous available through inorganic phosphate solubilization (Table 3). Bacteria of the genus Bacillus, including B. amyloliquefaciens, B. megaterium, and B. subtilis, isolated from maize [9, 10, 89], rice (Oryza sativa L.) [90, 91], Medicago polymorpha [92], and soil [93] are phosphate solubilizers, and these bacteria can solubilize phosphate even under salinity stress.
Biological nitrogen fixation is relevant to plant growth. Several bacterial genera can fix nitrogen [11,12,13,14,15,16,17,18]. In the present study, only LGMB417 (Stenotrophomonas sp.) showed this possible ability, as evidenced by the detection of the nifH gene and the results of biochemical tests. Bacteria of the genus Stenotrophomonas are present in different types of soil and rhizospheres of different crops, and they can promote plant growth through biological nitrogen fixation [94].
Iron is another essential nutrient for plant development. Therefore, siderophore production is critical. Iron acts as an enzyme cofactor in biochemical pathways involved in several plant physiological processes, such as respiration, photosynthesis, and biological nitrogen fixation [23]. In the present study, over half of the isolates evaluated [LGMB12, LGMB273, LGMB319, LGMB426, LGMB444 (Bacillus spp.), and LGMB324 (Lysinibacillus sp.)] could produce siderophores, indicating their significance in the context of plant growth. Tropical soils are characterized by intense geochemical weathering of primary minerals in the substratum rock, leading to the formation of stable secondary minerals, such as hematite and goethite (iron oxides) [95]; thus, Brazilian soils are rarely iron deficient. PGPB that can produce siderophores are capable of biocontrol, since the siderophores produced by these isolates have a greater affinity toward iron present in the soil [96]. This gives rise to competition among microorganisms for establishment in the rhizosphere, which can further prevent the proliferation of harmful microbes due to iron deficiency and loss of their pathogenicity [97]. Bacillus spp. and Enterobacter spp. isolated from iron-enriched soils were proven promising candidates for siderophore production [98]. Moreover, bacteria of the genus Stenotrophomonas play pivotal roles in biogeochemical processes, such as sulfur and nitrogen cycle [99,100,101]. In addition, Stenotrophomonas spp. can produce different forms of siderophore [102, 103].
IAA production is crucial for plant development [102]. Bacteria of the genera Enterobacter, Bacillus, and Pseudomonas can produce IAA [84]. This phytohormone promotes cell stretching, division, and differentiation [104]. However, optimum levels of IAA are critical, as the roots are extremely sensitive to auxin. As such, when present in trace amounts, IAA can activate plant responses, but at high concentrations, it can produce inhibitory effects [105]. In the present study, variations in IAA production were observed among the tested isolates. However, in some cases, we noted discordance between the results of biochemical and molecular analyses. For instance, LGMB444 could produce IAA according to the results of biochemical test, but the gene encoding the precursor of this phytohormone was not detected in molecular assays, perhaps because of the lack of primer specificity or the presence of an IAA residue in the bacteria itself [106, 107]. The opposite trends were observed for LGMB12, LGMB426, and LGMB429, which showed positive results of gene amplification but negative results in the biochemical tests (no pellicle formation in the semi-solid medium); this may be attributed to the incompatibility between the species analyzed and the conditions of biochemical assays [108].
Genes involved in the biological control of phytopathogens, such as phlD and acdS [109, 110], were also investigated. However, these genes were not detected in any isolate. According to Bruto et al. [111], not all PGPB possess the phlD gene, and the acdS gene is rather rare in genera of the order Enterobacterales (e.g., Enterobacter, among others). Few genera that have evolved close relationships with plants (e.g., Dickeya) express these genes [112].
Another mechanism underlying plant growth promotion includes the production of enzymes, such as amylases, which represent one of the most important groups of enzymes [113]. In the present study, LGMB12, LGMB273, LGMB319, LGMB426, LGMB444 (Bacillus spp.), LGMB45, LGMB324 (Lysinibacillus spp.), LGMB417 (Stenotrophomonas sp.), LGMB429 (Paenibacillus sp.), and LGMB456 (Pseudomonas sp.) produced amylase, indicating their ability to degrade and process raw materials or synthesize related products [114].
Hydrolytic enzymes directly affect the activity of soil microbiota (microbial biomass and basal respiration, among others). These enzymes are involved in the biocontrol of pathogens as well as the decomposition of organic compounds and release of nutrients in the soil, which can enhance soil quality and ultimately improve crop fields [115, 116]. In addition, other enzymes, such as chitinase, cellulase, and protease, among others, play vital roles in agriculture. For instance, these enzymes can enable biocontrol against many fungi through cell wall lysis [25, 26, 117]. The second most frequently detected enzyme in the present study was protease, produced by LGMB12, LGMB273, LGMB319, LGMB426, LGMB444 (Bacillus spp.), LGMB45, LGMB324 (Lysinibacillus spp.), LGMB417 (Stenotrophomonas sp.), and LGMB456 (Pseudomonas sp.). Proteases break down disrupted proteins to recycle their amino acids for use in other functions related to the regulation of plant growth, development, and defense [118].
Furthermore, LGMB12, LGMB273, LGMB319, LGMB426, LGMB444 (Bacillus spp.), LGMB417 (Stenotrophomonas sp.), LGMB429 (Paenibacillus sp.), and LGMB456 (Pseudomonas sp.) produced both cellulase and pectinase, which are involved in the lysis of pathogen cell wall, inducing plant systemic resistance during pathogen colonization [119]. LGMB23 (Lysinibacillus sp.), LGMB417 (Stenotrophomonas sp.), LGMB444 (Bacillus sp.), and LGMB456 (Pseudomonas sp.) produced esterase, which can inhibit the growth of pathogens, thus minimizing their detrimental effects on plants [120]. Only LGMB273 and LGMB444 (Bacillus spp.) produced chitinase. Isolates that can produce chitinase are potential biocontrol agents [121, 122]. Urease is another enzyme of extreme importance, and half of the isolates tested in the present study [LGMB45, LGMB324 (Lysinibacillus spp.), LGMB273, LGMB444 (Bacillus spp.), LGMB417 (Stenotrophomonas sp.), and LGMB456 (Pseudomonas sp.)] produced this enzyme. Through the inoculation of PGPB in the rhizosphere, urease is involved in the conversion of nitrogen for assimilation by plants, promoting growth and biocontrol activity [123].
Conclusion
In the present study, we explored the diversity of bacteria isolated from maize rhizosphere to evaluate their plant growth-promoting potential. Gene sequencing revealed the presence of the genera Atlantibacter, Bacillus, Enterobacter, Lysinibacillus, Paenibacillus, Pseudomonas, Rhizobium, and Stenotrophomonas. Eleven bacteria stood out in in vivo assays. Among these, five isolates [LGMB12, LGMB273, LGMB426 (Bacillus spp.), LGMB417 (Stenotrophomonas sp.), and LGMB456 (Pseudomonas sp.)] showed the most promising results and the greatest potential for plant growth promotion in maize owing to their biochemical and enzymatic characteristics. Thus, further research is warranted to test the biotechnological potential of these isolates under field conditions, exploring their utility as alternative biofertilizers for these crops. Moreover, it would be interesting to identify the bacteria to the species level, to exclude the ones pathogenic to plants, humans, and other non-human animals and elucidate the growth-promoting mechanisms within the plants.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
USDA - United States Department of Agriculture (2020) Grain: world markets and trade. https://apps.fas.usda.gov/psdonline/circulars/grain-corn-coarsegrains.pdf. Accessed 05 Oct. 2020
García-Lara S, Serna-Saldivar SO (2019) Corn history and culture. In: Serna-Saldivar SO (ed) Corn: chemistry and technology. Elsevier, pp 1-18. https://doi.org/10.1016/B978-0-12-811971-6.00001-2
FAO - Food and agriculture organization (2015) Status of the world’s soil resources: Main report. http://www.fao.org/documents/card/en/c/c6814873-efc3-41db-b7d3-2081a10ede50/pdf. Accessed 06 Oct. 2020
Rashmi I, Roy T, Kartika KS, Pal R, Coumar V, Kala S, Shinoji KC (2020) Organic and inorganic fertilizer contaminants in agriculture: impact on soil and water resources. In: Naeem M, Ansari A, Gil S (eds) Contaminants in Agriculture. Springer, Cham, pp 3-41. https://doi.org/10.1007/978-3-030-41552-5_1
Harman GE, Uphoff N (2019) Symbiotic root-endophytic soil microbes improve crop productivity and provide environmental benefits. Scientifica 9106395. https://doi.org/10.1155/2019/9106395
Hungria M, Campo RJ, Souza EM, Pedrosa FO (2010) Inoculation with selected strains of Azospirillum brasilense and A. lipoferum improves yields of maize and wheat in Brazil. Plant Soil 331:413–425. https://doi.org/10.1007/s11104-009-0262-0
Videira SS, Oliveira DM, Morais RF, Borges WL, Baldani VLD, Baldani JI (2012) Genetic diversity and plant growth promoting traits of diazotrophic bacteria isolated from two Pennisetum purpureum Schum. genotypes grown in the field. Plant Soil 356:51–66. https://doi.org/10.1007/s11104-011-1082-6
Cavalcanti MIP, Nascimento RC, Rodrigues DR, Escobar IEC, Fraiz ACR, Souza AP, Freitas ADS, Nóbrega RSA, Fernandes-Júnior PI (2020) Maize growth and yield promoting endophytes isolated into a legume root nodule by a cross-over approach. Rhizosphere 15:100211. https://doi.org/10.1016/j.rhisph.2020.100211
Bomfim CSG, Silva VB, Cursino LHS, Mattos WS, Santos JCSS, Souza LSB, Dantas BF, Freitas ADS, Fernandes-Júnior PI (2020) Endophytic bacteria naturally inhabiting commercial maize seeds occupy different niches and are efficient plant growth-promoting agents. Symbiosis 81:255–269. https://doi.org/10.1007/s13199-020-00701-z
Nascimento RC, Cavalcanti MIP, Correia AJ, Escobar IEC, Freitas ADS, Nóbrega RSA, Fernandes-Júnior PI (2021) Maize-associated bacteria from the Brazilian semiarid region boost plant growth and grain yield. Symbiosis 83:347–359. https://doi.org/10.1007/s13199-021-00755-7
Alves GC, Santos CLR, Zilli JE, Reis Junior FB, Marriel IE, Breda FAF, Boddey RM, Reis VM (2021) Agronomic evaluation of Herbaspirillum seropedicae strain ZAE94 as an inoculant to improve maize yield in Brazil. Pedosphere 31(4):583–595. https://doi.org/10.1016/S1002-0160(21)60004-8
Benami M, Gross A, Herzberg M, Prlofsky E, Vonshak A, Gillor O (2013) Assessment of pathogenic bacteria in treated graywater and irrigated soils. Sci Total Environ 1(458–460):298–302. https://doi.org/10.1016/j.scitotenv.2013.04.023
Etesami H, Alikhani HA (2019) Halotolerant plant growth-promoting fungi and bacteria as an alternative strategy for improving nutrient availability to salinity-stressed crop plants. In: Kumar M, Etesami H, Kumar V (eds) Saline soil-based agriculture by halotolerant microorganisms. Springer, Singapore, pp 103-146. https://doi.org/10.1007/978-981-13-8335-9_5
Crema A, Vandini G, Boschetti M, Nutini F, Cillis D, Casa R (2019) Interaction between soil variability and maize nitrogen status assessment from Sentinel-2. Precis Agric 19:453–459. https://doi.org/10.3920/978-90-8686-888-9_56
Carciochi WD, Sadras VO, Pagani A, Ciampitti IA (2020) Co-limitation and stoichiometry capture the interacting effects of nitrogen and sulfur on maize yield and nutrient use efficiency. Eur J Agron 113:125973. https://doi.org/10.1016/j.eja.2019.125973
Figueiredo LHM, Vasconcellos AG, Prado GS, Grossi-de-Sa MF (2019) An overview of intellectual property within agricultural biotechnology in Brazil. Biotechnol Res Innov 3(1):69–79. https://doi.org/10.1016/j.biori.2019.04.003
Azabou MC, Gharbi Y, Medhioub I, Ennouri K, Barham H, Tounsi S, Triki MA (2020) The endophytic strain Bacillus velezensis OEE1: an efficient biocontrol agent against Verticillium wilt of olive and potential plant growth promoting bacteria. Biol Control 142:104168. https://doi.org/10.1016/j.biocontrol.2019.104168
Kour D, Rana KL, Yadav N, Yadav AN, Kumar A, Meena VS, Singh B, Chauhan VS, Dhaliwal HS, Saxena AK (2019) Rhizospheric microbiomes: biodiversity, mechanisms of plant growth promotion, and biotechnological applications for sustainable agriculture. In: Kumar A, Meena V (eds) Plant growth promoting rhizobacteria agricultural sustainability, Springer, Singapore, pp 19-65. https://doi.org/10.1007/978-981-13-7553-8_2
Pii Y, Aldrighetti A, Valentinuzzi F, Mimmo T, Cesco S (2019) Azospirillum brasilense inoculation counteracts the induction of nitrate uptake in maize plants. J Exp Bot 70:1313–1324. https://doi.org/10.1093/jxb/ery433
Gunnabo AH, Geurts R, Wolde-Meskel E, Degefu T, Giller KE, Heerwaarden JV (2019) Genetic interaction studies reveal superior performance of Rhizobium tropici CIAT899 on a range of diverse east African common bean (Phaseolus vulgaris L) genotypes. Appl Environ Microbiol 85(24):e01763-19. https://doi.org/10.1128/AEM.01763-19
Fernandez-Göbel TF, Deanna R, Muñoz NB, Robert G, Asurmendi S, Lascano R (2019) Redox systemic signaling and induced tolerance responses during soybean – Bradyrhizobium japonicum interaction: involvement of nod factor receptor and autoregulation of nodulation. Front Plant Sci 10:141. https://doi.org/10.3389/fpls.2019.00141
Nehl DB, Allen SJ, Brown JF (1997) Deleterious rhizosfere bacteria: an integrating perspective. Appl Soil Ecol 5(1):1–20. https://doi.org/10.1016/S0929-1393(96)00124-2
Sansinenea E (2019) Bacillus spp.: as plant growth-promoting bacteria. In: Singh HB, Keswani C, Reddy MS, Sansinenea E, García-Estrada C (eds) Secondary metabolites of plant growth promoting rhizomicroorganisms. Springer, Singapore, pp 225-237. https://doi.org/10.1007/978-981-13-5862-3_11
Levy A, Conway JM, Dangl JL, Woyke T (2018) Elucidating bacterial gene functions in the plant microbiome. Cell Host Microbe 24(4):475–485. https://doi.org/10.1016/j.chom.2018.09.005
Reis Junior FB, Mendes IC, Teixeira KRS, Reis VM (2002) Uso de ferramentas moleculares em estudos da diversidade de microrganismos do solo. Embrapa, Distrito Federal
Zanini SF, Mussi JMS, Zanini MS, Sousa DR, Pessotti BMS, Damasceno JDLM, Silva MAS (2012) Identificação bioquímica e molecular de Lactobacillus spp. Isolados do íleo de frangos de corte tratados ou não com antimicrobianos. Cienc Rural 42(9):1648-1654 https://doi.org/10.1590/S0103-84782012000900021
Bull CT, Shetty KG, Subbarao KV (2002) Interactions between myxobacteria, plant pathogenic fungi, and biocontrol agents. Plant Dis 86:889–896. https://doi.org/10.1094/PDIS.2002.86.8.889
Malusà E, Pinzari F, Canfora L (2016) Efficacy of biofertilizers: challenges to improve crop production. In: Singh DP, Singh HB, Prabha R (eds) Microbial inoculants in sustainable agricultural productivity. Springer, New Delhi, 17-40. https://doi.org/10.1007/978-81-322-2644-4_2
Motoyama AB, Venancio EJ, Brandão GO, Petrofeza-Silva S, Pereira IS, Soares CMA, Felipe MSS (2000) Molecular identification of Paracoccidioides brasiliensis by PCR amplification of ribosomal DNA J Clin Microbiol. 38(8):3106–3109. https://doi.org/10.1128/JCM.38.8.3106-3109.2000
Ahemad M, Kibret M (2014) Mechanisms and applications of plant growth promoting rhizobacteria: current perspective. J King Saud Univ Sci 26:1–20. https://doi.org/10.1016/j.jksus.2013.05.001
Zuluaga MYA, Milani KML, Gonçalves LSA, Oliveira ALM (2020) Diversity and plant growth-promoting functions of diazotrophic/N-scavenging bacteria isolated from the soils and rhizospheres of two species of Solanum. PLoS ONE 15(1):e0227422. https://doi.org/10.1371/journal.pone.0227422
Vassilev N, Vassileva A, Lopez A, Martos V, Reyes A, Maksimovic I, Eichler-Löbermann B, Malusà E (2015) Unexploited potential of some biotechnological techniques for biofertilizer production and formulation. Appl Microbiol Biotechnol 99:4983–4996. https://doi.org/10.1007/s00253-015-6656-4
Ikeda AC, Bassani LL, Adamoski D, Stringari D, Cordeiro VK, Glienke C, Steffens MBR, Hungria M, Galli-Terasawa LV (2013) Morphological and genetic characterization of endophytic bacteria isolated from roots of different maize genotypes. Microb Ecol 65(1):154–160. https://doi.org/10.1007/s00248-012-0104-0
Szilagyi-Zecchin VJ, Ikeda AC, Hungria M, Adamoski D, Kava-Cordeiro V, Glienke C, Galli-Terasawa LV (2014) Identification and characterization of endophytic bacteria from corn (Zea mays L) roots with biotechnological potential in agriculture AMB. Express 4(1):1–9. https://doi.org/10.1186/s13568-014-0026-y
Versalovic J, Schneider M, Bruijin F, Lupski JR (1994) Genomic fingerprinting of bacteria using repetitive sequence-based polymerase chain reaction. Method Mol Cell Biol 5(1):25-40. ISSN: 0898-7750
Kaschuk G, Hungria M, Andrade DS, Campo RJ (2006) Genetic diversity of rhizobia associated with common bean grown under the no-tillage and conventional systems in South Brazil. Appl Soil Ecol 32(2):210–220. https://doi.org/10.1016/j.apsoil.2005.06.008
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principles and practice of numeral classification. WH Freeman & Company, San Francisco
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2019) Vegan: community ecology package. R package version 2: 5-6. https://cran.r-project.org, https://github.com/vegandevs/vegan
Suzuki R, Shimodaira H (2006) Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22(12):1540–1542. https://doi.org/10.1093/bioinformatics/btl117
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY (2017) Ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol 8(1):28–36. https://doi.org/10.1111/2041-210X.12628
Yu G, Lam TTL, Zhu H, Guan Y (2018) Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol Biol Evol 35(12):3041–3043. https://doi.org/10.1093/molbev/msy194
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173(2):697–703. https://doi.org/10.1128/jb.173.2.697-703.1991
Menna P, Hungria M, Barcellos FG, Bangel EV, Hess PN, Martínez-Romero E (2006) Molecular phylogeny based on the 16S rRNA gene of elite rhizobial strains used in Brazilian commercial inoculants. Syst Appl Microbiol 29(4):315–332. https://doi.org/10.1016/j.syapm.2005.12.002
Kimoto M, Soh SHG, Hirao I (2020) Sanger gap sequencing for genetic alphabet expansion of DNA. ChemBioChem 21(16):2287–2296. https://doi.org/10.1002/cbic.202000057
Euzéby JP (1997) List of bacterial names with standing in nomenclature: a folder available on the internet. Int J Syst Evol Microbiol 47(2):590–592. https://doi.org/10.1099/00207713-47-2-590
Parte AC (2013) LPSN – list of prokaryotic names with standing in nomenclature. Nucleic Acids Res 42(D1):613–616. https://doi.org/10.1093/nar/gkt1111
Parte AC (2018) LPSN – list of prokaryotic names with standing in nomenclature (bacterio.net), 20 years on. Int J Syst Evol Microbiol 68(6):1825–1829. https://doi.org/10.1099/ijsem.0.002786
Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Shäffer AA, Yu YK (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272(20):5101–5109. https://doi.org/10.1111/j.1742-4658.2005.04945.x
Edgar RC (2004) Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In: Nucleic acids symposium series, Information Retrieval Ltd., London. 41:95-98. https://doi.org/10.14601/Phytopathol_Mediterr-14998u1.29
Kumar S, Singh R, Williams CP (1863) Van Der Klei IJ (2016) Stress exposure results in increased peroxisomal levels of yeast Pnc1 and Gpd1, which are imported via a piggy-backing mechanism. BBA - Mol Cell Res 1:148–156. https://doi.org/10.1016/j.bbamcr.2015.10.017
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539-542 https://doi.org/10.1093/sysbio/sys029
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum-likelihood. Syst Biol 52(5):696–704. https://doi.org/10.1080/10635150390235520
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9(8):772. https://doi.org/10.1038/nmeth.2109
Hoagland DR, Arnon DI (1950) The water-culture method for growing plants without soil, 2nd edn. Circular, California, 347:1884-1949
Török I, Kondorosi A (1981) Nucleotide sequence of the R. meliloti nitrogenase reductase (nifH) gene. Nucleic Acids Res 9(21):5711-5724. https://doi.org/10.1093/nar/9.21.5711
Gardener BBM, Mavrodi DV, Thomashow LS, Weller DM (2001) A rapid polymerase chain reaction-based assay characterizing rhizosphere populations of 2,4-diacetylphloroglucionol-producing bacteria. Phytopathology 91(1):44–54. https://doi.org/10.1094/PHYTO.2001.91.1.44
Meyer JB, Frapolli M, Keel C, Maurhofer M (2011) Pyrroloquinoline quinone biosynthesis gene pqqC, a novel molecular marker for studying the phylogeny and diversity of phosphate-solubilizing pseudomonads. Appl Environ Microbiol 77(20):7345–7354. https://doi.org/10.1128/AEM.05434-11
Patten CL, Glick BR (2002) Role of Pseudomonas putida indoleacetic acid in development of the host plant root system. Appl Environ Microbiol 68(8):3795–3801. https://doi.org/10.1128/aem.68.8.3795-3801.2002
Braker G, Fesefeldt A, Witzel KP (1998) Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl Environ Microbiol 64(10):3769–3775. https://doi.org/10.1128/AEM.64.10.3769-3775.1998
Blaha D, Prigent-Combaret C, Mirza MS, Moënne-Loccoz Y (2006) Phylogeny of the 1-aminocyclopropane-1-carboxylic acid deaminase-encoding gene acdS in phytobeneficial and pathogenic Proteobacteria and relation with strain biogeography. FEMS Microbiol Ecol 56(3):455–470. https://doi.org/10.1111/j.1574-6941.2006.00082.x
Schwyn B, Neilands JB (1987) Universal chemical assay for the detection and determination of siderophores. Anal Biochem 160(1):47–56. https://doi.org/10.1016/0003-2697(87)90612-9
Sylvester-Bradley R, Asakawa N, La Torraca S, Magalhães FM, Oliveira LA, Pereira RM (1982) Levantamento quantitativo de microrganismo solubilizadores de fosfatos na rizosfera de gramíneas e forrageiras na Amazônia. Acta Amaz 12(1):15–22. https://doi.org/10.1590/1809-43921982121015
Araújo LM, Monteiro RA, Souza EM, Steffens MBR, Rigo LU, Pedrosa FO, Chubatsu LS (2004) GlnB is specifically required for Azospirillum brasilense NifA activity in Escherichia coli. Res Microbiol 155(6):491–495. https://doi.org/10.1016/j.resmic.2004.03.002
Kuss AV, Kuss VV, Lovato T, Flôres ML (2007) Fixação de nitrogênio e produção de ácido indol acético in vitro por bactérias diazotróficas endofíticas. Pesq Agropec Bras 42(10):1459–1465. https://doi.org/10.1590/S0100-204X2007001000013
Mandels M, Weber J (1969) The production of cellulases Adv Chem 95(23):937–414. https://doi.org/10.1021/ba-1969-0095.ch023
Hankin L, Anagnostakis SL (1975) The use of solid media for detection of enzyme production by fungi. Mycologia 67(3):597–607. https://doi.org/10.1080/00275514.1975.12019782
Renwick A, Campbell R, Coe S (1991) Assessment of in vivo screening systems for potential biocontrol agents of Gaeumannomyces graminis. Plant Pathol 40(4):524–532. https://doi.org/10.1111/j.1365-3059.1991.tb02415.x
Moura JM, Ferreira AF, Silva FMM, Rizzi J, Pinto LAA (2005) Obtenção de quitina a partir de carapaças de siri (Maia squinado): uso de um planejamento experimental na etapa de desmineralização. Vetor 15(1):7–17
Reid JD, Ogrydziak DM (1981) Chitinase-overproducing mutant of Serratia marcescens. Appl Environ Microbiol. 41(3):664–669. https://doi.org/10.1128/AEM.41.3.664-669.1981
Sierra GA (1957) A simple method for the detection of lypolytic activity of micro-organisms and some observations on the influence of the contact between cells and fatty substracts. Antonie Van Leeuwenhoek 23(1):15–22. https://doi.org/10.1007/bf02545855
Berg G, Roskot N, Steidle A, Eberl L, Zock A, Smalla K (2002) Plant-dependent genotypic and phenotypic diversity of antagonistic rhizobacteria isolated from different Verticillium host plants. Appl Environ Microbiol 68(7):3328–3338. https://doi.org/10.1128/AEM.68.7.3328-3338.2002
Dye DW (1968) A taxonomic study of the genus Erwinia 1. The “amylovora” group. J Sci 11:590–607. https://doi.org/10.1080/00288233.1981.10420894
Fukami J, Cerezini P, Hungria M (2018) Azospirillum: benefits that go far beyond biological nitrogen fixation. AMB Express 8:73. https://doi.org/10.1186/s13568-018-0608-1
Naseer I, Ahmad M, Nadeem SM, Ahmad I, Seher N, Zahir ZA (2019) Rhizobial inoculants for sustainable agriculture: prospects and applications. In: Giri B, Prasad R, Wu QS, Varma A (eds) Biofertilizers for sustainable agriculture and environment. Springer, Cham, pp 245-283. https://doi.org/10.1007/978-3-030-18933-4_11
Santos MS, Nogueira MA, Hungria M (2019) Microbial inoculants: reviewing the past, discussing the present and previewing an outstanding future for the use of beneficial bacteria in agriculture. AMB Express 9(1):205. https://doi.org/10.1186/s13568-019-0932-0
Kim W, Hong Y-p, Yoo J-h, Lee W-b, Choi C-s, Chung S-i (2002) Genetic relationship of Bacillus anthracis and closely related species based on variable-number tandem repeat analysis and BOX-PCR genomic fingerprinting. FEMS Microbiol Lett 207(1):21-27. 10. 1111/j.1574-6968.2002.tb11022.x
Gardan L, Shafik H, Belouin S, Brosch R, Grimont F, Grimont PAD (1999) DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson 1959). Int J Syst Bacteriol 49(2):469–478. https://doi.org/10.1099/00207713-49-2-469
Marques ASA, Marchaison A, Gardan L, Samson R (2008) BOX-PCR-based identification of bacterial species belonging to Pseudomonas syringae – P. viridiflava group. Genet Mol Biol 31(1):106-115. https://doi.org/10.1590/S1415-47572008000100019
Viana TFC, Campelo APS, Baldani JI, Fernandes Júnior PI, Baldani VLD, Silva WM, Paggi GM, Brasil MS (2020) Cultivable bacterial diversity associated with bromeliad roots from ironstone outcrops in central Brazil. Braz J Biol 80(4):872–880. https://doi.org/10.1590/1519-6984.224982
Borah A, Das R, Mazumdar R, Thakur D (2019) Culturable endophytic bacteria of Camellia species endowed with plant growth promoting characteristics. J Appl Microbiol 127(3):825–844. https://doi.org/10.1111/jam.14356
Ikeda AC, Szilagyi-Zecchin VJS, Savi DC, Kava V, Glienke C, Hungria M, Galli-Terasawa LV (2018) Bio prospecting plant growth-promoting bacteria isolated from maize (Zea mays L.) roots. J Biotech Res Biochem 1(1):10003. https://doi.org/10.24966/BRB-0019/100002
Khan AAH (2019) Plant-bacterial association and their role as growth promoters and biocontrol agents. In: Sayyed R (ed) Plant growth promoting rhizobacteria for sustainable stress management. Springer, Singapore, pp 389-419. 10. https://doi.org/10.1007/978-981-13-6986-5_16
Maela PM, Serepa-Dlamini MH (2019) Current understanding of bacterial endophytes, their diversity, colonization and their roles in promoting plant growth. Appl Microbiol 5(1):1000157. https://doi.org/10.4172/2471-9315.1000157
Maheshwari DK, Saraf M, Dheeman S (2019) Plant growth-promoting rhizobacteria (PGPR) as protagonist of ever-sustained agriculture: an introduction. In: Maheshwar D, Dheeman S (eds) Field crops: sustainable management by PGPR. Springer, Cham, pp 1-10. https://doi.org/10.1007/978-3-030-30926-8_1
Su W, Kamran M, Xie J, Meng X, Han Q, Liu T, Han J (2019) Shoot and root traits of summer maize hybrid varieties with higher grain yields and higher nitrogen use efficiency at low nitrogen application rates. Peer J 7:e7294. https://doi.org/10.7717/peerj.7294
Gao J, Luo Y, Wei Y, Huang Y, Zhang H, He W, Sheng H, An L (2019) Screening of plant growth promoting bacteria (PGPB) from rhizosphere and bulk soil of Caragana microphylla in different habitats and their effects on the growth of Arabidopsis seedlings. Biotechnol Biotechnol Equip 33(1):921–930. https://doi.org/10.1080/13102818.2019.1629841
Lobo CB, Tomás MSJ, Viruel E, Ferrero MA, Lucca ME (2019) Development of low-cost formulations of plant growth-promoting bacteria to be used as inoculants in beneficial agricultural technologies. Microbiol Res 219:12–25. https://doi.org/10.1016/j.micres.2018.10.012
Babu SV, Triveni S, Reddy RS, Sathyanarayana J (2017) Screening of maize rhizosperic phosphate solubilizing isolates for plant growth promoting characteristics. Int J Curr Microbiol App Sci 6:2090-2101. https://doi.org/10.20546/ijcmas.2017.610.249
Nautiyal CS, Srivastava S, Chauhan PS, Seem K, Mishra A, Sopory SK (2013) Plant growth-promoting bacteria Bacillus amyloliquefaciens NBRISN13 modulates gene expression profile of leaf and rhizosphere community in rice during salt stress. Plant Physiol Biochem 66:1–9. https://doi.org/10.1016/j.plaphy.2013.01.020
Borah M, Das P, Pathak SS, Boro RC, Barooah M (2017) Phosphate solubilization by endophytic bacteria isolated from Oryza sativa. Int. J. Curr Microbiol App Sci 6(10):2713-2721. https://doi.org/10.20546/ijcmas.2017.610.319
Chinnaswamy A, Peña TC, Stoll A, Rojo DP, Bravo J, Ricón A, Lucas MM, Pueyo JJ (2018) A nodule endophytic Bacillus megaterium strain isolated from Medicago polymorpha enhances growth, promotes nodulation by Ensifer medicae and alleviates salt stress in alfalfa plants. Ann Appl Biol 172:295–308. https://doi.org/10.1111/aab.12420
Kim MJ, Radhakrishnan R, Kang SM, You YH, Jeong EJ, Kim JG, Lee IJ (2017) Plant growth promoting effect of Bacillus amyloliquefaciens H-2-5 on crop plants and influence on physiological changes in soybean under soil salinity. Physiol Mol Biol Plants 23(3):571–580. https://doi.org/10.1007/s12298-017-0449-4
Perez-Perez R, Oudot M, Hernandez I, Napoles MC, Perez-Martinez S, Castillo DSD (2020) Isolation and characterization of Stenotrophomonas associated to maize (Zea mays L.) rhizosphere. Cultivos Tropicales 41(2). ISSN 1819-4087.
Cornell RM, Schwertmann U (2003) The iron oxides: structure, properties, reactions, occurrences and uses, 2nd edn. Wiley-VCH, Weinheim. https://doi.org/10.1002/3527602097
Shen X, Hu H, Peng H, Wang W, Zhang X (2013) Comparative genomic analysis of four representative plant growth-promoting rhizobacteria in Pseudomonas. BMC Genomics 14(1):271. https://doi.org/10.1186/1471-2164-14-271
Olanrewaju OS, Glick BR, Babalola OO (2017) Mechanisms of action of plant growth promoting bacteria. World J Microbiol Biotechnol 33(11):197. https://doi.org/10.1007/s11274-017-2364-9
Kumar VS, Menon S, Agarwal H, Gopalakrishnan D (2017) Characterization and optimization of bacterium isolated from soil samples for the production of siderophores. Resour Efficient Technologies 3(4):434–439. https://doi.org/10.1016/j.reffit.2017.04.004
Banerjee M, Yesmin L (2002) Sulfur-oxidizing plant growth promoting rhizobacteria for enhanced canola performance. US Patent 07491535.
Park M, Kim C, Yang J, Lee H, Shin W, Kim S, Sa T (2005) Isolation and characterization of diazotrophic growth promoting bacteria from rhizosphere of agricultural crops of Korea. Microbiol Res 160(2):127–133. https://doi.org/10.1016/j.micres.2004.10.003
Ramos PL, Moreira-Filho CA, Trappen SV, Swings J, Vos PD, Barbosa HR, Thompson CC, Vasconcelos ATR, Thompson FL (2011) An MLSA-based online scheme for the rapid identification of Stenotrophomonas isolates. Mem Inst Oswaldo Cruz 106(4):394–399. https://doi.org/10.1590/S0074-02762011000400003
Chhibber S, Gupta A, Sharan R, Gautam V, Ray P (2008) Putative virulence characteristics of Stenotrophomonas maltophilia: a study on clinical isolates. World J Microbiol Biotechnol 24(12):2819–2825. https://doi.org/10.1007/s11274-008-9812-5
Ryan RP, Monchy S, Cardinale M, Taghavi S, Crossman L, Avison MB, Berg G, Van der Lelie D, Dow JM (2009) The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat Rev Microbiol 7(7):514–525. https://doi.org/10.1038/nrmicro2163
Mayer E, Quadros PD, Fulthorpe R (2019) Plantibacter flavus, Curtobacterium herbarum, Paenibacillus taichungensis, and Rhizobium selenitireducens endophytes provide host-specific growth promoting of Arabidopsis thaliana, basil, lettuce, and bok choy plants. Appl Environ Microbiol 85(19):e00383-e419. https://doi.org/10.1128/AEM.00383-19
Pandey H, Pandey V, Nandi SK, Palni LMS (2019) Role of plant growth substances in regulating pseudomonocotyly and correlative inhibition in some alpine Himalayan rosettes. S Afr J Bot 125:493–498. https://doi.org/10.1016/j.sajb.2019.08.017
Defez R, Valenti A, Andreozzi A, Romano S, Ciaramella M, Pesaresi P, Fornali S, Bianco C (2019) New insights into structural functional roles of indole-3-acetic acid (IAA): changes in DNA topology and gene expression in bacteria. Biomolecules 9(10):522. https://doi.org/10.3390/biom9100522
Orzinska A, Guz K, Mikula M, Kulecka M, Kluska A, Balabas A, Pelc-Klopotowska M, Ostrowski J, Brojer E (2018) A preliminary evaluation of next-generation sequencing as a screening tool for target genotyping of erythrocyte and platelet antigens in blood donors. Blood Transfus 16(3):285–292. https://doi.org/10.2450/2017.0253-16
Kell DB, Kaprelyants AS, Weichart DH, Harwood CR, Barer MR (1998) Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Van Leeuwenhoek 73(169):169–187. https://doi.org/10.1023/a:1000664013047
Glick BR, Todorovic B, Czarny J, Cheng Z, Duan J, McConkey B (2007) Promotion of plant growth by bacterial ACC deaminase. Crit Rev Plant Sci 26(5–6):227–242. https://doi.org/10.1080/07352680701572966
Hernández-León R, Rojas-Solís D, Contreras-Pérez M, Orozco-Mosqueda MC, Macías-Rodríguez LI, Cruz HR, Valencia-Cantero E, Santoyo G (2015) Characterization of the antifungal and plant growth-promoting effects of diffusible and volatile organic compounds produce by Pseudomonas fluorescens strains. Biol Control 81:83–92. https://doi.org/10.1016/j.biocontrol.2014.11.011
Bruto M, Prigent-Combaret C, Muller D, Moënne-Loccoz Y (2014) Analysis of genes contributing to plant-beneficial functions in plant growth-promoting rhizobacteria and related Proteobacteria. Sci Rep 4:6261. https://doi.org/10.1038/srep06261
Nascimento FX, Rossi MJ, Soares CRFS, McConkey BJ, Glick BR (2014) New insights into 1-aminocyclopropane-1-carboxylate (ACC) desaminase phylogeny, evolution and ecological significance. PLoS ONE 9(6):e99168. https://doi.org/10.1371/journal.pone.0099168
Souza PM, Magalhães PO (2010) Application of microbial α-amylase in industry – a review. Braz J Microbiol 41(4):850–861. https://doi.org/10.1590/S1517-83822010000400004
Hasan F, Shah AA, Hameed A (2006) Industrial applications of microbial lipases. Enzyme Microb Technol 39(2):235–251. https://doi.org/10.1016/j.enzmictec.2005.10.016
Trasar-Cepeda C, Leers MC, Gil-Sotres (2008) Hydrolytic enzyme activities in agricultural and forest soils. Some implications for their use as indicators. Soil Biol Biochem 40(9):2146-2155 https://doi.org/10.1016/j.soilbio.2008.03.015
Inayati A, Sulistyowati L, Aini LQ, Yusnawan E (2020) Mycoparasitic activity of indigenous Trichoderma virens strains against mungbeen soil borne pathogen Rhizoctonia solani: hyperparasite and hydrolytic enzyme production. J Agric Sci 42(2):229-242 https://doi.org/10.17503/agrivita.v0i0.2514
Mahanty T, Bhattacharjee S, Goswami M, Bhattacharyya P, Das B, Ghosh A, Tribedi P (2017) Biofertilizers: a potential approach for sustainable agriculture development. Environ Sci Pollut Res 24:3315–3335. https://doi.org/10.1007/s11356-016-8104-0
Mouayed H, Wardy GA, Seddeq ASS (2019) Isolation and identification of Pseudomonas putida from soils of plant roots and determine the ability to produce hydrolases enzymes. Iraqi J Sci 60:228-233. https://doi.org/10.24996/ijs.2019.60.2.4
Ashok G, Nambirajan G, Baskaran K, Viswanathan C, Alexander X (2020) Phylogenetic diversity of epiphytic pink-pigmented methylotrophic bacteria and role in alleviation of abiotic stress in plants. In: Yadav AN, Singh J, Rastegari AA, Yadav N (eds) Plants microbiomes for sustainable agriculture. Springer, Cham, pp 245-262. https://doi.org/10.1007/978-3-030-38453-1_8
Shah SB, Hu H, Wang W, Liu Y, Ali F, Xu P, Tang H (2019) Evaluation of plant growth-promoting activity of strain HBCD-sjtu. J Biol Regul Homeost Agents 33(4):1187–1192
Singh RK, Kumar DP, Solanki MK, Singh P, Srivastva AK, Kumar S, Kashyap PL, Saxena AK, Singhal PK, Arora DK (2013) Optimization of media components for chitinase production by chickpea rhizosphere associated Lysinibacillus fusiformis B-CM18. J Basic Microbiol 53(5):451–460. https://doi.org/10.1002/jobm.201100590
Asraoui M, Zanella F, Marcato S, Squartini A, Amzil J, Hamdache A, Baldan B, Ezziyyani M (2018) Bacillus amyloliquefaciens enhanced strawberry plants defense responses, upon challenge with Botrytis cinerea. In: Ezziyyani M (ed) Advanced intelligent systems for sustainable development. Springer, Cham, pp 46-53. https://doi.org/10.1007/978-3-030-11878-5_5
Goswami D, Patel K, Parmar S, Vaghela H, Muley N, Dhandhukia P, Thakker JN (2015) Elucidating multifaceted urease producing marine Pseudomonas aeruginosa BG as a cogent PGPR and bio-control agent. Plant Growth Regul 75(1):253–263. https://doi.org/10.1007/s10725-014-9949-1
Acknowledgements
The authors would like to thank the Vértika Agropecuária Ltda, Total Biotecnologia Ltda, and EMBRAPA Soja for the material made available to carry out this work and the Phytopathology Department – UFPR (Universidade Federal do Paraná) – Setor de Ciências Agrárias, for providing space to develop part of the work.
Funding
This work was financed by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and by the INCT – Plant-Growth Promoting Microorganisms for Agricultural Sustainability and Environmental Responsibility (CNPq 465133/2014–4, Fundação Araucária-STI 043/2019, CAPES).
Author information
Authors and Affiliations
Contributions
Conceptualization, Lygia Vitória Galli-Terasawa and Mariangela Hungria; data curation, formal analysis, and investigation, Tairine Graziella Ercole, Daiani Cristina Savi, and Douglas Adamoski; funding acquisition and resources, Lygia Vitória Galli-Terasawa, Vanessa Merlo Kava, and Mariangela Hungria; methodology, Daiani Cristina Savi and Tairine Graziella Ercole; project administration and supervision, Lygia Vitória Galli-Terasawa; writing — original draft preparation, Tairine Graziella Ercole and Daiani Cristina Savi; writing — review and editing, all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ercole, T.G., Savi, D.C., Adamoski, D. et al. Diversity of maize (Zea mays L.) rhizobacteria with potential to promote plant growth. Braz J Microbiol 52, 1807–1823 (2021). https://doi.org/10.1007/s42770-021-00596-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42770-021-00596-y