
Abstract
Purpose
The complexities of pathological changes in Alzheimer’s disease (AD) and their relationships with associated risk factors and clinical symptoms, and the development of disease-modifying therapy or preventive intervention for AD are being intensively investigated. The emerging advances in brain positron emission tomography (PET) imaging allow for in vivo visualization and quantitation of specific neurochemical and molecular pathophysiologic changes in the brain tissue. This review is focused on the recent advances in molecular PET imaging of the brain and its clinical applications in AD.
Methods
The development in PET radiopharmaceuticals targeting brain glucose metabolism, amyloid accumulation, tau protein aggregation, neuroinflammation, acetylcholine system, and synaptic density are discussed along with their potential clinical applications in AD.
Results
PET imaging studies can provide diagnostic and prognostic biomarkers of AD, as well as for selection and monitoring of novel therapies. Given the complexity and potential overlapping pathologies and comorbidities, a single biomarker can neither provide the diagnostic certainty required for early detection of AD nor the identification of presymptomatic at-risk individuals. Therefore, multimodal studies are required to better understand the relationships between different biomarkers, answer the controversial issues, and incorporate new diagnostic criteria for the various stages in the continuum of AD.
Conclusion
Molecular PET imaging studies have significant roles in the clinical practice and the clinical trials of novel therapeutic agents in AD. However, standardization and validation across multiple participating sites are still required for the subsequent transition from clinical research into clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease and the most common cause (60–80%) of dementia [1]. In more than half of cases, AD is associated with other neurodegenerative co-morbidities [2]. AD affects millions of people and leads to a tremendous socioeconomic burden worldwide [3]. Current mainstream therapies of AD have limited efficacy and cannot effectively decelerate the progression of this disease.
The definitive pathomorphological diagnosis of AD requires brain autopsy or biopsy with extracellular amyloid-beta (Aß) plaque and intracellular neurofibrillary tangles (NFTs) composed of filamentous tau proteins. Other frequent pathological findings in the AD brain include dystrophic neurites, activated microglia, reactive astrocyte, eosinophilic Hirano bodies, granulovacuolar degeneration, cerebral amyloid angiopathy with synaptic and neuronal damage in vulnerable regions [4,5,6] (Fig. 1. Various hypotheses regarding the etiologies of AD include the amyloid, tau propagation, cholinergic, inflammatory, mitochondrial cascade, calcium homeostasis, neurovascular, metal ion, and lymphatic system hypotheses [7]. Recently, it was proposed that AD has multiple and complex causalities which can overlap [8]. However, the link between these multiple pathological changes, the sequences, and possible overlap of occurrence, their relationships with associated risk factors and clinical symptoms, and the development of disease-modifying therapy (or preventive intervention) for AD are yet to be established.

Proposed mechanisms and pathologies in AD
Current recommendations for defining the preclinical and early symptomatic stages of AD help to improve early and accurate clinical diagnosis, select the candidates for potential therapeutic intervention, and monitor treatment efficacy [9, 10]. The emerging advances in brain imaging using high-resolution PET/CT and PET/MRI instruments, new molecular-targeted radiopharmaceuticals, and quantitative analysis software, allow for in vivo visualization and quantitation of specific pathomorphological (i.e., neurodegeneration) and pathophysiological (i.e., altered function of neurotransmitters) changes in the brain tissue, including Aß and Tau protein depositions, neuroinflammation, and in both clinical and research settings [11, 12]. Advanced molecular, functional, and morphologic imaging enables a holistic understanding of the spatial and temporal dynamics of multiple processes involved in the progression of AD. These imaging technologies enable a more accurate diagnosis and prognosis of AD and facilitate the development of more effective therapies [6, 13,14,15,16,17]. Here, we reviewed the recent advances in molecular PET imaging of the brain and its clinical applications in AD.
2 Development in PET Radiopharmaceuticals
The ideal characteristics of radiopharmaceuticals for imaging molecular targets in the brain include (a) easy labeling with radionuclides; (b) sufficient lipid-solubility (LogP 0.8–2.4) to facilitate passive diffusion cross the blood–brain barrier (BBB); (c) high affinity and selectivity for the target (i.e., for amyloid plaques, tau protein or receptors); (d) slow dissociation from the target binding site; (e) rapid clearance from the blood and tissues that don’t express the target; (f) resistance to systemic metabolism (i.e., liver microsomal enzymes) to prevent the contribution of radiolabeled metabolites to the background radioactivity in tissues [18]. Several PET radiotracers have been developed for imaging of various molecular targets in AD, which can be divided into 6 categories according to pathophysiologic mechanisms of AD: (a) brain glucose metabolism, (b) Aß deposition, (c), tau protein aggregation (d) neuroinflammation, (e) acetylcholine transport, and (f) synaptic density. The chemical structures of PET radiopharmaceuticals used in AD are summarized in Fig. 2, while the examples of normal scans compared to abnormal scans in AD are provided in Fig. 3.

Examples of radiopharmaceuticals used for brain PET imaging studies in AD; A FDG, B amyloid-beta tracers, C tau tracers, D TSPO tracers, E acetylcholine transporter tracers, and F synaptic density tracers

Examples of normal PET scans in cognitively normal subjects vs. abnormal scans in patients with AD
2.1 Radiopharmaceuticals for Brain Glucose Metabolism
Neurons in the human brain demand glucose as an obligatory energy resource for physiological brain function to generate ATP and neurotransmitters [19]. Ido et al. developed the first 18F-2-deoxy-2-fluoro-d-glucose (18F-FDG) synthesis to mimic glucose in 1978 [20]. 18F-FDG was modified from 14C-2-deoxyglucose [21], an analog of D-glucose, by substituting carbon-2 (C-2) in which equatorial hydroxy with fluorine-18 (Fig. 2). The rationale of the 18F-FDG design [22] is based on the fact that the fluorine atom has a Van der Waals radius that is smaller but still sufficiently similar to that of the hydroxyl group, but is more electro-negative; it has a strong C–F bond on C-2 which preserves the pharmacological properties of 14C-2-deoxyglucose [23] required for glucose transporter (Glut-1) binding and hexokinase (HK) enzyme affinity. After the uptake in neurons facilitated by Glut-1, the hydroxyl group on carbon-6 of 18F-FDG can be phosphorylated by hexokinase to become 18F-FDG-6-P, which is not a substrate for glucose 6-phosphatase (EC 3.1.3.9, G6Pase) due to the presence of fluorine in the C-2 position instead of a hydroxyl group. Therefore, it is metabolically trapped inside the neurons proportionally to glucose metabolic activity. The 110-min half-life of fluorine-18 allows for efficient washout of non-metabolized 18F-FDG from the tissues and its renal clearance from the blood, which results in a high target to background tissue ratios in PET images [24].
2.2 Radiopharmaceuticals for Imaging Amyloid Accumulation
The radiopharmaceuticals targeted to bind to insoluble Aβ40 and Aβ42 isoforms can be divided into two classes, based on the pharmacophore:
-
1.
Benzothiazole class Previously, thioflavin-T, a common in vitro fluorescent probe known to bind Aβ protein, was used for in situ staining of amyloid plaques based on its ability to bind diverse types of amyloid fibrils [25]. This led to the development of 11C-Pittsburg Compound B (11C-PIB)—the first in humans in vivo Aβ imaging tracer [26]. The F-18 styryl benzoxazole derivative of PIB was introduced as 18F-Flutemetamol (previously 18F-GE067) to overcome the limitation of the short half-life of C-11 [27]. 18F-Flutemetamol was approved by both FDA in 2013 and EMA in 2014 [28, 29]. Later, Swahn et al. reported 18F-NAV4694, which has lower lipophilicity, decreased non-specific binding, and increased washout rate from non-target tissues [30].
-
2.
Stilbene class Polyethyleneglycol stilbene serendipitously showed excellent binding affinity to Aβ aggregates similar to thioflavin-T and its derivatives [31]. To date, the FDA approved two tracers in this class, including 18F-Florbetaben and 18F-Florbetapir [32, 33]. The 18F-Florbetaben (previously 18F-BAY94-9172) or 18F-AV-1 shares common planar structure to 11C-PIB with extended aromatic systems and alkylamino substitution [34]. The 18F-Florbetapir (18F-AV-45) contains a pyridine ring substituted at position 2 of 18F-Florbetaben [35].
2.3 Radiopharmaceuticals for Imaging Tau Protein Aggregation
Another pathology pertinent for AD diagnosis is the intraneuronal aggregation of tau protein [36]. Primary challenges in the development of tau-targeted radiotracers included: (a) the ability to cross the BBB neuronal cell membrane; (b) high selectivity to specific or multiple tau isoforms at least 10-fold, as compared to the selectivity of Aβ-targeted radiotracer, due to 5–20 times lower abundance than Aβ plaques [37]. Tau-targeted radiotracers can be grouped into two generations:
-
1.
The first-generation The 18F-FDDNP represents the first milestone in the development of the tau-targeted radiotracers [38]. 18F-FDDNP binds to both Aβ plaques and NFTs. Despite its high non-selective in vivo binding, it encouraged extensive efforts to develop the first selective tau-targeted radiotracer in the arylquinoline class, 18F-THK523 [39]. The THK family tracers were synthesized with modified substitutions to improve in vivo potency and pharmacokinetic properties. 18F-THK-5105 and 18F-THK-5117 are racemic mixtures, while the S-enantiomers, 18F-THK-5317 and 18F-THK-5351, showed superior 4R affinity to R-enantiomer, 18F-THK-5451 [40,41,42,43]. The 18F-T807 (AKA 18F-AV-1451 or 18F-Flortaucipir) and 18F-T808 are two representative tracers in the pyridoindole class [44, 45]. The core structure of 18F-T808 is benzo[4,5]imidozol[1,2-a]pyrimidine with aliphatic fluorine. While 18F-T807 is a pyrido[4,3-b]indole with 18F-substitution on pyridine ring, it is more stable to oxidative defluorination. To date, 18F-T807 is the only FDA-approved tau tracer [46].
Another direction in the development of tau-targeted radiotracers was based on the chemical structure of an Aβ-targeted radiotracer 11C-PBB3 (pyridinyl-butadienyl-benzothiazole 3) [47]. However, its butadiene bridge can undergo photoisomerization, resulting in low radiochemical purity.
-
2.
The second-generation Several new tau-targeted radiotracers are designed focusing on higher affinity and specificity to tau aggregates, improved pharmacokinetic profiles, and minimizing off-target binding to circumvent drawbacks observed in the first-generation of tau-targeted radiotracers. The 18F-RO-948 was modified from 18F-flortaucipir to lessen the lipophilicity and lower plasma-free F-18 fraction. It shows excellent kinetics without off-target retention [48]. Further SAR exploring the 4-pyridine side chain of 18F-RO-948 resulted in 18F-PI2620 [49]. With the same pyrolo[2,3-b:4,5-c’]dipyridine core structure, both tracers have a high affinity for aggregated tau and significantly reduced MAO-A binding properties. To prevent the defluorination observed in 18F-T808, two geminal deuterium atoms were incorporated in the benzimidazopyrimidine core of 18F-GTP-1 [50]. Later, using intensive SAR studies the 18F-MK6240 containing an azaindole core was designed to minimize the effect of fluorine on isoquinoline ring with the primary amine, which improved high affinity to NFT [51].
2.4 Radiopharmaceuticals for Imaging Neuroinflammation
Although neuroinflammatory response mechanisms in neurodegeneration are unknown, translocator protein (TSPO), previously known as peripheral benzodiazepine receptor [52] is upregulated in neuroinflammation and is considered a potential biomarker [53]. 11C-PK11195, a lipid-soluble isoquinoline carboxamide with R-enantiomer at N-sec-butyl side chain, was developed as the first radioligand targeting TSPO [54].
More than 50 new tracers have been developed to date aiming to increase the specific binding to TSPO by reducing lipophilicity, including: 11C-PBR28 [55], 11C-DAA1106 [56], 18F-FEDAA1106 [57], 18F-FEPPA [58], 18F-FEMPA [59], and 18F-DPA714 [60]. Recently, two novel tracers, 11C-ER176 and 18F-Ge180, showed an improved selectivity to TSPO [61, 62]. The 11C-ER176 is the quinazoline analog of 11C-PK11195 [61], while the 18F-Ge180 possesses a tricyclic indole core structure [62].
2.5 Radiopharmaceuticals for Imaging Acetylcholine System
Significant achievements have been made in the development of PET radiotracers for imaging cholinergic system functions in the brain, including radiotracers targeted to:
2.5.1 Muscarinic Acetylcholine Receptors (mAChR)
The mAChR subtypes 1, 4, 5 are mainly found in CNS [63]. The 11C-Xanomeline, an M1/M4 preferring orthosteric agonist, was investigated using SAR to optimize the side chain [64]. Searching for a more selective M1 mAChR agonist, the 11C-AF150 was developed as an S-enantiomer rigid analog of Ach [65]. The 11C-GSK1034702 is a novel M1 mAChR agonist with N-substituted benzimidazolone moiety with a robust in vivo activity [66]. By changing to indolinone moiety, the 11C-LSN3172176 has lower lipophilicity, rapid metabolism, although a moderate affinity [67]. 11C-MK6884 is the only M4 specific allosteric modulator developed to date [68].
2.5.2 Nicotinic Acetylcholine Receptors (nAChR)
A primary subtype of nAChR is the heteromeric α4β2 receptor—the binding target for radiotracer development. Several radiotracers were demonstrated to bind to α4β2 receptors. Highly-specific α4β2 nAChR targeted tracers include: the 18F-Nifene, which was modified from a previous prototype, 18F-2FA, employing a 3-pyrroline ring connecting to pyridine [69]; the 18F-Flubatine, with an azabicyclic core structure [70]; and 18F-AZAN, an azabicyclo [2.2.1]heptane [71].
Moreover, an attempt to target another major subtype of homomeric α7 receptor was first reported by Hashimoto et al. [72]. The 11C-Chiba1001 contains a diazabicyclo[3.2.2]nonane. A further structural modification was reported as 18F-ASEM, which replaced phenylformate in 11C-Chiba1001 with dibenzo[b,d]thiophene moiety [73].
2.5.3 Cholinergic Enzymes (AChE)
Acetylcholinesterase, known as AChE, catalyzes the cleavage of acetylcholine in synapses [52]. Various compounds have been investigated over the past three decades to quantify AChE. The 11C-AMP [74] and 11C-PMP [75] are simple esters that cross BBB and serve as substrates of AChE. The AChE-hydrolyzed products of these radiotracers become more polar and thus are metabolically trapped in the brain. Similarly, 11C-BMP [76] is cleaved by the butyrylcholinesterase (BChE), the second cholinesterase exhibiting an increased expression/activity in AD.
2.5.4 Vesicular Acetylcholine Transporters (VAChT)
VAChT is a major vesicular acetylcholine transporter. In 1969, Vesamicol (2-[4-phenylpiperidino] cyclohexanol) was reported to inhibit the transport of acetylcholine into synaptic vesicles in cholinergic nerve terminals [77]. Subsequently, 18F-FEOBV [78], and 18F-VAT [79] have been developed based on the SAR of Vesamicol to visualize the regulation of acetylcholine uptake at the synaptic vesicular level.
2.6 Radiopharmaceuticals for Imaging Synaptic Density
Synaptic vesicle glycoprotein 2A (SV2A) is pervasively expressed throughout the brain. It is also the binding site of Levetiracetam, an antiepileptic drug [80]. Towards the development of SV2A-targeted radiotracers, an acetamide moiety in the structure of Levetiracetam was replaced either by pyridine or imidazole moieties, and alkyl substitution on lactam moiety was explored as well. Resulting various derivatives of Levetiracetam have been labelled with C-11 or F-18, resulting in 18F-UCB-H [81], 11C-UCB-A [82], 11C-UCB-J [83], 18F-SynVest1 [84], 18F-SynVest2 [85], and 18F-1 [86], respectively.
3 Clinical Applications of PET Imaging in AD
3.1 AD Diagnosis
The NIA-AA research criteria using combined fluid and imaging biomarkers to diagnose AD have been proposed in 2018 [10], reflecting dynamic changes that relate disease stage to AD biomarkers, in which Aβ biomarkers become abnormal first. Neurodegenerative biomarkers become abnormal later and correlate with the severity of clinical symptoms [87, 88]. The biomarkers included in these recent criteria and the diagnostic categories are summarized in Table 1. Amyloid PET, tau PET, and FDG PET imaging studies play crucial roles as the biomarkers for amyloid (A) and tau (T) proteinopathies and neurodegeneration (N), respectively, as in Fig. 4. Figure 5 shows two examples of using AT(N) criteria in clinical cases.

Proposed changes in biomarkers and physiology in AD and the potential use of neuroimaging to detect the abnormality in each phase

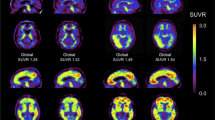
Example cases of A a patient with MCI who showed positive results for Aß and tau deposition and glucose hypometabolism compatible with AD pattern [A + T + (N) +] and B a patient with subjective cognitive impairment (SCI) who showed negative results for Aß and tau deposition and normal FDG PET [A − T − (N)−], which can be excluded from being in AD continuum
It has been well-established that high concentrations of Aβ cause dendritic and axonal atrophy, leading to neuronal death. Aβ also inhibits the long-term potentiation of the hippocampus. It has been shown that individuals with elevated Aβ in the brain are prone to cognitive decline and symptomatic AD and that the severity of AD correlates with the levels of insoluble Aβ detected by either CSF or PET [89, 90]. This correlation is more pronounced when amyloid PET is combined with FDG PET and MRI volumetric analysis of brain structures [91, 92]. The regions with a high Aβ deposition rate detected by 11C-PiB PET are posterior cingulate > frontal > supramarginal/orbitofrontal > lateral temporal > superior parietal, temporooccipital regions, and are significantly higher in APOEe4 carriers [93]. At the same time, the hippocampus shows a relatively slower rate of Aβ deposition [94]. Aβ deposition can also be found in approximately 30% of cognitively normal elderly [95] and can appear as long as 20 years preceding the clinical symptoms of AD [94] (Fig. 4). Aβ PET can facilitate differential diagnosis of AD from other neurodegenerative diseases rarely show high Aβ deposition (e.g., non-fluent aphasia, progressive supranuclear palsy, and frontotemporal dementia) or demonstrate the different location of cortical Aβ deposition (e.g., cerebral amyloid angiopathy). However, some neurodegenerative diseases may demonstrate a high Aβ deposition pattern indistinguishable from AD, such as dementia with Lewy’s body (DLB) [96]. Therefore, positive Aβ accumulation alone is sufficient evidence of a disease belonging to the AD continuum but not sufficient for a definitive diagnosis of AD [10, 18, 97]. Another challenge in the interpretation of results of amyloid PET imaging is the heterogeneity in interpretation criteria across different Aβ–targeted radiotracers, PET imaging instrumentation, and methods used at different imaging centers. These factors limit the diagnostic and prognostic standardization and comparison of the effectiveness of therapies, particularly in a large multi-center clinical study setting [98,99,100]. Attempts to reach the universal read-out, either qualitatively or quantitatively, have been made to overcome this challenge [98, 100, 101].
PET imaging of tau proteins has recently become a promising technology for assessing tauopathy—an essential marker for various neurodegenerative disorders, including AD. The patterns of tau-targeted radiotracer deposition correspond well with Braak staging of NFTs pathology [102]. There is a strong evidence to support the relationship between amyloid and tau proteinopathies. Worse cognitive performance is related to mesial temporal and neocortical tau load, even in amyloid-β–negative cognitively normal individuals [14, 103]. The topographic distribution of pathologic tau deposits defines pathologic subtypes [104], clinical phenotype [105], patterns of brain atrophy [106], and correlates with cognitive decline [107]. Tau deposition can also predict further neurodegeneration, as assessed by FDG PET or structural MRI [108]. However, some issues still exist. First, aggregated tau isoforms are heterogeneous in different neurodegenerative disorders [37]. Second, tau PET deposition restricted mainly to the medial temporal lobe can be noted in healthy aging, possibly consistent with the “primary age-related tauopathy” (PART). This finding reflects neuropathological findings of NFTs in neurons in the medial temporal lobe, which has been shown to result in hippocampal atrophy and impaired episodic memory performance that are Aβ-independent [109, 110]. Third, the off-target binding of tau–targeted PET radiotracers in areas such as the choroid plexus, basal ganglia, longitudinal sinuses, meninges, or structures expressing monoamine oxidase B, limit the specificity and accuracy of the first-generation radiotracers, although are less pronounced in the second-generation of radiotracers [111,112,113]. However, there is a debate that the suspected “off-target” binding may be reflecting true tau-binding, or binding to some not yet identified molecular targets [114]. These issues altogether lead to challenges in interpretation of tau PET imaging results, when comparing different tracers and establishing standard criteria for a cut-off standard uptake values (SUV) of normal versus abnormal tau PET imaging results [113, 115].
18F-FDG reflects the glucose metabolic activity predominantly in neurons. Patterns of altered 18F-FDG uptake reflecting local neuronal dysfunction and synapses in remote areas connected to the cortical projection neurons in the primary lesion can provide a differential diagnosis of AD from the other causes for dementia [116]. Criteria for AD diagnosis using FDG PET is well established with glucose hypometabolism in the posterior cingulate, precuneus, parietal and temporal association cortices, and also frontal association cortex in advanced cases, with relatively preserved metabolism at sensorimotor and primary visual cortices, basal ganglia, thalamus, brainstem, and cerebellum [117]. The severity of glucose hypometabolism in these brain structures is more pronounced in early-onset patients (EOAD) [118]. This pattern can be used for the differential diagnosis of AD from FTD, DLB, and normal aging [119]. FDG PET has a better specificity of 89% compared to amyloid PET (58–85%) in differentiating AD from normal controls, although the sensitivity is similar (90%) or even lower than amyloid PET (90–96%) [120]. However, overlapping patterns of accumulation of radiotracers in the brain in AD other neurodegenerative disorders may affect the accuracy of FDG PET [148]. PET imaging of amyloid in the brain may have a potential role to aid in accurate diagnosis of AD, particularly in differentiating cases of suspected AD variants with atypical presentation who have equivocal FDG PET results [121].
PET imaging of TSPO (initially described as the peripheral benzodiazepine receptor, PBR) can assess the processes of neuroinflammation mediated by activated microglia, pro-inflammatory cytokines, in disease-relevant areas across a broad spectrum of neurodegenerative diseases, including AD [122]. Neuroinflammation has been reported in the early stages of AD before the onset of dementia and its magnitude correlates with clinical severity [123], which may have a diagnostic value. However, it is not clear whether the increased TSPO signals reflect the primary cause or secondary results from other pathological insults in neurodegeneration and whether the signals indicate the presence of destructive pro-inflammatory (M1) or protective anti-inflammatory (M2) cells [124]. Monitoring the spatial and temporal dynamics of neuroinflammation in neurodegenerative diseases will help to understand the degree to which neuroinflammation is a causative or reactive process and justify the development of radiotracers that specifically target M1 or M2 inflammatory cells in the brain. The limited clinical utility of TSPO-targeted PET tracers is currently due to their low signal-to-noise ratio, making subtle neuroinflammation challenging to detect in the brain. Current TSPO-targeted PET radiotracers have a relatively low-affinity binding to a prevalent form of polymorphic TSPO (A147T), as compared to wild-type TSPO. Moreover, TSPO-targeted PET radiotracers show substantial differences in affinity between subjects classified as high-affinity binders (HABs) and low-affinity binders (LABs). These points should be considered in the development of novel TSPO-targeted radiotracers with increased sensitivity and specificity. Furthermore, PET imaging of TSPO as a biomarker of neuroinflammation in the brain, cannot be used to differentiate AD from normal controls [125] or other neurodegenerative diseases, such as Parkinson’s disease, amyotrophic lateral sclerosis, MS, and FTD [54]. Therefore, additional studies to better understand the role of TSPO in AD and other neurodegenerative diseases are required to improve the interpretation of the PET images acquired with TSPO-targeted radiotracers and for establishing its role in the diagnosis of AD [51, 126].
Acetylcholine (ACh) has a crucial role in the nervous systems and is an essential factor in many forms of dementia, including AD [127]. Cholinergic synapses are particularly affected by Aβ causing early neurotoxicity and resulting in synaptic loss and cognitive impairment [128]. Loss of cholinergic neurons in the basal forebrain, including the neurons that form the nucleus basalis of Meynert, contributes to memory and attention deficits in AD [129]. PET radiotracers targeted to either presynaptic AChE or the vesicle ACh transporter (VAChT) or postsynaptic nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs)—are promising for imaging neuronal cholinergic activity. Previous studies demonstrated a reduction of cortical binding of AChE-targeted tracers in AD and MCI patients, especially in the brain regions innervated with cholinergic projections [130], in parallel with the decrease in the degree of binding VAChT/nAChR-targeted radiotracers [131, 132]. The degree of binding of these radiotracers in the AD-affected brain regions significantly correlates to both global and respective regional cognitive functions [133]. A recent study has demonstrated that a novel VAChT-targeted radiotracer has a higher sensitivity than FDG PET in discriminating AD from normal controls [131].
The loss of synapses is one of the pathologic hallmarks of AD, which leads to a deficit in neurotransmission in neuron-neuron interaction and strongly correlates with cognitive decline [134]. Aβ and tau mediated toxicities are believed to cause the loss of synapses and presynaptic proteins in patients with MCI and dementia [134, 135], although a recent study found the association between PiB PET and SV2A PET only in patients with MCI but not AD [136]. PET imaging of SV2A expression in synapses provides noninvasive detection of synaptic density and its loss during the pathogenesis and progression of AD. There was a significant reduction of synaptic density in the hippocampus and entorhinal cortex in AD/MCI patients compared to age-matched controls [137]. In patients with AD and MCI, PET imaging of SV2A demonstrated a significant loss of synaptic density in the hippocampus and entorhinal cortex, followed by the parahippocampal cortex, amygdala, lateral temporal cortex, pre-frontal cortex, posterior cingulate cortex/precuneus, lateral parietal cortex, and pericentral cortex, which correlated with the magnitude of brain atrophy and cognitive decline [138, 139]. The cognitive decline was more pronounced when the loss of synaptic density in the medial temporal cortex was associated with the regional accumulation of tau protein [140].
3.2 Prediction of Prognosis
Mild cognitive impairment (MCI) is a syndrome defined by a cognitive decline that is more significant than expected for an individual's age and education level but does not interfere with daily life activities. Prevalence of MCI ranges from 3 to 19% in adults older than 65 years and progresses to AD at a rate of approximately 15% per year, which is in turn described as a prodromal or transitional stage between normal aging to AD [141, 142].
In patients with MCI, neurofibrillary tangles (NFTs) are initially detected in the hippocampus, and other medial temporal regions, then spread to the parietal and frontal cortices as MCI progresses to AD [143]. Although the previous studies on the correlation between Aβ deposition with the cognitive decline in AD showed heterogeneous results [92, 94], the correlation between Aβ deposition with memory impairment and the rate of memory decline in MCI and healthy elderly subjects are well accepted [18]. Previous data showed a strong relationship between Aβ deposition in PET and CSF biomarkers [144] and episodic memory performance. MCI “converters” to AD showed a higher Aβ deposition than “non-converters” [18, 145,146,147]. There are correlations between Aβ and CSF Aβ1-42, total tau, and episodic memory [146]. The rates of neocortical Aβ deposition also impact disease progression [94], and earlier onset of Aβ deposition may therefore lead to earlier disease onset [93].
The PET imaging of tau allows to assess the density, extension, and regional distribution of tau deposition and could be helpful to predict the progression of cognitive decline. Tau imaging might be more important than Aβ imaging in the assessment of neurodegeneration and cognitive decline. Increasing levels of cortical tau deposition in individuals with Aβ pathology were associated with growing impairment in several cognitive domains, which led to its potential applications, including disease staging, tracking progression, and use as a surrogate marker of cognitive status [14, 102, 148].
Although high deposition of Aβ plaques and NFTs correlates with clinical status at autopsy, neuronal loss is necessary for developing clinical dementia [149]. FDG PET is a sensitive measure of cognition and functional ability change in MCI and has value in predicting future cognitive decline [150]. MCI-converters demonstrate lower glucose metabolism in the temporoparietal cortex [151]. Previous studies showed that overall, amyloid PET has higher sensitivity while FDG PET has higher specificity in predicting MCI-converters [152]. Figure 6 shows examples of multimodal imaging in predicting a neurocognitive decline in three patients with MCI, which was superior to the results of a single modality imaging [92].

Example cases of MCI subjects who are A converter with PET evidence of Aß deposition and neurodegeneration, B non-converter with neither PET nor MRI evidence of Aß deposition and neurodegeneration and C non-converter with evidence of mild neurodegeneration from PET and MRI without Aß deposition on PET
PET imaging demonstrated a lower density of nicotinic AChR in MCI converters to AD dementia, as compared to non-converters. Also, episodic memory, working memory, and executive functions are strongly correlated with reduction of α4β2 nAChR binding in the basal forebrain-cortical and septohippocampal cholinergic projection [153]. More studies are required to establish the prognostic value of PET imaging of acetylcholine receptors in MCI conversion to AD.
3.3 Selection and Development of Novel Therapies
A deeper understanding of the molecular mechanism of Aβ formation, degradation, and neurotoxicity is being translated into new therapeutic approaches. The currently approved palliative treatment regimens involve acetylcholinesterase inhibitors, glutamatergic agents, nonsteroidal anti-inflammatory drugs, and antioxidants. The most recent approaches focus on the amyloid hypothesis (22.3% of trials) and are aimed at increasing the removal of Aβ or blocking the formation of Aβ oligomers and fibrils, thus inhibiting neurotoxicity [7, 154, 155]. Other therapeutic trials focus on neurotransmitter hypothesis (19%), mitochondrial cascade and related hypotheses (17%), tau propagation hypothesis (12.7%), and less than 10% of trials on other hypotheses of pathogenesis and progression of AD [7]. Combining disease-specific with non-specific therapeutic agents and lifestyle interventions might be the concept for a successful treatment strategy for AD. Identification of abnormalities at the molecular level during the early stages of neurodegenerative processes could enable the development of disease-modifying medications that can be administered during the presymptomatic period to achieve a maximal benefit in terms of preventing neuronal loss. Among possible new drug targets, α7 nAChR and M1 muscarinic acetylcholine receptors are gaining increasing focus, which is due to their potential role in preventing amyloid toxicity and tau hyper-phosphorylation, increasing synaptic strength and stability, modulating neuroinflammation by acting on non-neuronal cells, and improving cholinergic deficit and cognitive dysfunction [89,90,91,92,93, 129]. Therefore, molecular imaging with PET has become increasingly crucial in therapeutic trials, for the selection of suitable candidates, enabling the determination of a personalized optimal window for therapeutic intervention, providing proof of target engagement, establishing the risk of disease progression, and monitoring treatment effectiveness as a surrogate outcome measure [14].
3.4 Monitoring Treatment Response
Quantitative analysis in PET studies is necessary for longitudinal observational studies and intervention trials to identify a specific endpoint as the therapeutic effect (i.e., changes in Aß plaque or tau deposition), which may be modest and not clinically apparent. Phase 2 and 3 trials in AD typically involve multiple centers’ and require standardized protocols for the acquisition and analysis of data to minimize the variability across different centers [156]. Although several biological factors influence the interpretation of the PET images, such as age, genetic background, and comorbid conditions. A drop in the PET signal in the absence of change in either amyloid or tau deposition throughout AD trials may result from progressive cortical atrophy or altered uptake by a drug treatment that competes with tracer binding. Significantly reduced blood flow in AD, also limits tracer delivery to and clearance from the brain, particularly in gray matter regions, known to be susceptible to neurodegeneration and atrophy [156]. These issues are still challenging for monitoring the effect of disease-modifying treatments for AD. The variability of results and their interpretation can be minimized by study design, subject selection, and stratification.
FDG PET could be a potential imaging biomarker for selecting patients and assessing the outcome of AD-modifying treatments in clinical trials, particularly in the preclinical and early clinical studies [157]. Several interventional trials presented an excellent correlation between clinical outcomes and FDG PET findings [157,158,159,160,161]. However, there are significant variations of the study design and data analysis methods used in these clinical trials. Therefore, the feasibility of using FDG PET as an imaging biomarker in multi-center therapeutics trials still requires the standardization of largely user-independent methods to quantify regional metabolic impairment. Moreover, the possible confounding effects on FDG PET unrelated to disease progression or regression should also be considered, such as synaptic activity, metabolism, or density unrelated to synaptic loss.
PET imaging of TSPO expression density can be used as a biomarker to monitor response in clinical trials of novel neurodegenerative therapeutics focusing on neuroinflammation. Monitoring neuroinflammation could allow tracking of disease progression and indicate responses to therapeutic trials. Evidence of decreased TSPO PET signal in preclinical studies of novel therapeutics in AD models suggests that it could be used to monitor successful treatment responses in clinical trials [162]. Nevertheless, the challenges in using TSPO PET for monitoring treatment responses are similar to its use for diagnostic purposes in AD, as described above.
The reduction of cortical binding of AChE-targeted radiotracers in AD patients shows a further decrease after treatment with AChE inhibitors [62, 64]. The degree of treatment-induced reduction in AChE binding is prominent in the frontal cortex and correlates with the improvement in frontal lobe functions such as execution and attention. Such treatment-induced inhibition of AChE binding is not observed in PET imaging studies of nAChR binding [65], which might be explained, at least in part, by the modulation of different cholinergic receptors playing different roles in AD pathology. The potential role of SV2A PET imaging as an endpoint measure of therapeutic efficacy of drugs affecting brain synaptic density is being determined in other clinical trials [163].
3.5 Identification of At-Risk Individuals
Several studies in the presymptomatic population with genetic risk factors provide information about presymptomatic AD-related brain changes and identify the at-risk individuals for further developing symptoms of AD [164]. Early identification of the at-risk individuals may enable therapeutic interventions and lifestyle modifications at a time when the disease burden is mild, which may prevent or delay functional and irreversible cognitive losses [18].
High Aß deposition is associated with a significant risk for developing cognitive decline in asymptomatic elderly individuals with subjective cognitive impairment (SCI). Although amyloid-positive asymptomatic subjects have a higher life-long risk of converting to AD, its short-term conversion predictive value is low. In contrast, tau deposition in asymptomatic elderly subjects or primary age-related tauopathy (PART) in the absence of amyloid is possibly insufficient to develop memory decline [165], although controversy still exists that PART may be an early phase of AD [166]. The relationship between PART and suspected non-amyloid pathology (SNAP) has been proposed as the preclinical abnormalities during the development of AD. Individuals with amyloid depositions on PET show evidence of worse clinical and cognitive outcomes as compared to individuals with SNAP [167]. Ongoing studies are aimed to understand these controversies.
Glucose hypometabolism on FDG PET in the same regions as in patients with clinical manifestations of AD has been observed before the onset of the disease in several groups of at-risk individuals, including carriers of autosomal dominant mutations responsible for early-onset familial AD (FAD), e.g., amyloid precursor protein (APP), presenilin 1 (PS1), and presenilin 2 (PS2); apolipoprotein E (APOE) E4 allele carriers; SCD; and normal elderly subjects who declined to MCI and AD several years after PET [142, 168, 169]. However, additional carefully designed longitudinal studies are needed to conclude the relationship between cerebral hypometabolism and time-to conversion. Table 2 summarizes the potential applications of PET radiopharmaceuticals in AD.
4 Conclusion
Molecular PET imaging studies with different radiotracers targeted to key molecular mechanisms of AD can provide promising diagnostic and prognostic biomarkers for diagnosis, staging, and prognosis of AD, as well as for selection and monitoring of novel therapies of AD. It has become more obvious that given the complexity and potential overlapping pathologies and comorbidities, a single biomarker can neither provide the diagnostic certainty required for early detection of AD nor the identification of presymptomatic at-risk individuals. Therefore, multimodal studies are required to better understand the relationships between different biomarkers, answer the controversial issues, and incorporate new diagnostic criteria for the various stages in the continuum of AD. Moreover, PET imaging studies should be made mandatory in clinical trials of novel therapeutic agents in AD, including the standardization and validation across multiple participating sites of radiotracer production, imaging protocols, interpretation, and quantification of images, for the subsequent transition from clinical research into clinical practice.
References
Schultz, C., Del Tredici, K., & Braak, H. (2004). Neuropathology of Alzheimer’s disease. In R. W. Richter & B. Z. Richter (Eds.), Alzheimer’s disease. Current clinical neurology (pp. 21–31). Humana Press. https://doi.org/10.1007/978-1-59259-661-4_2
Barker, W. W., Luis, C. A., Kashuba, A., Luis, M., Harwood, D. G., Loewenstein, D., et al. (2002). Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Disease & Associated Disorders, 16(4), 203–212.
Wortmann, M. (2012). Dementia: A global health priority—highlights from an ADI and World Health Organization report. Alzheimer’s Research & Therapy, 4(5), 40.
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & Dementia, 8(1), 1–13.
DeTure, M. A., & Dickson, D. W. (2019). The neuropathological diagnosis of Alzheimer’s disease. Molecular Neurodegeneration, 14(1), 1–18.
De Strooper, B., & Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell, 64(4), 603–615.
Liu, P. P., Xie, Y., Meng, X. Y., & Kang, J. S. (2019). History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduction and Targeted Therapy, 4, 29.
Hascup, E. R., & Hascup, K. N. (2020). Toward refining Alzheimer’s disease into overlapping subgroups. Alzheimer’s & Dementia (N Y), 6(1), e12070.
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia, 7(3), 280–292.
Jack, C. R., Jr., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & Dementia, 14(4), 535–562.
Chételat, G., Arbizu, J., Barthel, H., Garibotto, V., Law, I., Morbelli, S., et al. (2020). Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. The Lancet Neurology, 19(11), 951–962.
Chételat, G., Arbizu, J., Barthel, H., Garibotto, V., Lammertsma, A. A., Law, I., et al. (2021). Finding our way through the labyrinth of dementia biomarkers. European Journal of Nuclear Medicine and Molecular Imaging, 48, 2320–2324.
Doré, V., Krishnadas, N., Bourgeat, P., Huang, K., Li, S., Burnham, S., et al. (2021). Relationship between amyloid and tau levels and its impact on tau spreading. European Journal of Nuclear Medicine and Molecular Imaging, 48(7), 2225–2232.
Villemagne, V. L., Doré, V., Burnham, S. C., Masters, C. L., & Rowe, C. C. (2018). Imaging tau and amyloid-β proteinopathies in Alzheimer disease and other conditions. Nature Reviews Neurology, 14(4), 225–236.
Nordberg, A., Rinne, J. O., Kadir, A., & Långström, B. (2010). The use of PET in Alzheimer disease. Nature Reviews Neurology, 6(2), 78–87.
Jack, C. R., Jr. (2012). Alzheimer disease: New concepts on its neurobiology and the clinical role imaging will play. Radiology, 263(2), 344–361.
Shaffer, J. L., Petrella, J. R., Sheldon, F. C., Choudhury, K. R., Calhoun, V. D., Coleman, R. E., et al. (2013). Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology, 266(2), 583–591.
Villemagne, V. L., Fodero-Tavoletti, M. T., Pike, K. E., Cappai, R., Masters, C. L., & Rowe, C. C. (2008). The ART of loss: Aβ imaging in the evaluation of Alzheimer’s disease and other dementias. Molecular Neurobiology, 38(1), 1–15.
Mergenthaler, P., Lindauer, U., Dienel, G. A., & Meisel, A. (2013). Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends in Neurosciences, 36(10), 587–597.
Ido, T., Wan, C. N., Casella, V., Fowler, J., Wolf, A., Reivich, M., et al. (1978). Labeled 2-deoxy-D-glucose analogs. 18F-labeled 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose and 14C-2-deoxy-2-fluoro-D-glucose. Journal of Labelled Compounds and Radiopharmaceuticals, 14(2), 175–183.
Sokoloff, L. (1979). Mapping of local cerebral functional activity by measurement of local cerebral glucose utilization with [14C] deoxyglucose. Brain, 102(4), 653–668.
Yu, S. (2006). Review of 18F-FDG synthesis and quality control. Biomedical imaging and intervention Journal, 2(4), e57.
Sokoloff, L., Reivich, M., Kennedy, C., Rosiers, M. D., Patlak, C., Pettigrew, K., et al. (1977). The [14C] deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat 1. Journal of Neurochemistry, 28(5), 897–916.
Fowler, J. S., & Ido, T. (2002). Initial and subsequent approach for the synthesis of 18FDG. Seminars in Nuclear Medicine, 32(1), 6–12.
Biancalana, M., & Koide, S. (2010). Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1804(7), 1405–1412.
Klunk, W. E., Engler, H., Nordberg, A., Wang, Y., Blomqvist, G., Holt, D. P., et al. (2004). Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Annals of Neurology, 55(3), 306–319.
Koole, M., Lewis, D. M., Buckley, C., Nelissen, N., Vandenbulcke, M., Brooks, D. J., et al. (2009). Whole-body biodistribution and radiation dosimetry of 18F-GE067: A radioligand for in vivo brain amyloid imaging. Journal of Nuclear Medicine, 50(5), 818–822.
Retrieved January 2, 2022, from https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203137s008lbl.pdf.
Retrieved January 2, 2022, from https://www.ema.europa.eu/en/medicines/human/EPAR/vizamyl.
Swahn, B.-M., Sandell, J., Pyring, D., Bergh, M., Jeppsson, F., Juréus, A., et al. (2012). Synthesis and evaluation of pyridylbenzofuran, pyridylbenzothiazole and pyridylbenzoxazole derivatives as 18F-PET imaging agents for β-amyloid plaques. Bioorganic & Medicinal Chemistry Letters, 22(13), 4332–4337.
Zhang, W., Oya, S., Kung, M.-P., Hou, C., Maier, D. L., & Kung, H. F. (2005). F-18 polyethyleneglycol stilbenes as PET imaging agents targeting Aβ aggregates in the brain. Nuclear Medicine and Biology, 32(8), 799–809.
Retrieved January 2, 2022, from https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204677s000lbl.pdf.
Retrieved January 2, 2022, from https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202008_Florbetapir_Orig1s000TOC.cfm..
Rowe, C. C., Ackerman, U., Browne, W., Mulligan, R., Pike, K. L., O’Keefe, G., et al. (2008). Imaging of amyloid β in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. The Lancet Neurology, 7(2), 129–135.
Choi, S. R., Golding, G., Zhuang, Z., Zhang, W., Lim, N., Hefti, F., et al. (2009). Preclinical properties of 18F-AV-45: A PET agent for Aβ plaques in the brain. Journal of Nuclear Medicine, 50(11), 1887–1894.
Guo, T., Noble, W., & Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathologica, 133(5), 665–704.
Villemagne, V. L., Fodero-Tavoletti, M. T., Masters, C. L., & Rowe, C. C. (2015). Tau imaging: Early progress and future directions. The Lancet Neurology, 14(1), 114–124.
Agdeppa, E. D., Kepe, V., Liu, J., Flores-Torres, S., Satyamurthy, N., Petric, A., et al. (2001). Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for β-amyloid plaques in Alzheimer’s disease. Journal of Neuroscience, 21(24), RC189-RC.
Okamura, N., Suemoto, T., Furumoto, S., Suzuki, M., Shimadzu, H., Akatsu, H., et al. (2005). Quinoline and benzimidazole derivatives: Candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. Journal of Neuroscience, 25(47), 10857–10862.
Harada, R., Okamura, N., Furumoto, S., Furukawa, K., Ishiki, A., Tomita, N., et al. (2016). 18F-THK5351: A novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. Journal of Nuclear Medicine, 57(2), 208–214.
Jonasson, M., Wall, A., Chiotis, K., Saint-Aubert, L., Wilking, H., Sprycha, M., et al. (2016). Tracer kinetic analysis of (S)-18F-THK5117 as a PET tracer for assessing tau pathology. Journal of Nuclear Medicine, 57(4), 574–581.
Lemoine, L., Gillberg, P.-G., Svedberg, M., Stepanov, V., Jia, Z., Huang, J., et al. (2017). Comparative binding properties of the tau PET tracers THK5117, THK5351, PBB3, and T807 in postmortem Alzheimer brains. Alzheimer’s Research & Therapy, 9(1), 1–13.
Okamura, N., Harada, R., Ishiki, A., Kikuchi, A., Nakamura, T., & Kudo, Y. (2018). The development and validation of tau PET tracers: Current status and future directions. Clinical and Translational Imaging, 6(4), 305–316.
Xia, C. F., Arteaga, J., Chen, G., Gangadharmath, U., Gomez, L. F., Kasi, D., et al. (2013). [18F] T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s & Dementia, 9(6), 666–676.
Zhang, W., Arteaga, J., Cashion, D. K., Chen, G., Gangadharmath, U., Gomez, L. F., et al. (2012). A highly selective and specific PET tracer for imaging of tau pathologies. Journal of Alzheimer’s Disease, 31(3), 601–612.
Jie, C. V., Treyer, V., Schibli, R., & Mu, L. (2021). Tauvid™: The First FDA-approved PET tracer for imaging tau pathology in Alzheimer’s disease. Pharmaceuticals, 14(2), 110.
Kimura, Y., Ichise, M., Ito, H., Shimada, H., Ikoma, Y., Seki, C., et al. (2015). PET quantification of tau pathology in human brain with 11C-PBB3. Journal of Nuclear Medicine, 56(9), 1359–1365.
Gobbi, L. C., Knust, H., Körner, M., Honer, M., Czech, C., Belli, S., et al. (2017). Identification of three novel radiotracers for imaging aggregated tau in Alzheimer’s disease with positron emission tomography. Journal of Medicinal Chemistry, 60(17), 7350–7370.
Kroth, H., Oden, F., Molette, J., Schieferstein, H., Capotosti, F., Mueller, A., et al. (2019). Discovery and preclinical characterization of [18 F] PI-2620, a next-generation tau PET tracer for the assessment of tau pathology in Alzheimer’s disease and other tauopathies. European Journal of Nuclear Medicine and Molecular Imaging, 46(10), 2178–2189.
Bohórquez, S. S., Marik, J., Ogasawara, A., Tinianow, J. N., Gill, H. S., Barret, O., et al. (2019). [18 F] GTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging, 46(10), 2077–2089.
Walji, A. M., Hostetler, E. D., Selnick, H., Zeng, Z., Miller, P., Bennacef, I., et al. (2016). Discovery of 6-(Fluoro-18 F)-3-(1 H-pyrrolo [2, 3-c] pyridin-1-yl) isoquinolin-5-amine ([18F]-MK-6240): A positron emission tomography (PET) imaging agent for quantification of neurofibrillary tangles (NFTs). Journal of Medicinal Chemistry, 59(10), 4778–4789.
Tu, L. N., Morohaku, K., Manna, P. R., Pelton, S. H., Butler, W. R., Stocco, D. M., et al. (2014). Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. Journal of Biological Chemistry, 289(40), 27444–27454.
Pasqualetti, G., Brooks, D. J., & Edison, P. (2015). The role of neuroinflammation in dementias. Current Neurology and Neuroscience Reports, 15(4), 17.
Camsonne, R., Crouzel, C., Comar, D., Mazière, M., Prenant, C., Sastre, J., et al. (1984). Synthesis of 1-(2-chlorophenyl)-N-[11C] methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide (PK 11195): A new ligand for peripheral benzodiazepine receptors. Journal of Labelled Compound Radiopharmaceuticals, 21, 985–991.
Damont, A., Boisgard, R., Kuhnast, B., Lemée, F., Raggiri, G., Scarf, A. M., et al. (2011). Synthesis of 6-[18F] fluoro-PBR28, a novel radiotracer for imaging the TSPO 18 kDa with PET. Bioorganic & Medicinal Chemistry Letters, 21(16), 4819–4822.
Maeda, J., Suhara, T., Zhang, M. R., Okauchi, T., Yasuno, F., Ikoma, Y., et al. (2004). Novel peripheral benzodiazepine receptor ligand [11C] DAA1106 for PET: An imaging tool for glial cells in the brain. Synapse (New York, N. Y.), 52(4), 283–291.
Wang, M., Gao, M., & Zheng, Q.-H. (2012). Fully automated synthesis of PET TSPO radioligands [11C] DAA1106 and [18F] FEDAA1106. Applied Radiation and Isotopes, 70(6), 965–973.
Wilson, A. A., Garcia, A., Parkes, J., McCormick, P., Stephenson, K. A., Houle, S., et al. (2008). Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nuclear Medicine and Biology, 35(3), 305–314.
Varrone, A., Oikonen, V., Forsberg, A., Joutsa, J., Takano, A., Solin, O., et al. (2015). Positron emission tomography imaging of the 18-kDa translocator protein (TSPO) with [18F] FEMPA in Alzheimer’s disease patients and control subjects. European Journal of Nuclear Medicine and Molecular Imaging, 42(3), 438–446.
James, M. L., Fulton, R. R., Vercoullie, J., Henderson, D. J., Garreau, L., Chalon, S., et al. (2008). DPA-714, a new translocator protein–specific ligand: Synthesis, radiofluorination, and pharmacologic characterization. Journal of Nuclear Medicine, 49(5), 814–822.
Castellano, S., Taliani, S., Milite, C., Pugliesi, I., Da Pozzo, E., Rizzetto, E., et al. (2012). Synthesis and biological evaluation of 4-phenylquinazoline-2-carboxamides designed as a novel class of potent ligands of the translocator protein. Journal of Medicinal Chemistry, 55(9), 4506–4510.
Wadsworth, H., Jones, P., Chau, W., Durrant, C., Fouladi, N., Passmore, J., et al. (2012). [18F] GE-180: A novel fluorine-18 labelled PET tracer for imaging translocator protein 18 kDa (TSPO). Bioorganic & Medicinal Chemistry Letters, 22(3), 1308–1313.
Vuckovic, Z., Gentry, P. R., Berizzi, A. E., Hirata, K., Varghese, S., Thompson, G., et al. (2019). Crystal structure of the M5 muscarinic acetylcholine receptor. Proceedings of the National Academy of Sciences, 116(51), 26001–26007.
Bonifazi, A., Yano, H., Del Bello, F., Farande, A., Quaglia, W., Petrelli, R., et al. (2014). Synthesis and biological evaluation of a novel series of heterobivalent muscarinic ligands based on xanomeline and 1-[3-(4-butylpiperidin-1-yl) propyl]-1, 2, 3, 4-tetrahydroquinolin-2-one (77-LH-28-1). Journal of Medicinal Chemistry, 57(21), 9065–9077.
Buiter, H. J., Leysen, J. E., Schuit, R. C., Fisher, A., Lammertsma, A. A., & Windhorst, A. D. (2012). Radiosynthesis and biological evaluation of the M1 muscarinic acetylcholine receptor agonist ligand [11C] AF150 (S). Journal of Labelled Compounds and Radiopharmaceuticals, 55(7), 264–273.
Budzik, B., Garzya, V., Shi, D., Walker, G., Woolley-Roberts, M., Pardoe, J., et al. (2010). Novel N-substituted benzimidazolones as potent, selective, CNS-penetrant, and orally active M1 mAChR agonists. ACS Medicinal Chemistry Letters, 1(6), 244–248.
Nabulsi, N. B., Holden, D., Zheng, M.-Q., Bois, F., Lin, S.-F., Najafzadeh, S., et al. (2019). Evaluation of 11C-LSN3172176 as a novel PET tracer for imaging M1 muscarinic acetylcholine receptors in nonhuman primates. Journal of Nuclear Medicine, 60(8), 1147–1153.
Tong, L., Li, W., Lo, M.M.-C., Gao, X., Wai, J.M.-C., Rudd, M., et al. (2020). Discovery of [11C] MK-6884: A positron emission tomography (PET) imaging agent for the study of M4Muscarinic receptor positive allosteric modulators (PAMs) in neurodegenerative diseases. Journal of Medicinal Chemistry, 63(5), 2411–2425.
Pichika, R., Easwaramoorthy, B., Collins, D., Christian, B. T., Shi, B., Narayanan, T. K., et al. (2006). Nicotinic α4β2 receptor imaging agents: Part II. Synthesis and biological evaluation of 2-[18F] fluoro-3-[2-((S)-3-pyrrolinyl) methoxy] pyridine (18F-nifene) in rodents and imaging by PET in nonhuman primate. Nuclear Medicine and Biology, 33(3), 295–304.
Sabri, O., Becker, G.-A., Meyer, P. M., Hesse, S., Wilke, S., Graef, S., et al. (2015). First-in-human PET quantification study of cerebral α4β2* nicotinic acetylcholine receptors using the novel specific radioligand (−)-[18F] Flubatine. NeuroImage, 118, 199–208.
Gao, Y., Kuwabara, H., Spivak, C. E., Xiao, Y., Kellar, K., Ravert, H. T., et al. (2008). Discovery of (−)-7-methyl-2-exo-[3′-(6-[18F] fluoropyridin-2-yl)-5′-pyridinyl]-7-azabicyclo [2.2. 1] heptane, a radiolabeled antagonist for cerebral nicotinic acetylcholine receptor (α4β2-nAChR) with optimal positron emission tomography imaging properties. Journal of Medicinal Chemistry, 51(15), 4751–4764.
Hashimoto, K., Nishiyama, S., Ohba, H., Matsuo, M., Kobashi, T., Takahagi, M., et al. (2008). [11C] CHIBA-1001 as a novel PET ligand for α7 nicotinic receptors in the brain: A PET study in conscious monkeys. PLoS ONE, 3(9), e3231.
Horti, A. G., Gao, Y., Kuwabara, H., Wang, Y., Abazyan, S., Yasuda, R. P., et al. (2014). 18F-ASEM, a radiolabeled antagonist for imaging the α7-nicotinic acetylcholine receptor with PET. Journal of Nuclear Medicine, 55(4), 672–677.
Irie, T., Fukushi, K., Akimoto, Y., Tamagami, H., & Nozaki, T. (1994). Design and evaluation of radioactive acetylcholine analogs for mapping brain acetylcholinesterase (AchE) in vivo. Nuclear Medicine and Biology, 21(6), 801–808.
Snyder, S. E., Tluczek, L., Jewett, D. M., Nguyen, T. B., Kuhl, D. E., & Kilbourn, M. R. (1998). Synthesis of 1-[11C] methylpiperidin-4-yl propionate ([11C] PMP) for in vivo measurements of acetylcholinesterase activity. Nuclear Medicine and Biology, 25(8), 751–754.
Snyder, S. E., Gunupudi, N., Sherman, P. S., Butch, E. R., Skaddan, M. B., Kilbourn, M. R., et al. (2001). Radiolabeled cholinesterase substrates: In vitro methods for determining structure-activity relationships and identification of a positron emission tomography radiopharmaceutical for in vivo measurement of butyrylcholinesterase activity. Journal of Cerebral Blood Flow & Metabolism, 21(2), 132–143.
Brittain, R. T., Levy, G. P., & Tyers, M. B. (1969). The neuromuscular blocking action of 2-(4-phenylpiperidino) cyclohexanol (AH 5183). European Journal of Pharmacology, 8(1), 93–99.
Mulholland, G. K., Wieland, D. M., Kilbourn, M. R., Frey, K. A., Sherman, P. S., Carey, J. E., et al. (1998). [18F] fluoroethoxy-benzovesamicol, a PET radiotracer for the vesicular acetylcholine transporter and cholinergic synapses. Synapse (New York, N. Y.), 30(3), 263–274.
Tu, Z., Zhang, X., Jin, H., Yue, X., Padakanti, P. K., Yu, L., et al. (2015). Synthesis and biological characterization of a promising F-18 PET tracer for vesicular acetylcholine transporter. Bioorganic & Medicinal Chemistry, 23(15), 4699–4709.
Boschi, F., Camps, P., Comes-Franchini, M., Muñoz-Torrero, D., Ricci, A., & Sánchez, L. (2005). A synthesis of levetiracetam based on (S)-N-phenylpantolactam as a chiral auxiliary. Tetrahedron Asymmetry, 16(22), 3739–3745.
Warnier, C., Lemaire, C., Becker, G., Zaragoza, G., Giacomelli, F., Jl, A., et al. (2016). Enabling efficient positron emission tomography (PET) imaging of synaptic vesicle glycoprotein 2A (SV2A) with a robust and one-step radiosynthesis of a highly potent 18F-labeled ligand ([18F] UCB-H). Journal of Medicinal Chemistry, 59(19), 8955–8966.
Estrada, S., Lubberink, M., Thibblin, A., Sprycha, M., Buchanan, T., Mestdagh, N., et al. (2016). [11C] UCB-A, a novel PET tracer for synaptic vesicle protein 2 A. Nuclear Medicine and Biology, 43(6), 325–332.
Nabulsi, N. B., Mercier, J., Holden, D., Carré, S., Najafzadeh, S., Vandergeten, M.-C., et al. (2016). Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. Journal of Nuclear Medicine, 57(5), 777–784.
Li, S., Cai, Z., Wu, X., Holden, D., Pracitto, R., Kapinos, M., et al. (2018). Synthesis and in vivo evaluation of a novel PET radiotracer for imaging of synaptic vesicle glycoprotein 2A (SV2A) in nonhuman primates. ACS Chemical Neuroscience, 10(3), 1544–1554.
Cai, Z., Li, S., Finnema, S., Lin, S.-f, Zhang, W., Holden, D., et al. (2017). Imaging synaptic density with novel 18F-labeled radioligands for synaptic vesicle protein-2A (SV2A): Synthesis and evaluation in nonhuman primates. Journal of Nuclear Medicine, 58(supplement 1), 547.
Trump, L., Lemos, A., Jrm, J., Pasau, P., Bnd, L., Mercier, J., et al. (2020). Development of a general automated flow photoredox 18F-difluoromethylation of N-heteroaromatics in an AllinOne synthesizer. Organic Process Research & Development, 24(5), 734–744.
Jack, C. R., Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology, 9(1), 119–128.
Zhang, D., Wang, Y., Zhou, L., Yuan, H., Shen, D., Alzheimer’s Disease Neuroimaging Initiative. (2011). Multimodal classification of Alzheimer’s disease and mild cognitive impairment. NeuroImage, 55(3), 856–867.
Thientunyakit, T., Sethanandha, C., Muangpaisan, W., & Minoshima, S. (2021). 3D-SSP analysis for amyloid brain PET imaging using 18F-florbetapir in patients with Alzheimer’s dementia and mild cognitive impairment. Medical Journal of Malaysia, 76(4), 493–501.
Thientunyakit, T., Thongpraparn, T., Sethanandha, C., Yamada, T., Kimura, Y., Muangpaisan, W., et al. (2021). Relationship between F-18 florbetapir uptake in occipital lobe and neurocognitive performance in Alzheimer’s disease. Japanese Journal of Radiology, 39(10), 984–993.
Thientunyakit, T., Sethanandha, C., Muangpaisan, W., Chawalparit, O., Arunrungvichian, K., Siriprapa, T., et al. (2018). Implementation of [18F]-labeled amyloid brain PET imaging biomarker in the diagnosis of Alzheimer’s disease: First-hand experience in Thailand. Nuclear Medicine Communications, 39(2), 186–192.
Thientunyakit, T., Sethanandha, C., Muangpaisan, W., Chawalparit, O., Arunrungvichian, K., Siriprapa, T., et al. (2020). Relationships between amyloid levels, glucose metabolism, morphologic changes in the brain and clinical status of patients with Alzheimer’s disease. Annals of Nuclear Medicine, 34(5), 337–348.
Burnham, S. C., Laws, S. M., Budgeon, C. A., Doré, V., Porter, T., Bourgeat, P., et al. (2020). Impact of APOE-ε4 carriage on the onset and rates of neocortical Aβ-amyloid deposition. Neurobiology of Aging, 95, 46–55.
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., et al. (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. The Lancet Neurology, 12(4), 357–367.
Braak, H., Braak, E., Bohl, J., & Reintjes, R. (1996). Age, neurofibrillary changes, Aβ-amyloid and the onset of Alzheimer’s disease. Neuroscience Letters, 210(2), 87–90.
Rowe, C. C., Ng, S., Ackermann, U., Gong, S. J., Pike, K., Savage, G., et al. (2007). Imaging β-amyloid burden in aging and dementia. Neurology, 68(20), 1718–1725.
Rowe, C. C., & Villemagne, V. L. (2013). Brain amyloid imaging. Journal of Nuclear Medicine Technology, 41(1), 11–18.
Klunk, W. E., Koeppe, R. A., Price, J. C., Benzinger, T. L., Devous, M. D., Sr., Jagust, W. J., et al. (2015). The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s & Dementia, 11(1), 1-15.e4.
Rowe, C. C., Jones, G., Doré, V., Pejoska, S., Margison, L., Mulligan, R. S., et al. (2016). Standardized expression of 18F-NAV4694 and 11C-PiB β-amyloid PET results with the Centiloid Scale. Journal of Nuclear Medicine, 57(8), 1233–1237.
Bischof, G. N., Bartenstein, P., Barthel, H., van Berckel, B. N., Doré, V., van Eimeren, T., et al. (2021). Towards a universal readout for fluorine-18 labelled amyloid tracers: The CAPTAINs Study. Journal of Nuclear Medicine, 62(7), 999–1005.
Amadoru, S., Doré, V., McLean, C. A., Hinton, F., Shepherd, C. E., Halliday, G. M., et al. (2020). Comparison of amyloid PET measured in Centiloid units with neuropathological findings in Alzheimer’s disease. Alzheimer’s Research & Therapy, 12(1), 1–8.
Schöll, M., Lockhart, S. N., Schonhaut, D. R., O’Neil, J. P., Janabi, M., Ossenkoppele, R., et al. (2016). PET imaging of tau deposition in the aging human brain. Neuron, 89(5), 971–982.
Groot, C., Doré, V., Robertson, J., Burnham, S. C., Savage, G., Ossenkoppele, R., et al. (2021). Mesial temporal tau is related to worse cognitive performance and greater neocortical tau load in amyloid-β–negative cognitively normal individuals. Neurobiology of Aging, 97, 41–48.
Murray, M. E., Graff-Radford, N. R., Ross, O. A., Petersen, R. C., Duara, R., & Dickson, D. W. (2011). Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. The Lancet Neurology, 10(9), 785–796.
Ossenkoppele, R., Schonhaut, D. R., Schöll, M., Lockhart, S. N., Ayakta, N., Baker, S. L., et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain, 139(5), 1551–1567.
Ossenkoppele, R., Lyoo, C. H., Sudre, C. H., van Westen, D., Cho, H., Ryu, Y. H., et al. (2020). Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer’s disease. Alzheimer’s & Dementia, 16(2), 335–344.
Hanseeuw, B. J., Betensky, R. A., Jacobs, H. I., Schultz, A. P., Sepulcre, J., Becker, J. A., et al. (2019). Association of amyloid and tau with cognition in preclinical Alzheimer disease: A longitudinal study. JAMA Neurology, 76(8), 915–924.
Chiotis, K., Saint-Aubert, L., Rodriguez-Vieitez, E., Leuzy, A., Almkvist, O., Savitcheva, I., et al. (2018). Longitudinal changes of tau PET imaging in relation to hypometabolism in prodromal and Alzheimer’s disease dementia. Molecular Psychiatry, 23(7), 1666–1673.
Crary, J. F., Trojanowski, J. Q., Schneider, J. A., Abisambra, J. F., Abner, E. L., Alafuzoff, I., et al. (2014). Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathologica, 128(6), 755–766.
Josephs, K. A., Murray, M. E., Tosakulwong, N., Whitwell, J. L., Knopman, D. S., Machulda, M. M., et al. (2017). Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: A clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathologica, 133(5), 705–715.
Betthauser, T. J., Cody, K. A., Zammit, M. D., Murali, D., Converse, A. K., Barnhart, T. E., et al. (2019). In vivo characterization and quantification of neurofibrillary tau PET radioligand 18F-MK-6240 in humans from Alzheimer disease dementia to young controls. Journal of Nuclear Medicine, 60(1), 93–99.
Smith, R., Schöll, M., Leuzy, A., Jögi, J., Ohlsson, T., Strandberg, O., et al. (2020). Head-to-head comparison of tau positron emission tomography tracers [18F] flortaucipir and [18F] RO948. European Journal of Nuclear Medicine and Molecular Imaging, 47(2), 342–354.
Leuzy, A., Chiotis, K., Lemoine, L., Gillberg, P.-G., Almkvist, O., Rodriguez-Vieitez, E., et al. (2019). Tau PET imaging in neurodegenerative tauopathies—still a challenge. Molecular Psychiatry, 24(8), 1112–1134.
Ikonomovic, M. D., Abrahamson, E. E., Price, J. C., Mathis, C. A., & Klunk, W. E. (2016). [F-18] AV-1451 PET retention in choroid plexus: More than “off-target” binding. Annals of Neurology, 80(2), 307–308.
Villemagne, V. L., Lopresti, B. J., Doré, V., Tudorascu, D., Ikonomovic, M. D., Burnham, S., et al. (2021). What is T+? A gordian knot of tracers, thresholds, and topographies. Journal of Nuclear Medicine, 62(5), 614–619.
Minoshima, S., Mosci, K., Cross, D., & Thientunyakit, T. (2021). Brain [F-18] FDG PET for Clinical Dementia Workup: Differential diagnosis of Alzheimer’s Disease and other types of dementing disorders. Seminars in Nuclear Medicine, 51(3), 230–240.
Minoshima, S., Frey, K. A., Koeppe, R. A., Foster, N. L., & Kuhl, D. E. (1995). A diagnostic approach in Alzheimer’s disease using three-dimensional stereotactic surface projections of fluorine-18-FDG PET. Journal of Nuclear Medicine, 36(7), 1238–1248.
Sakamoto, S., Ishii, K., Sasaki, M., Hosaka, K., Mori, T., Matsui, M., et al. (2002). Differences in cerebral metabolic impairment between early and late onset types of Alzheimer’s disease. Journal of The Neurological Sciences, 200(1–2), 27–32.
Mosconi, L., Tsui, W. H., Herholz, K., Pupi, A., Drzezga, A., Lucignani, G., et al. (2008). Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. Journal of Nuclear Medicine, 49(3), 390–398.
Morris, E., Chalkidou, A., Hammers, A., Peacock, J., Summers, J., & Keevil, S. (2016). Diagnostic accuracy of 18F amyloid PET tracers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. European Journal of Nuclear Medicine and Molecular Imaging, 43(2), 374–385.
Brendel, M., Schnabel, J., Schönecker, S., Wagner, L., Brendel, E., Meyer-Wilmes, J., et al. (2017). Additive value of amyloid-PET in routine cases of clinical dementia work-up after FDG-PET. European Journal of Nuclear Medicine and Molecular Imaging, 44(13), 2239–2248.
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. The Lancet Neurology, 14(4), 388–405.
Yokokura, M., Mori, N., Yagi, S., Yoshikawa, E., Kikuchi, M., Yoshihara, Y., et al. (2011). In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. European Journal of Nuclear Medicine and Molecular Imaging, 38(2), 343–351.
Geloso, M. C., Corvino, V., Marchese, E., Serrano, A., Michetti, F., & D’Ambrosi, N. (2017). The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Frontiers in Aging Neuroscience, 9, 242.
Stefaniak, J., & O’Brien, J. (2016). Imaging of neuroinflammation in dementia: A review. Journal of Neurology, Neurosurgery & Psychiatry, 87(1), 21–28.
Song, Y. S. (2019). Perspectives in TSPO PET imaging for neurologic diseases. Nuclear Medicine and Molecular Imaging, 53(6), 382–385.
Wilcock, G. K., Esiri, M. M., Bowen, D. M., & Smith, C. C. (1982). Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. Journal of the Neurological Sciences, 57(2–3), 407–417.
Bell, K. F., Ducatenzeiler, A., Ribeiro-da-Silva, A., Duff, K., Bennett, D. A., & Cuello, A. C. (2006). The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiology of Aging, 27(11), 1644–1657.
Ferreira-Vieira, T. H., Guimaraes, I. M., Silva, F. R., & Ribeiro, F. M. (2016). Alzheimer’s disease: Targeting the cholinergic system. Current Neuropharmacology, 14(1), 101–115.
Kuhl, D. E., Minoshima, S., Frey, K. A., Foster, N. L., Kilbourn, M. R., & Koeppe, R. A. (2000). Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Annals of Neurology, 48(3), 391–395.
Aghourian, M., Legault-Denis, C., Soucy, J., Rosa-Neto, P., Gauthier, S., Kostikov, A., et al. (2017). Quantification of brain cholinergic denervation in Alzheimer’s disease using PET imaging with [18F]-FEOBV. Molecular Psychiatry, 22(11), 1531–1538.
Schmitz, T. W., Mur, M., Aghourian, M., Bedard, M.-A., Spreng, R. N., Alzheimer’s Disease Neuroimaging Initiative. (2018). Longitudinal Alzheimer’s degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Reports, 24(1), 38–46.
Okada, H., Ouchi, Y., Ogawa, M., Futatsubashi, M., Saito, Y., Yoshikawa, E., et al. (2013). Alterations in α4β2 nicotinic receptors in cognitive decline in Alzheimer’s aetiopathology. Brain, 136(10), 3004–3017.
Robinson, J. L., Molina-Porcel, L., Corrada, M. M., Raible, K., Lee, E. B., Lee, V.M.-Y., et al. (2014). Perforant path synaptic loss correlates with cognitive impairment and Alzheimer’s disease in the oldest-old. Brain, 137(9), 2578–2587.
Pooler, A. M., Noble, W., & Hanger, D. P. (2014). A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology, 76, 1–8.
O’Dell, R. S., Mecca, A. P., Chen, M.-K., Naganawa, M., Toyonaga, T., Lu, Y., et al. (2021). Association of Aβ deposition and regional synaptic density in early Alzheimer’s disease: A PET imaging study with [11C] UCB-J. Alzheimer’s Research & Therapy, 13(1), 1–12.
Chen, M.-K., Mecca, A. P., Naganawa, M., Finnema, S. J., Toyonaga, T., Lin, S.-f, et al. (2018). Assessing synaptic density in Alzheimer disease with synaptic vesicle glycoprotein 2A positron emission tomographic imaging. JAMA Neurology, 75(10), 1215–1224.
De Wilde, M. C., Overk, C. R., Sijben, J. W., & Masliah, E. (2016). Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimer’s & Dementia, 12(6), 633–644.
Mecca, A. P., Chen, M. K., O’Dell, R. S., Naganawa, M., Toyonaga, T., Godek, T. A., et al. (2020). In vivo measurement of widespread synaptic loss in Alzheimer’s disease with SV2A PET. Alzheimer’s & Dementia, 16(7), 974–982.
Vanhaute, H., Ceccarini, J., Michiels, L., Koole, M., Sunaert, S., Lemmens, R., et al. (2020). In vivo synaptic density loss is related to tau deposition in amnestic mild cognitive impairment. Neurology, 95(5), e545–e553.
Gauthier, S., Reisberg, B., Zaudig, M., Petersen, R. C., Ritchie, K., Broich, K., et al. (2006). Mild cognitive impairment. The Lancet, 367(9518), 1262–1270.
Petersen, R. C., Smith, G. E., Waring, S. C., Ivnik, R. J., Tangalos, E. G., & Kokmen, E. (1999). Mild cognitive impairment: Clinical characterization and outcome. Archives of Neurology, 56(3), 303–308.
Small, G. W., Kepe, V., Ercoli, L. M., Siddarth, P., Bookheimer, S. Y., Miller, K. J., et al. (2006). PET of brain amyloid and tau in mild cognitive impairment. New England Journal of Medicine, 355(25), 2652–2663.
Doecke, J. D., Ward, L., Burnham, S. C., Villemagne, V. L., Li, Q.-X., Collins, S., et al. (2020). Elecsys CSF biomarker immunoassays demonstrate concordance with amyloid-PET imaging. Alzheimer’s Research & Therapy, 12(1), 1–10.
Okello, A., Koivunen, J., Edison, P., Archer, H., Turkheimer, F., Någren, Ku., et al. (2009). Conversion of amyloid positive and negative MCI to AD over 3 years: An 11C-PIB PET study. Neurology, 73(10), 754–760.
Forsberg, A., Engler, H., Almkvist, O., Blomquist, G., Hagman, G., Wall, A., et al. (2008). PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiology of Aging, 29(10), 1456–1465.
Doraiswamy, P. M., Sperling, R., Johnson, K., Reiman, E. M., Wong, T., Sabbagh, M., et al. (2014). Florbetapir F 18 amyloid PET and 36-month cognitive decline: A prospective multicenter study. Molecular Psychiatry, 19(9), 1044–1051.
Pontecorvo, M. J., Devous, M. D., Sr., Navitsky, M., Lu, M., Salloway, S., Schaerf, F. W., et al. (2017). Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain, 140(3), 748–763.
Price, J. L., Ko, A. I., Wade, M. J., Tsou, S. K., McKeel, D. W., & Morris, J. C. (2001). Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Archives of Neurology, 58(9), 1395–1402.
Landau, S. M., Harvey, D., Madison, C. M., Koeppe, R. A., Reiman, E. M., Foster, N. L., et al. (2011). Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiology of Aging, 32(7), 1207–1218.
Chetelat, G., Desgranges, B., De La Sayette, V., Viader, F., Eustache, F., & Baron, J.-C. (2003). Mild cognitive impairment: Can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology, 60(8), 1374–1377.
Mallik, A., Drzezga, A., & Minoshima, S. (2017). Clinical amyloid imaging. Seminars in Nuclear Medicine, 47(1), 31–43.
Sabri, O., Meyer, P. M., Gräf, S., Hesse, S., Wilke, S., Becker, G.-A., et al. (2018). Cognitive correlates of α4β2 nicotinic acetylcholine receptors in mild Alzheimer’s dementia. Brain, 141(6), 1840–1854.
Panza, F., Solfrizzi, V., Frisardi, V., Imbimbo, B. P., Capurso, C., D’Introno, A., et al. (2009). Beyond the neurotransmitter-focused approach in treating Alzheimer’s disease: Drugs targeting β-amyloid and tau protein. Aging Clinical and Experimental Research, 21(6), 386–406.
Zhang, F., Zhong, R.-j, Cheng, C., Li, S., & Le, W.-d. (2021). New therapeutics beyond amyloid-β and tau for the treatment of Alzheimer’s disease. Acta Pharmacologica Sinica, 42(9), 1382–1389.
Schmidt, M. E., Chiao, P., Klein, G., Matthews, D., Thurfjell, L., Cole, P. E., et al. (2015). The influence of biological and technical factors on quantitative analysis of amyloid PET: Points to consider and recommendations for controlling variability in longitudinal data. Alzheimer’s & Dementia, 11(9), 1050–1068.
Tzimopoulou, S., Cunningham, V. J., Nichols, T. E., Searle, G., Bird, N. P., Mistry, P., et al. (2010). A multi-center randomized proof-of-concept clinical trial applying [18F] FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer’s disease. Journal of Alzheimer’s Disease, 22(4), 1241–1256.
Craft, S., Baker, L. D., Montine, T. J., Minoshima, S., Watson, G. S., Claxton, A., et al. (2012). Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Archives of Neurology, 69(1), 29–38.
Smith, G. S., Laxton, A. W., Tang-Wai, D. F., McAndrews, M. P., Diaconescu, A. O., Workman, C. I., et al. (2012). Increased cerebral metabolism after 1 year of deep brain stimulation in Alzheimer disease. Archives of Neurology, 69(9), 1141–1148.
Teipel, S. J., Drzezga, A., Bartenstein, P., Möller, H.-J., Schwaiger, M., & Hampel, H. (2006). Effects of donepezil on cortical metabolic response to activation during 18FDG-PET in Alzheimer’s disease: A double-blind cross-over trial. Psychopharmacology (Berl), 187(1), 86–94.
Mega, M. S., Dinov, I. D., Porter, V., Chow, G., Reback, E., Davoodi, P., et al. (2005). Metabolic patterns associated with the clinical response to galantamine therapy: A fludeoxyglucose f 18 positron emission tomographic study. Archives of Neurology, 62(5), 721–728.
James, M. L., Belichenko, N. P., Shuhendler, A. J., Hoehne, A., Andrews, L. E., Condon, C., et al. (2017). [(18)F]GE-180 PET detects reduced microglia activation after LM11A-31 therapy in a mouse model of Alzheimer’s disease. Theranostics, 7(6), 1422–1436.
Bao, W., Xie, F., Zuo, C., Guan, Y., & Huang, Y. H. (2021). PET neuroimaging of Alzheimer’s disease: Radiotracers and their utility in clinical research. Frontiers in Aging Neuroscience, 13, 114.
Bateman, R. J., Aisen, P. S., De Strooper, B., Fox, N. C., Lemere, C. A., Ringman, J. M., et al. (2011). Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Research & Therapy, 3(1), 1–13.
Harrison, T. M., La Joie, R., Maass, A., Baker, S. L., Swinnerton, K., Fenton, L., et al. (2019). Longitudinal tau accumulation and atrophy in aging and Alzheimer disease. Annals of Neurology, 85(2), 229–240.
Duyckaerts, C., Braak, H., Brion, J.-P., Buée, L., Del Tredici, K., Goedert, M., et al. (2015). PART is part of Alzheimer disease. Acta Neuropathologica, 129(5), 749–756.
Jack, C. R., Knopman, D. S., Chételat, G., Dickson, D., Fagan, A. M., Frisoni, G. B., et al. (2016). Suspected non-Alzheimer disease pathophysiology—concept and controversy. Nature Reviews Neurology, 12(2), 117–124.
Mosconi, L., De Santi, S., Brys, M., Tsui, W. H., Pirraglia, E., Glodzik-Sobanska, L., et al. (2008). Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biological Psychiatry, 63(6), 609–618.
Mosconi, L., Sorbi, S., de Leon, M. J., Li, Y., Nacmias, B., Myoung, P. S., et al. (2006). Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. Journal of Nuclear Medicine, 47(11), 1778–1786.
Acknowledgements
The patients’ images in this work are from the research projects partially funded by National Research Council of Thailand through Health System Research Institute (Grant Nos. 58–036, 60–065, and 62–044), Research Development Fund, Faculty of Medicine Siriraj Hospital (Grant Nos. R016236003 and R016134004) and the International Atomic Energy Agency under CRP E13043 (Grant No. 19256).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflicts of interest related to any aspect of this study.
Rights and permissions
About this article

Cite this article
Thientunyakit, T., Shiratori, S., Ishii, K. et al. Molecular PET Imaging in Alzheimer’s Disease. J. Med. Biol. Eng. 42, 301–317 (2022). https://doi.org/10.1007/s40846-022-00717-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40846-022-00717-4