
Abstract
Amyotrophic lateral sclerosis (ALS), a complicated neurodegenerative disorder affected by hereditary and environmental variables, is a condition. In this study, the genetic makeup of ALS is investigated, with a focus on the SOD1 gene’s single-nucleotide polymorphisms (SNPs) and their ability to affect disease risk. Eleven high-risk missense variations that may impair the functionality of the SOD1 protein were discovered after a thorough examination of SNPs in the SOD1 gene. These mutations were chosen using a variety of prediction approaches, highlighting their importance in the aetiology of ALS. Notably, it was discovered that the stability of the SOD1 wild-type protein structure was compromised by the G38R and G42D SOD1 variants. Additionally, Edaravone, a possible ALS medication, showed a greater affinity for binding mutant SOD1 structures, pointing to potential personalised treatment possibilities. The high-risk SNPs discovered in this investigation seem to have functional effects, especially on the stability of proteins and their interactions with other molecules. This study clarifies the complex genetics of ALS and offers insights into how these genetic variations may affect the effectiveness of therapeutic interventions, particularly in the context of edaravone. In this study advances our knowledge of the genetic mechanisms causing ALS vulnerability and prospective therapeutic strategies. Future studies are necessary to confirm these results and close the gap between individualised clinical applications and improved ALS care.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease, stands as a devastating neurodegenerative affliction characterized by the progressive degeneration of both upper and lower motor neurons within the cerebral cortex, brainstem, and spinal cord. This intricate pathology gives rise to profound morbidity and mortality, chiefly attributed to respiratory insufficiency, muscular rigidity, and complete paralysis (Rahman et al. 2023; Ruffo et al. 2021). Global prevalence of ALS hovers around 2.7 cases per 100,000 individuals (Kumar et al. 2023), underscored by an intricate interplay between genetic predisposition and environmental factors. The disease may manifest sporadically or manifest within familial contexts, further accentuating its multifaceted etiology (Bonifacino et al. 2021).
The identification of genetic mutations has emerged as a pivotal factor in unraveling the intricate molecular underpinnings of this affliction. Genetic mutations residing within the superoxide dismutase 1 (SOD1) gene emerge as a predominant driver behind familial ALS (Candelise et al. 2022), constituting approximately 10–20% of ALS cases (Daud and Yousafzai 2021), specifically known as ALS1. This genetic landscape highlights over 150 distinct mutations embedded within the SOD1 gene (Deneault et al. 2022), each exerting a distinctive influence on the disease. This genomic tapestry is especially pronounced in over 100 mutations involving the copper-zinc SOD1 gene variant (Maung et al. 2021), encapsulating the disease’s hereditary dimension. The emergence of mutant SOD1 proteins creates an intricate cascade of pathogenic events, instigating a complex interplay within the cellular environment (Lünemann et al. 2021). These atypical proteins act as catalysts for protein misfolding, fostering aggregation and the formation of intracellular inclusions that intricately associate with vulnerable motor neurons (Chaudhary et al. 2022). These complex events inevitably combine, leading to various difficulties including, excitotoxicity, neuroinflammation, impaired axonal transport and disrupted mitochondrial functionality, which will inevitably culminate in the degradation of the motor neurons and weakening of muscles (Mead et al. 2023); both of which are prominent signs of ALS.
Recent genomic studies have expanded our knowledge of the genetic aspects of ALS, going beyond SOD1 mutations, as demonstrated by extensive genome-wide association studies (GWAS) (McDonough 2022). These studies have identified single nucleotide polymorphisms (SNPs) and other genetic variances with potential implications for ALS vulnerability and disease trajectory (Wei et al. 2022). However, the reported SNP-based heritability estimates for ALS are relatively low compared to other diseases, suggesting that DNA variants not tagged by common SNPs may be more important for ALS (Pan et al. 2023).
The genetics of ALS exhibits intricacy, with common and rare genetic variations converging to influence disease susceptibility (Apte and Kumar 2023). Within these variations, non-deleterious single nucleotide polymorphisms (nsSNPs) are genetic changes (Imran et al. 2021) that do not tend to cause harmful effects in individuals who carry them (Sivaramakrishnan and Kumar 2022). These SNPs have the potential to be situated within, or in close proximity to, genes that participate in diverse ALS-related biological processes, including but not limited to RNA processing (Wang et al. 2021), protein homeostasis, and neuronal function (Krokidis et al. 2022; Zhou and Xu 2023). These SNPs possess the capacity to engage in interactions with pathogenic mutations or alternative genetic variants, thereby exerting an influence on the alteration of disease risk or its level of severity. Contemporary investigations have unveiled multiple risk loci affiliated with ALS, notably encompassing genes intricately interwoven with protein homeostasis and RNA processing (Oroian et al. 2021). In a recent investigation, next-generation sequencing (NGS) was employed to delineate infrequent genetic variants linked to the susceptibility of ALS (Mathioudakis et al. 2023). Notably, this study demonstrated that employing routine genetic assessment through NGS exhibited superior efficacy in the identification of mutations responsible for disease causation compared to conventional genetic testing approaches. In a separate study, the identification of 15 risk loci pertaining to ALS unfolded, each characterized by unique genetic frameworks and neuron-specific biological attributes. These findings accentuate the intricate nature of ALS genetics (Gao 2020), emphasizing the significance of identifying both common and rare genetic variants associated with disease susceptibility. A holistic grasp of the complex synergy between genetic predispositions and environmental influences in ALS mandates a continued exploration of the functional ramifications of these genetic disparities, along with their cascading effects on gene expression, protein functionality, and intricate cellular mechanisms. The discernment of non-synonymous single nucleotide polymorphism (nsSNPs) holds the potential to facilitate a more profound comprehension among researchers of the genetic underpinnings inherent to the ailment. This extends to delineating the specific roles of designated genes and intricate pathways within its pathogenesis, culminating in the formulation of customized risk evaluation paradigms and therapeutic protocols (Bamshad et al. 2022; Meijboom and Brown 2022; Dang et al. 2023).
A clinical study involving both familial and sporadic ALS cases highlighted SOD1 as the most frequently mutated gene among Caucasian ALS patients (Gagliardi et al. 2023). The study revealed mutation rates of 78.9% in familial cases and 21.1% in sporadic cases. Additionally, in a separate study involving 915 Polish ALS patients, 21.1% were familial cases, and 2.3% were sporadic ALS cases (Berdyński et al. 2022). The genetic mutations, particularly within the SOD1 gene, play a prominent role in familial ALS cases. However, there remains a research gap in understanding the full spectrum of genetic variations contributing to ALS susceptibility. This study aims to bridge this gap through in-silico analysis, focusing on identifying SNPs and elucidating their interactions with pathogenic mutations. By comprehensively assessing the genetic intricacies, this research strives to unravel the underlying mechanisms, offering insights into disease development and potential therapeutic avenues for ALS.
Materials and methods
Table 1 outlines the various databases and tools utilized in the analysis of the SOD1 gene in the context of ALS. The investigation primarily focused on identifying high-risk missense variations in the SOD1 gene and assessing their impact on protein functionality. These tools played crucial roles in tasks such as retrieving genetic variations, predicting amino acid replacements, evaluating effects on native protein functions, and assessing structural and stability changes.
Sequence retrieval and nsSNP mining in SOD1 gene
The UniProt database (UniProt ID: P00441) provides crucial insight into the human SOD1 fasta sequence, an enzyme pivotal in mitigating oxidative stress. Considering SOD1's role in neurodegenerative disorders like ALS, investigating missense single nucleotide variations (nsSNVs) within the SOD1 gene is paramount. These genetic anomalies can profoundly impact protein structure and function, potentially contributing to disease pathogenesis. To unravel these intricacies, we sourced nsSNVs from the SNP database (https://www.ncbi.nlm.nih.gov/snp/), setting the foundation for a comprehensive molecular analysis.
Computational assessment of detrimental SNPs
In our current investigation, we employed a systematic screening approach to identify highly impactful single-nucleotide polymorphisms (SNPs) associated with the SOD1 gene. Out of the 599 SNPs initially identified through the dbSNP database, we utilized the SIFT prediction tool to filter out deleterious and tolerated non-synonymous SNPs. Subsequently, these SNPs underwent additional screening using SNAP, AlignGVGD, and PANTHER to predict their functional effects. Only those SNPs consistently predicted as deleterious, effect-inducing, C65, probably damaging, and destabilizing across all applied tools underwent further evaluation. To enhance the reliability of our predicted results, these selected SNPs were subjected to the PredictSNP metapredictor, which integrates the outcomes of all the aforementioned prediction tools, and the results were meticulously compared.
Sorting intolerant from tolerant (SIFT) analysis
To discern the impact of SNVs based on sequence homology, the Sorting Intolerant from Tolerant (SIFT) tool was employed. By initially identifying functionally similar sequences and aligning them, SIFT predicts possible amino acid replacements through normalized probability calculations. Positions with normalized probabilities ≤ 0.05 are classified as detrimental, while those with probabilities > 0.05 are considered tolerable (Rafaee et al. 2022). This predictive method enables the assessment of the phenotypic and functional implications of amino acid alterations.
Screening for non-acceptable polymorphisms (SNAP2) analysis
The Screening for Non-acceptable Polymorphisms (SNAP2) program was employed to evaluate the effects of genetic variants based on diverse sequences and variant characteristics. Utilizing a protein sequence in FASTA format as input, SNAP2 generates scores that gauge the likelihood of a mutation altering the function of the native protein. Scores range from 0 (highly impactful) to 100 (strongly neutral). The outcomes are visually represented, with color-coded indications of low, weak, or high effect likelihood (Wanarase et al. 2023). SNAP2 facilitates the prediction and visualization of potential substitutions at each protein location, fostering insights into the structural and functional consequences of alterations.
Align GVGD analysis
The Align GVGD web server facilitates the distinction between detrimental and neutral effects of missense substitutions. This predictive tool scrutinizes protein sequence alignments, assessing mutations through the lens of amino acid biophysical characteristics. Projections are classified into subcategories, ranging from C0 (least likely impact) to C65 (A table would fit the methods part better. Listing every tool that you have utilized is of great importance. However, a table would ease the comparison of the used software for the reader.most severe). Align GVGD harnesses amino acid characteristics and alignment patterns to provide a comprehensive evaluation of the potential harm posed by missense mutations (Rozario et al. 2021). This approach enhances our comprehension of the functional implications of genetic variations.
PANTHER cSNP analysis
The PANTHER cSNP (Protein analysis through evolutionary relationship-codingSNP) classification system amalgamates molecular functions, evolutionary relationships, and protein–protein interactions to evaluate the effects of amino acid substitutions. This method generates position-specific evolutionary conservation (PSEC) scores by analyzing aligned evolutionarily related proteins, shedding light on the functional repercussions of these changes (Falahi et al. 2021). By incorporating protein sequences, amino acid substitutions, and the human body, the PANTHER cSNP system provides a holistic evaluation of alterations' impact on protein structure and function, contributing to an improved understanding of their influence.
PredictSNP and MUpro analysis
The PredictSNP tool was employed to validate mutations and assess their effect on protein stability, offering robust consensus predictions. Additionally, MUpro estimated changes in protein stability resulting from single-point mutations. By estimating alterations in protein stability and accompanying Delta G values, MUpro provides insights into the effects of mutations on protein stability. The integration of PredictSNP and MUpro offers an in-depth examination of the mutations (Shinwari et al. 2023), enriching our understanding of their functional implications and their influence on protein structure and function.
Determining evolutionary conservation profiles through ConSurf analysis
Assessing the evolutionary conservation of amino acid positions within a protein is facilitated by the ConSurf service (https://consurf.tau.ac.il/). This resource employs advanced phylogenetic analysis techniques, including empirical Bayesian and maximum likelihood methods, to calculate evolutionary rates for specific sites. Through a color-coded scheme representing conservation scores ranging from 1 to 9, positions are classified as variable, moderately conserved, or highly conserved (Frlan 2022). This valuable insight aids in identifying residues and regions of the protein critical for specific functions. To initiate analysis, the protein sequence is submitted to the ConSurf server in FASTA format.
Relative Surface Accessibility (RSA) is a measurement that expresses how much a residue is exposed to the solvent around it. It offers a rough estimate of a residue's accessibility or depth inside the protein structure. The total accessible surface area (ASA) of the residues in the protein structure is compared to the ASA of the residues in the most exposed state to a solvent molecule to normalise the RSA values. It is possible to learn more about the structural and functional significance of particular residues by examining the conservation grades and RSA values. While residues with high RSA values are more likely to be involved in interactions with other molecules or solvent molecules, highly conserved residues are frequently linked to important functional or structural roles in the protein (Besterman et al. 2021).
Predicting solvent accessibility with NetSurfP-2.0
The NetSurfP-2.0 server (http://www.cbs.dtu.dk/services/NetSurfP/) serves as a powerful tool for forecasting diverse structural and functional attributes of amino acids within a protein sequence. This resource provides comprehensive insights into amino acid surface accessibility, secondary structure, disorder tendencies, and phi/psi dihedral angles. Through the analysis of the protein's FASTA sequence using NetSurfP, researchers gain valuable information about the structural characteristics of the SOD1 protein. This encompasses the identification of buried and exposed regions and the evaluation of amino acid accessibility within the protein structure (Li et al. 2021).
Investigating the impact of nsSNPs on protein structure
This computational resource, the SAAFEC-SEQ tool (Sequence-Based Stabilisation Analysis of Amino Acid Changes) (http://compbio.clemson.edu/SAAFEC-SEQ/), is devoted to examining the structural effects of mutations on protein stability. To forecast the possible effects of altering amino acids, SAAFEC-SEQ incorporates a variety of variables, including physicochemical features, sequence traits, and evolutionary insights. SAAFEC-SEQ provides insightful information on how particular amino acid substitutions may affect the stability of protein structures, illuminating the understanding of their functional implications. It does this by using protein sequence and mutation data as inputs (Zaji et al. 2023).
Exploring protein–protein interactions
A vital resource is the STRING database (Search Tool for the Retrieval of Interacting Genes/Proteins), which can be accessed through (https://string-db.org/). Through the collection of information from physical interaction databases and carefully maintained biological pathway databases, it plays a crucial part in unravelling and assembling knowledge about protein–protein interactions. This database not only enables a thorough analysis of protein–protein interactions, but it also offers information on their functional ramifications. The STRING database enables researchers to investigate potential linkages and interactions between the query protein and other proteins of interest by utilising the protein sequence of the target protein as input (Vora et al. 2023).
Predicting and evaluating SOD1 protein’s 3D structure
Homology-based modeling with SWISS-MODEL
Utilizing the SWISS-MODEL server (https://swissmodel.expasy.org), UniProtKB proteome data enables template-based homology modeling of protein structures. Accurate predictions are generated using the protein's FASTA sequence as input. This approach is supplemented by Ramachandran plot analysis, and Qualitative model energy analysis—Z (QMEAN-Z) scoring, assessment tools to validate and ensure the reliability of the predicted protein structures (Stitou et al. 2021) for further analysis.
Assessing point mutation impact
Through targeted alteration of the native protein sequence, point mutations were introduced to evaluate their structural effects. SWISS-MODEL was employed for analyzing structural changes in the modified protein. The same methodology applied to wild-type protein structures was used for the modified model, offering insights into potential shifts in protein conformation, stability, and interactions.
Stereochemical quality evaluation
The stereochemical quality of the protein structure was assessed using PROCHECK and ERRAT tools (https://saves.mbi.ucla.edu/). PROCHECK examines overall structure geometry and individual residue details, providing precise insights into bond lengths, angles, and torsion angles (Park et al. 2023). ERRAT assesses non-bonded interactions by comparing with high-resolution protein structures, offering an evaluation of model quality. These tools provide comprehensive insights into stereochemical accuracy and overall structure quality, enhancing validation and refinement processes (Ullah et al. 2022). Predicted models in protein data bank (PDB) format are used as input.
Molecular docking of a Edaravone with the SOD1 protein and its mutant forms
The PyRx tool (https://pyrx.sourceforge.io/) was employed for docking analysis to assess the binding activity of the chosen ligand with the SOD1 wild type mutant and the two distinct mutated protein structures (G38R and G42D). Each mutated protein structure was individually examined against Edaravone to investigate their respective affinities of binding. This investigation facilitated the exploration of Edaravone's binding mechanisms and potential interactions with the SOD1 protein and its mutant variants, providing valuable insights into Edaravone’s efficacy. Energy minimization was achieved through blind docking with grid box dimensions X: 69.8102 Å, Y: 48.0051 Å, and Z: 42.3756 Å, applying Gasteiger charges. Additionally, interactions were visualized using BIOVIA Discovery Studio (https://discover.3ds.com/discovery-studio-visualizer-download), contributing to further considerations for ALS treatment strategies.
Results
Retrival of SNPs
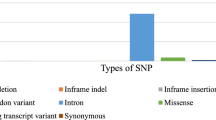
The human SOD1 gene's single nucleotide polymorphisms (SNPs) were retrieved from the dbSNP database. A total of 19,593 SNPs were found, including different kinds of variations. There were 14,063 intron variants, 599 missense variants (non-synonymous SNPs), 320 synonymous variants, 12 inframe deletions, 2 inframe insertions, and 4597 initiator codon variants and non-coding transcript variants left over. Only missense variations (nsSNPs) of the SOD1 gene, which make up 3.06% of all known SNPs in the human SOD1 gene, were chosen for this investigation in Fig. 1. A more thorough research into the potential functional effects of these sequence changes on the SOD1 protein is possible because to our analysis's focus on nsSNPs.

Insights from the dbSNP database on the functional classification and distribution of SNPs in the SOD1 gene
Computational evaluation of nsSNPs
Based on the results of the initial screening of 599 nsSNPs in the SOD1 gene, it was possible to infer functional effects. 77 nsSNPs were divided into two categories using the SIFT analysis: harmful and tolerated. Additional filtering was applied using the SNAP2, Align GVGD, and PANTHER processes to further narrow the selection. 34 variations (44.16%) that were anticipated to have functional impacts were found by SNAP2 analysis, indicating a potential impact on protein function. A total of 35 variations (45.45%) had no effect, whereas eight variants (10.39%) were categorised as neutral, indicating no substantial functional alterations. This suggests a subset of nsSNPs that could affect how proteins function.
Additional information about the potential consequences of the nsSNPs was revealed through Align GVGD study. 16 (20.78%) of the 77 nsSNPs were classified as having the greatest chance of being affected (Class C65), which suggests that functional effects are more likely to occur. Additionally, 24 nsSNPs (31.17%) (Class C15, C25, C35, C45, and C55) were identified as being less likely to be impacted, indicating a lesser likelihood of functional impact. Finally, 37 mutations were ignored because they contained non-missense mutations, wrong wild-type amino acids, or unknown amino acids.
PANTHER analysis evaluated the nsSNPs’ anticipated functional effects. It determined that 33 nsSNPs (42.86%) were likely to be harmful, pointing to a high probability of functional disruption. Six nsSNPs (7.79%) were labelled as likely benign, indicating that they have no or little effect on protein function. A moderate likelihood of functional effects was found for three nsSNPs (3.9%). Further research is required because the remaining 35 residues could not be found.
11 nsSNPs (I113T, G38R, G42D, G86R, G94C, I114T, C7F, I152T, G13R, and F46C) were chosen as significant based on the thorough analysis and the reliable results from several techniques. These nsSNPs were classified as Class C65 in Align GVGD analysis, showed detrimental effects in SIFT and SNAP2 predictions analysis. This selection draws attention to particular nsSNPs that are probably to affect the SOD1 protein’s functionality.
Consequently, the investigation of nsSNPs in the SOD1 gene using a variety of prediction techniques revealed important information about their possible functional implications. Further research into the selected 11 significant nsSNPs has the potential to advance our knowledge of the functional effects of SOD1 gene alterations.
An additional study was carried out using the PredictSNP tool in Table 2 specifically for the chosen 11 nsSNPs in order to further validate the results. PredictSNP makes a determination as to whether a mutation is harmful or tolerable based on a comparison of the data from several techniques presented in the table. This thorough investigation offers more proof of the possible effects of these nsSNPs on the composition and operation of the SOD1 protein.
Analysis of conservation and protein structure stability of SOD1 protein nsSNPs
To evaluate the effects of 11 high-risk nsSNPs in SOD1, the study used MUpro. It was hypothesised that mutations that resulted in DDG levels below 0 would cause the protein to become unstable. Additionally, as shown in Table 3, mutations with DDG values below − 1 were found to significantly reduce protein stability.
The ConSurf server was used to analyse the functional, structural, and evolutionary conservation of amino acid residues in SOD1, taking phylogenetic links between homologous sequences into consideration. An empirical Bayesian method was used to determine predicted evolutionary conservation scores, which were then divided into three groups: variable (scores 1–4), intermediate (scores 5–6), and conserved (scores 7–9).
Seven of the 11 nsSNP variations (G38R, G42D, G86R, G94R, C7F, and F46C) had conservation values that were relatively preserved, ranging from − 0.613 to − 0.928. For three variations (I113T, I114T, and G13R), the estimated average conservation scores ranged from − 0.136 to 0.191. With a conservation value of 0.528, the mutation I152T was categorised as variable as shown in Table 3. Notably, the analysis showed that while the remaining seven chosen variations were predicted to be exposed, four variants were predicted to be placed within the protein structure as shown in Fig. 2.

Analysis of SOD1 protein conservation
Due to their accessibility to other molecules and probable involvement in crucial binding interactions or enzymatic activities, the 7 nsSNPs that were selected and labelled as exposed were of great interest for further research. These interactions can be hampered by mutations in these exposed locations, which may result in disease or altered biological processes.
Analysis of predicted solvent accessibility
The 7 selected variants (G38R, G42D, G94R, G94C, I114T, I152T, and G13R) had their solvent accessibility and stability evaluated using NetSurfP. The amino acids are exposed when the Relative Solvent Accessibility (RSA) value is high, generally near to 1 or 100%, and they frequently participate in interactions with other molecules or contribute to the function of the protein. The amino acids may be buried, making them less accessible and maybe playing a structural or stabilising role, as opposed to low RSA values that are close to zero.
Understanding the total surface area of amino acids that solvents can access requires an understanding of the absolute solvent accessible area (ASA) values shown in Table 4 and Fig. 3.

Solvent accessibility and stability prediction using NetSurfP
As a result, for furthering research concentrate on the three exposed variants (G38R, G42D, and G13R), as their accessibility to solvent and probable involvement in interactions make them particularly notable for in-depth exploration.
The effects of pathogenic SOD1 mutations on structure
Using SAAFEQ-SEQ, a technique that forecasts how mutations would alter protein structure based on delta G values, the stability of SOD1 mutations was evaluated. According to Table 5 of the analysis results, all changes that were looked into caused the protein structure to become unstable. If a mutation has a negative G value, which denotes a reduction in protein stability, it is often regarded as destabilising. The projected destabilising effect is expected to be more significant the lower the negative G value. In contrast, the mutations G38R and G42D, which have G values greater than those of G13R, which are close to or equivalent to -1, are anticipated to have a significant effect on the stability of the SOD1 protein for further study.
Researching protein–protein interactions
To carry out crucial biological processes, such as cell signalling, proteins cooperate and interact with one another. As a result, changes in one protein's amino acid composition can have an impact on the proteins in its network. The STRING tool predicts that SOD1’s functional partners include CCS (Copper chaperone for superoxide dismutase), BCL2 (Apoptosis regulator Bcl-2), PARK7 (Protein/nucleic acid deglycase DJ-1), VDAC1 (Voltage-dependent anion-selective channel protein 1), SOD2 (Superoxide dismutase [Mn], mitochondrial), FUS (RNA-binding.
With a high score of 0.999, SOD1 was determined to be the major partner of CCS in these interactions. Figure 4 shows the network of protein–protein interactions including SOD1. This network demonstrates how SOD1 and its related proteins interact, illuminating how the two work together inside the cell.

SOD1 interactome network analysis using STRING server
Validation of protein models for structural and functional analysis
Based on their delta G values, the G38R and G42D variations of SOD1 demonstrate destabilising effects, as seen in Table 5. SWISS-MODEL was used to create the three-dimensional structures of the human SOD1 protein as well as the mutant SOD1 protein variations. Using the UniProtKB proteome's best-aligned template, SWISS-MODEL produced two partially structural models of the SOD1 protein.
The servers SWISS-MODEL PROCHECK, and ERRAT were used for later quality evaluations. The values exceeded 90% for both the wild-type and the mutant structures, showing that a sizeable number of amino acid residues in both the wild-type and the modified structures adopt advantageous backbone conformations. Both the wild-type and mutant structures in this study attained QMEAN scores above 91%, indicating high-quality models. A QMEAN score near to 0 is thought to indicate superior structural quality as shown in Table 6.
As shown in Fig. 5, these findings imply that the protein models are trustworthy and appropriate for additional structural and functional investigations. The models’ high quality increases their reliability for a thorough investigation of protein structure and function.

Comparative structural models of A SOD1 wild type and mutated variants B G38R, C G42D
Edaravone's binding interactions with SOD1 wild type and mutant variants in the context of ALS treatment
Edaravone, also known as Radicava, is a well-known antioxidant and free radical scavenger that has a lot of promise for the treatment of ALS. Its main pharmacological action focuses on lowering oxidative stress, a significant contributor to the neurodegenerative processes seen in ALS. Figure 6 shows the results of a ligand extraction from the PubChem database using PubChem CID 4021 in order to study Edaravone. This investigation is an important step in learning more about Edaravone's molecular characteristics and its potential to lessen the consequences of ALS.

Edaravone Ligand (PubChem CID 4021) is retrieved from the PubChem database
However, it is clear that all three SOD1 variations, including the wild type and the G38R and G42D mutants, share similar types of interactions with edaravone, such as conventional hydrogen bonds, pi-donor hydrogen bonds, and pi-alkyl bonds, as depicted in Fig. 7 and described in Table 7. However, there are minute variations in bond lengths between the wild type and mutants.

Docking Structures of SOD1 A wild type, B mutant variant G38R, and C mutant variant G42D with Edanavore
The differences in bond distances reflect the strength and stability of these relationships, with closer distances indicating more stable interactions. Some bond distances in the G38R and G42D mutants are a little bit longer than in the wild type, suggesting that their interactions with edaravone may be weaker. According to these findings, the G38R and G42D mutations introduced into SOD1 may lead to modifications in the binding behavior of Edaravone.
Discussion
The severe neurodegenerative illness ALS has intricate genetic roots. With a focus on SNPs within the SOD1 gene, our work aims to close the knowledge gap in comprehending the whole range of genetic differences contributing to ALS risk. The aetiology of ALS is complex, with both genetic predisposition and environmental variables contributing to disease progression (Roberts et al. 2022). Our thorough examination of SOD1 SNPs sheds important light on putative functional effects on the SOD1 protein and the complex genetic landscape of ALS.
The copper-zinc SOD1 gene variation makes up a sizeable part of these, emphasising its hereditary nature and importance in ALS genetics (Goutman et al. 2023). A series of pathogenic events, including protein misfolding, aggregation, and the development of intracellular inclusions inside susceptible motor neurons, are triggered by mutations in the SOD1 gene. These complex interactions result in mitochondrial dysfunction, impaired axonal transport, excitotoxicity, neuroinflammation, motor neuron degeneration, and the distinctive muscle weakness seen in ALS (Jankovic et al. 2021). Understanding the genetic component of ALS, in particular the function of the SOD1 gene, is essential for understanding the complexities of the illness. In addition to offering insights into the intricate biology of ALS, it also prepares the way for specialised therapeutic approaches meant to lessen the suffering of ALS patients and their families.
With a focus on single nucleotide polymorphisms (SNPs) within the SOD1 gene, our work aims to close the knowledge gap in comprehending the whole range of genetic differences contributing to ALS risk. The aetiology of ALS is complex, with both genetic predisposition and environmental variables contributing to disease progression. Our thorough examination of SOD1 SNPs sheds important light on putative functional effects on the SOD1 protein and the complex genetic landscape of ALS.
19,593 SNPs from the human SOD1 gene, representing diverse genetic variants, were included in the initial dataset. The purpose of the study was to investigate the potential impact of missense variations (nsSNPs) on the SOD1 protein. Several prediction algorithms, including SIFT, SNAP2, Align GVGD, and PANTHER, were used to classify these variants as harmful, tolerated, or neutral out of the initial pool of 599 nsSNPs. 11 high-risk nsSNPs were found by a careful filtering method (I113T, G38R, G42D, G86R, G94C, I114T, C7F, I152T, G13R, F46C), all of which had negative consequences as indicated by numerous prediction algorithms.
The probable damage of these 11 nsSNPs was verified using the PredictSNP programme. Additionally, the study used MUpro to evaluate the stability of SOD1 mutations with an emphasis on delta G (DDG) values. There may be a connection between mutation-induced instability and the onset of ALS, as mutations with DDG values below − 1 were found to dramatically reduce the stability of the SOD1 protein.
Using the ConSurf server, the investigation also looked into the evolutionary conservation of amino acid residues in SOD1. Notably, seven of the 11 chosen nsSNPs were shown to have a high degree of projected conservation, suggesting that they may have functional importance. Four of these variants out of these seven were expected to be entrenched in the protein structure, indicating potential roles in binding interactions and enzymatic activity.
Three exposed nsSNPs (G38R and G42D) that were likely to interact with other molecules were highlighted by the solvent accessibility study. These publicly available nsSNPs might participate in protein–protein interactions and possibly influence the pathophysiology of ALS.
The clinical phenotypes associated with SOD1 genetic variants exhibit significant heterogeneity. Our analysis adhered to the Human Genome Variation Society recommendations (https://varnomen.hgvs.org), revealing that variants G38R and G42D in our study are linked to prolonged survival (Tasca et al. 2020).
Numerous functional studies have been conducted using transgenic mouse models with heightened expression levels of the G38R mutated gene. These investigations demonstrated variations in the age of onset, disease progression, and overall lifespan. Additionally, Drosophila melanogaster models confirmed a progressive deterioration of motor functions, presenting a distinct clinical phenotype associated with the G38R variant. The same experimental model further elucidated the impact of G38R variants, leading to a reduced lifespan in the midge accompanied by movement abnormalities (Ruffo et al. 2022).
The G42D mutation, frequently identified in familial amyotrophic lateral sclerosis (FALS) patients, is commonly associated with initial symptoms of lower limb weakness (mean onset age: 46.0 years) and a very slow, ascending paresis, resulting in a decade-long or more survival. Interestingly, our patient carrying the G42D mutation exhibited a different clinical presentation, characterized by a relatively rapid disease progression (16 months) in contrast to the typical slow progression (Wei et al. 2017). This discrepancy may be attributed to various factors, including the early age of onset predicting longer survival, as suggested by previous observations. Notably, the patient with the G42D mutation deviated from the expected slow progression, indicating potential influences from epigenetic factors such as sex, modifier genes, environmental elements, and other unidentified variables on disease phenotype and progression.
Furthermore, structural modelling of both wild-type and mutant SOD1 proteins validated the accuracy of these models, allowing for their use in later structural and functional studies.
In 2020, the estimated prevalence of Amyotrophic Lateral Sclerosis (ALS) in 22 European countries reached 121,028 cases, while the global incidence is documented to range from 1 to 2.6 cases per 100,000 individuals (Tzeplaeff et al. 2023). The study looked at Edaravone's binding affinity in order to shed light on prospective therapeutic approaches for ALS. Edaravone is an antioxidant that shows promise as a treatment for the disease (Mead et al. 2023). According to the docking research, Edaravone had a higher affinity for the mutant SOD1 structures (G38R and G42D) than it did for the wild-type structure. This implies that these SOD1 mutations have a major impact on the way Edaravone binds and may affect how effective it is as a potential treatment.
The nsSNPs in the SOD1 gene have been thoroughly analysed in this paper, along with any potential functional ramifications. The discovery of 11 high-risk nsSNPs provides insight into their functions in the stability of the SOD1 protein and their possible connections with other components. The results further highlight the possibility that SOD1 mutations affect the therapeutic effectiveness of edaravone, cause its antioxidative properties and its relevance in mitigating oxidative stress associated with ALS pathophysiology. This study adds to our knowledge of the genetic causes of ALS and sheds light on prospective treatment options for this debilitating condition. It is necessary to conduct additional research and clinical trials to confirm the practical applicability of these findings and investigate individualised treatment plans for ALS patients.
Conclusion
The SOD1 gene's single nucleotide polymorphisms (SNPs) were thoroughly examined in this study, revealing insight on their potential functional effects in relation to Amyotrophic lateral sclerosis (ALS). The study shed light on the genetic makeup of ALS by identifying 11 high-risk nsSNPs that negatively affect the SOD1 protein.
The analysis of SOD1 stability and its relationship to Edaravone in the study further highlights the potential applicability of these genetic variants to ALS treatments. The potential for individualised treatment plans depending on the genetic profile of the patient is suggested by Edaravone's increased binding affinity for mutant SOD1 structures.
In order to confirm the clinical implications of these findings and investigate customised therapy methods, more study will be necessary as we move forward. The complex genetics of ALS are still being uncovered, giving hope for better medicines and individualised care for ALS patients. Future research should work to close the knowledge gap between genetics and clinical use, thereby enhancing the lives of those suffering from this deadly neurodegenerative illness.
Data availability
All data generated or analysed during this study are included in this published article.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- ASA:
-
Accessible surface area
- cSNP:
-
Coding single nucleotide polymorphism
- GWAS:
-
Genome-wide association studies
- NGS:
-
Next generation sequencing
- nsSNPs:
-
Non-synonymous single nucleotide polymorphism
- nsSNVs:
-
Non-synonymous/missense single nucleotide variations
- PDB:
-
Protein data bank
- PSEC:
-
Position specific evolutionary conservation
- QMEAN:
-
Qualitative model energy analysis
- RSA:
-
Relative surface accessibility
- SA:
-
Surface area
- SAAFEC-SEQ:
-
Sequence-based stabilisation analysis of amino acid change
- SIFT:
-
Sorting intolerant from tolerant
- SNAP:
-
Screening for non-acceptable polymorphism
- SNPs:
-
Single nucleotide polymorphisms
- SOD1:
-
Superoxide dismutase 1
- STRING:
-
Search tool for the retrieval of interacting genes/proteins
References
Apte M, Kumar A (2023) Correlation of mutated gene and signalling pathways in ASD. IBRO Neurosci Rep. https://doi.org/10.1016/j.ibneur.2023.03.011
Bamshad C, Najafi-Ghalehlou N, Pourmohammadi-Bejarpasi Z, Tomita K, Kuwahara Y, Sato T et al (2022) Mitochondria: how eminent in ageing and neurodegenerative disorders? Hum Cell 36(1):41–61. https://doi.org/10.1007/s13577-022-00833-y
Berdyński M, Miszta P, Safranow K, Andersen PM, Morita M, Filipek S et al (2022) SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci Rep. https://doi.org/10.1038/s41598-021-03891-8
Besterman AD, Althoff T, Elfferich P, Gutierrez-Mejia I, Sadik J, Bernstein JA et al (2021) Functional and structural analyses of novel Smith-Kingsmore Syndrome-Associated MTOR variants reveal potential new mechanisms and predictors of pathogenicity. PLoS Genet 17(7):e1009651. https://doi.org/10.1371/journal.pgen.1009651
Bonifacino T, Zerbo RA, Balbi M, Torazza C, Frumento G, Fedele E et al (2021) Nearly 30 years of animal models to study amyotrophic lateral sclerosis: a historical overview and future perspectives. Int J Mol Sci 22(22):12236. https://doi.org/10.3390/ijms222212236
Candelise N, Salvatori I, Scaricamazza S, Nesci V, Zenuni H, Ferri A et al (2022) Mechanistic insights of mitochondrial dysfunction in amyotrophic lateral sclerosis: an update on a lasting relationship. Metabolites 12(3):233. https://doi.org/10.3390/metabo12030233
Chaudhary R, Agarwal V, Rehman M, Kaushik AS, Mishra V (2022) Genetic architecture of motor neuron diseases. J Neurol Sci 434(120099):120099. https://doi.org/10.1016/j.jns.2021.120099
Dang X, Zhang L, Franco A, Dorn GW II (2023) Activating mitofusins interrupts mitochondrial degeneration and delays disease progression in SOD1 mutant amyotrophic lateral sclerosis. Hum Mol Genet 32(7):1208–1222. https://doi.org/10.1093/hmg/ddac287
Daud S, Yousafzai A (2021) Study on amyotrophic lateral sclerosis: a disease of motor neuron in Pakistan. International Journal of Pathology 19(3):155–164
Deneault E, Chaineau M, Nicouleau M, Castellanos Montiel MJ, Franco Flores AK, Haghi G et al (2022) A streamlined CRISPR workflow to introduce mutations and generate isogenic iPSCs for modeling amyotrophic lateral sclerosis. Methods 203:297–310. https://doi.org/10.1016/j.ymeth.2021.09.002
Falahi S, Karaji AG, Koohyanizadeh F, Rezaiemanesh A, Salari F (2021) A comprehensive in Silico analysis of the functional and structural impact of single nucleotide polymorphisms (SNPs) in the human IL-33 gene. Comput Biol Chem 94(107560):107560. https://doi.org/10.1016/j.compbiolchem.2021.107560
Frlan R (2022) An evolutionary conservation and druggability analysis of enzymes belonging to the bacterial shikimate pathway. Antibiotics (basel) 11(5):675. https://doi.org/10.3390/antibiotics11050675
Gagliardi D, Ripellino P, Meneri M, Del Bo R, Antognozzi S, Comi GP et al (2023) Clinical and molecular features of patients with amyotrophic lateral sclerosis and SOD1 mutations: a monocentric study. Front Neurol 14:1169689. https://doi.org/10.3389/fneur.2023.1169689
Gao AY (2020) Investigating the loss of sodium/proton exchanger isoform 6 (nhe6) function in christianson syndrome. McGill University (Canada) ProQuest Dissertations Publishing, 2020. p 29274264
Goutman SA, Savelieff MG, Jang D-G, Hur J, Feldman EL (2023) The amyotrophic lateral sclerosis exposome: recent advances and future directions. Nat Rev Neurol 19(10):617–634. https://doi.org/10.1038/s41582-023-00867-2
Imran FS, Al-Thuwaini TM, Al-Shuhaib MBS, Lepretre F (2021) A novel missense single nucleotide polymorphism in the GREM1 gene is highly associated with higher reproductive traits in awassi sheep. Biochem Genet 59(2):422–436. https://doi.org/10.1007/s10528-020-10006-x
Jankovic M, Novakovic I, Gamil Anwar Dawod P, Gamil Anwar Dawod A, Drinic A, Abdel Motaleb FI et al (2021) Current concepts on genetic aspects of mitochondrial dysfunction in amyotrophic Lateral Sclerosis. Int J Mol Sci 22(18):9832. https://doi.org/10.3390/ijms22189832
Krokidis MG, Exarchos TP, Vlamos P (2022) Gene expression profiling and bioinformatics analysis in neurodegenerative diseases. Handbook of computational neurodegeneration. Springer International Publishing, Cham
Kumar M, Tyagi N, Faruq M (2023) The molecular mechanisms of spinocerebellar ataxias for DNA repeat expansion in disease. Emerg Topics Life Sci. https://doi.org/10.1042/ETLS20230013
Li B, Mendenhall J, Capra JA, Meiler J (2021) A multitask deep-learning method for predicting membrane associations and secondary structures of proteins. J Proteome Res 20(8):4089–4100. https://doi.org/10.1021/acs.jproteome.1c00410
Lünemann JD, Malhotra S, Shinohara ML, Montalban X, Comabella M (2021) Targeting inflammasomes to treat neurological diseases. Ann Neurol 90(2):177–188. https://doi.org/10.1002/ana.26158
Mathioudakis L, Dimovasili C, Bourbouli M, Latsoudis H, Kokosali E, Gouna G (2023) Study of Alzheimer’s disease-and frontotemporal dementia-associated genes in the Cretan Aging Cohort. Neurobiol Aging 123:111–128
Maung MT, Carlson A, Olea-Flores M, Elkhadragy L, Schachtschneider KM, Navarro-Tito N et al (2021) The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. https://doi.org/10.1096/fj.202100273rr
McDonough SI (2022) From calcium channels to new therapeutics. Voltage-gated calcium channels. Springer International Publishing, Cham, pp 687–706
Mead RJ, Shan N, Reiser HJ, Marshall F, Shaw PJ (2023) Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov 22(3):185–212. https://doi.org/10.1038/s41573-022-00612-2
Meijboom KE, Brown RH (2022) Approaches to gene modulation therapy for ALS. Neurotherapeutics 19(4):1159–1179. https://doi.org/10.1007/s13311-022-01285-w
Oroian BA, Ciobica A, Timofte D, Stefanescu C, Serban IL (2021) New metabolic, digestive, and oxidative stress-related manifestations associated with posttraumatic stress disorder. Oxid Med Cell Longev 2021:1–18. https://doi.org/10.1155/2021/5599265
Pan S, Kang H, Liu X, Lin S, Yuan N, Zhang Z et al (2023) Brain Catalog: a comprehensive resource for the genetic landscape of brain-related traits. Nucleic Acids Res 51(D1):D835–D844. https://doi.org/10.1093/nar/gkac895
Park SW, Lee BH, Song SH, Kim MK (2023) Revisiting the Ramachandran plot based on statistical analysis of static and dynamic characteristics of protein structures. J Struct Biol 215(1):107939. https://doi.org/10.1016/j.jsb.2023.107939
Rafaee A, Kashani-Amin E, Meybodi AM, Ebrahim-Habibi A, Sabbaghian M (2022) Structural modeling of human AKAP3 protein and in silico analysis of single nucleotide polymorphisms associated with sperm motility. Sci Rep. https://doi.org/10.1038/s41598-022-07513-9
Rahman MM, Islam MR, Alam Tumpa MA, Shohag S, Khan Shuvo S, Ferdous J et al (2023) Insights into the promising prospect of medicinal chemistry studies against neurodegenerative disorders. Chem Biol Interact 373:110375. https://doi.org/10.1016/j.cbi.2023.110375
Roberts B, Theunissen F, Mastaglia FL, Akkari PA, Flynn LL (2022) Synucleinopathy in amyotrophic lateral sclerosis: a potential avenue for antisense therapeutics? Int J Mol Sci 23(16):9364. https://doi.org/10.3390/ijms23169364
Rozario LT, Sharker T, Nila TA (2021) In silico analysis of deleterious SNPs of human MTUS1 gene and their impacts on subsequent protein structure and function. PLoS ONE 16(6):e0252932. https://doi.org/10.1371/journal.pone.0252932
Ruffo P, Strafella C, Cascella R, Caputo V, Conforti FL, Andò S et al (2021) Deregulation of ncRNA in neurodegenerative disease: focus on circRNA, lncRNA and miRNA in amyotrophic lateral sclerosis. Front Genet. https://doi.org/10.3389/fgene.2021.784996
Ruffo P, Perrone B, Conforti FL (2022) SOD-1 variants in amyotrophic lateral sclerosis: Systematic re-evaluation according to ACMG-AMP guidelines. Genes (basel) 13(3):537. https://doi.org/10.3390/genes13030537
Shinwari K, Rehman HM, Xiao N, Guojun L, Khan MA, Bolkov MA et al (2023) Novel high-risk missense mutations identification in FAT4 gene causing Hennekam syndrome and Van Maldergem syndrome 2 through molecular dynamics simulation. Inform Med Unlocked 37(101160):101160. https://doi.org/10.1016/j.imu.2023.101160
Sivaramakrishnan V, Kumar A (2022) Structural systems biology approach delineate the functional implications of SNPs in exon junction complex interaction network. J Biomol Struct Dyn 41(21):11969–11986. https://doi.org/10.1080/07391102.2022.2164355
Stitou M, Toufik H, Bouachrine M, Lamchouri F (2021) Quantitative structure–activity relationships analysis, homology modeling, docking and molecular dynamics studies of triterpenoid saponins as Kirsten rat sarcoma inhibitors. J Biomol Struct Dyn 39(1):152–170. https://doi.org/10.1080/07391102.2019.1707122
Tasca G, Lattante S, Marangi G, Conte A, Bernardo D, Bisogni G et al (2020) SOD1 p.D12Y variant is associated with amyotrophic lateral sclerosis/distal myopathy spectrum. Eur J Neurol 27(7):1304–1309. https://doi.org/10.1111/ene.14246
Tzeplaeff L, Wilfling S, Requardt MV, Herdick M (2023) Current state and future directions in the therapy of ALS. Cells 12(11):1523
Ullah S, Khan SU, Khan A, Junaid M, Rafiq H, Htar TT et al (2022) Prospect of Anterior Gradient 2 homodimer inhibition via repurposing FDA-approved drugs using structure-based virtual screening. Mol Divers 26(3):1399–1409. https://doi.org/10.1007/s11030-021-10263-x
Vora D, Kapadia H, Dinesh S, Sharma S, Manjegowda DS (2023) Gymnema sylvestre as a potential therapeutic agent for PCOS: insights from mRNA differential gene expression and molecular docking analysis. Futur J Pharm Sci. https://doi.org/10.1186/s43094-023-00529-6
Wanarase SR, Chavan SV, Sharma S, Susha (2023) Evaluation of SNPs from human IGFBP6 associated with gene expression: an in-silico study. J Biomol Struct Dyn 41(23):13937–13949. https://doi.org/10.1080/07391102.2023.2192793
Wang JC, Ramaswami G, Geschwind DH (2021) Gene co-expression network analysis in human spinal cord highlights mechanisms underlying amyotrophic lateral sclerosis susceptibility. Sci Rep. https://doi.org/10.1038/s41598-021-85061-4
Wei Q, Zhou Q, Chen Y, Ou R, Cao B, Xu Y et al (2017) Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: a case-control study and literature review. Sci Rep. https://doi.org/10.1038/srep44606
Wei T, Guo Z, Wang Z, Li X, Zheng Y, Hou H et al (2022) Exploring the causal relationship between dietary macronutrients and neurodegenerative diseases: a bi-directional two-sample Mendelian randomization study. Ageing Neur Dis 2(3):14. https://doi.org/10.20517/and.2022.12
Zaji HD, Seyedalipour B, Hanun HM, Baziyar P, Hosseinkhani S, Akhlaghi M (2023) Computational insight into in silico analysis and molecular dynamics simulation of the dimer interface residues of ALS-linked hSOD1 forms in apo/holo states: a combined experimental and bioinformatic perspective. 3 Biotech. https://doi.org/10.1007/s13205-023-03514-1
Zhou W, Xu R (2023) Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis. Front Neurosci 17:1189470. https://doi.org/10.3389/fnins.2023.1189470
Acknowledgements
We thank BioNome, Bangalore (www.bionome.in/) for providing insight in bioinformatics analysis and scientific article writing.
Funding
The present study is funded by the Department of Scientific Research and Education, BioNome (Funding ID: DSRE/BNM/SR/2023/A0115).
Author information
Authors and Affiliations
Contributions
All authors participated in the analysis, writing, reviewing, and editing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Declared none.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bhor, S., Tonny, S.H., Dinesh, S. et al. Computational screening of damaging nsSNPs in human SOD1 genes associated with amyotrophic lateral sclerosis identifies destabilising effects of G38R and G42D mutations through in silico evaluation. In Silico Pharmacol. 12, 20 (2024). https://doi.org/10.1007/s40203-024-00191-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-024-00191-7