
Abstract
The aggregation of misfolded SOD1 proteins in neurodegenerative illnesses is a key pathological hallmark in amyotrophic lateral sclerosis (ALS). SOD1 is stabilized and enzymatically activated after binding to Cu/Zn and forming intramolecular disulfide. SOD1 aggregation/oligomerization is triggered by the dissociation of Cu and/or Zn ions. Therefore, we compared the possible effects of ALS-associated point mutations of the holo/apo forms of WT/I149T/V148G SOD1 variants located at the dimer interface to determine structural characterization using spectroscopic methods, computational approaches as well as molecular dynamics (MD) simulations. Predictive results of computational analysis of single-nucleotide polymorphisms (SNPs) suggested that mutant SOD1 has a deleterious effect on activity and structure destabilization. MD data analysis indicated that changes in flexibility, stability, hydrophobicity of the protein as well as increased intramolecular interactions of apo-SOD1 were more than holo-SOD1. Furthermore, a decrease in enzymatic activity in apo-SOD1 was observed compared to holo-SOD1. Comparative intrinsic and ANS fluorescence results of holo/apo-WT-hSOD1 and mutants indicated structural alterations in the local environment of tryptophan residue and hydrophobic patches, respectively. Experimental and MD data supported that substitution effect and metal deficiency of mutants (apo forms) in the dimer interface may promote the tendency to protein mis-folding and aggregation, consequently disrupting the dimer–monomer equilibrium and increased propensity to dissociation dimer into SOD-monomer ultimately leading to loss of stability and function. Overall, data analysis of apo/holo SOD1 forms on protein structure and function using computational and experimental studies will contribute to a better understanding of ALS pathogenicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As a fatal disorder, amyotrophic lateral sclerosis (ALS) occurs as a consequence of the destruction of the spinal cord, motor neurons in the stem of the brain, and also the motor cortex (Krivickas 2003). Approximately 5–10% of cases are familial ALS (fALS), while most cases of ALS are sporadic (sALS) (Zarei, et al. 2015). Mutations in Cu/Zn human superoxide dismutase 1 (hSOD1) gene are one of the most common and important causes of ALS, accounting for 23% of fALS and ~ 7% of sALS (Tang et al. 2019). hSOD1 is known as a 32 kDa homodimeric protein. Structurally, hSOD1 is well organized by forming eight antiparallel β-strands. Electrostatic loop VII (residues 121–142) and metal-binding loop IV (49–83) are formed as two active site loops (Svensson et al. 2010). Some residues, including V5, V7, K9, I17, E49-T54, I113-R115 and V148-Q153 participate in SOD1 dimer interface (Ghosh et al. 2020). Hitherto, more than 200 mutations of hSOD1 have been reported to be ALS-associated (Bernard et al. 2020). fALS mutations are distributed all over the hSOD1 sequence with no pattern and all mutations lead to the same disease almost all the time and produce the same result. The disease progression rate is significantly different in the mutations, which promotes demetallation of hSOD1 and decreases the intra-subunit disulfide stabilizing bond, leading to hSOD1 misfolding, reduced stability of hSOD1, and/or destabilized dimer interface (Baziyar et al. 2022a; Famil Samavati et al. 2022; Bhatia et al. 2015). There is evidence to show that stabilization of the SOD1 dimer leads to the increased thermostability of protein, prevents monomerization, and inhibits aggregation. Studies point to the role of instability in the structure of hSOD1 (electrostatic loop and dimer interface) associated with fALS, which affects the interaction of monomers and the geometry of the active site and the folding of SOD1 (Khare et al. 2006); consequently, it may alter the stability and function of SOD1 structure even following minor perturbations in this region (Molnar et al. 2009). Dissociation of the oligomers into monomers instigates an aberrant exposure of hydrophobic regions of monomers to an aqueous environment, and considering the hydrophobic regions can be aggregation-prone, this causes aggregation of the proteins (Frieden 2007; Jamadagni et al. 2011). A recent finding in this regard indicates a sequence prone to aggregation of protein 147GVIGIAQ153 in β-8 (β strand), which causes changes in the structure and dynamics of the backbone (Ivanova et al. 2014). DNA sequencing analysis showed a heterozygous, c.446 T > G the substitution of Valine to Glycine in the hSOD1 mutation of GTA to GGA (V 148 to G) and c.449 T > C mutation of ATT to ACT, which leads to a substitution of Isoleucine to Threonine at codon 149. Both mutations are located in exon 5 (Deng et al. 1993; Fong et al. 2006). Currently, riluzole is prescribed for ALS to alleviate its pathology, which inhibits glutamatergic neurotransmission. Unfortunately, taking this drug only increases life expectancy by 2 to 3 months. Recently, another drug called edaravone (which delays disease progression by reducing reactive oxygen species) has been approved by the FDA to effectively reduce the pathology ALS-associated. However, reports have shown that taking therapeutic doses of edaravone can cause dangerous side effects that will lead to disease progression as well as unanticipated diseases. Therefore, broad endeavors should be made to extend new drug molecules that are necessarily convenient and cost-effective, not only to improve survival time but as well to decrease side effects (Jackson et al. 2019; Jaiswal 2019). The V148 is conserved between seven species from humans to mycobacteria. Residues (I113 and V148) form two-way symmetric dimer interactions and significantly destabilize the mutant homodimer structure in FALS mutants (Deng et al. 1993). V148 is essential for maintaining the dimer interface, and substitution with either amino acid leads to a lower propensity to dimerization than WT SOD1. Specifically, between the SOD1 monomers, V148–V148 and F50–I151 are important hydrophobic contacts. Previous studies showed that mutation in A 4 residue (A4V) leads to increased flexibility in V148 and I 151 residues and possibly induces steric clash at the interface or weakens the interactions (Fay et al. 2016). The side chain Cδ1 of I149 has Van der Waals interactions with different amino acids including V47, I112, V148, and L117. Mutants, A4V is repulsed around the mutation site, but Van der Waals interactions are disrupted by I113T and Ile149Thr at the dimer interface and the SOD1 core, respectively (Cardoso et al. 2002; Wright et al. 2019). Understanding the role of hSOD1 mutants in protein activities/aggregation at the molecular level requires information on their dynamic properties, which can be enhanced at the molecular level alongside experimental methods. Molecular dynamics (MD) simulations are an alternative technique to investigate conformational changes induced by mutations and provide atomic-level insights that relate protein dynamics and structure to its biological functions. Computer simulations, known as in silico analysis, have become an efficient way to better understand disease-associated mutations and their consequences for protein structures. In silico analysis effectively predicts the impacts caused by non-synonymous single-nucleotide variants (nsSNVs) (Krebs and Mesquita 2016; Kumar et al. 2014). In fact, this is necessary for understanding the pathology of fALS illnesses. Accordingly, this study was designed to investigate the possible effects of apo/holo hSOD1 point mutations V148G and I149T located at the dimer interface, and we addressed the dynamic properties of these mutant systems using MD simulations alongside experimental methods to describe their properties and their relation and protein aggregation on a molecular level.
Methods and materials
Computational methods
Sequence analysis and dataset
The hSOD1 sequence was obtained from the UniProt database (ID: P00441) (Silva et al. 2019). The protein wild-type hSOD1 structure was achieved using Protein Data Bank [PDB ID: 2C9V] (Berman et al. 2000). This PDB ID was used because it had the highest atomic resolution (1.07 Å) among the structure types (Rose et al. 2016).
Functional and stability prediction analysis
The i-Stable integrated server was applied for evaluating the stability of the structure of the mutants in comparison to the WT. The i-Stable predicts the alteration in the stability of protein resulting from single amino acid residue changes utilizing a support vector machine (SVM) (Chen et al. 2013). The results acquired from i-Stable incorporate the prediction from the different other algorithms including I-mutant2.0 PDB, I-mutant2.0 SEQ (Capriotti et al. 2005), AUTO-MUTE RF, and AUTO-MUTE SVM (Masso and Vaisman 2014). In addition, the DUET algorithm (Pires et al. 2014a) was used to predict the alteration in the conformational stability of hSOD1 when mutated. There are two complementary approaches for DUET as a web server for an integrated computational method of studying missense mutations within proteins: mCSM (Pires et al. 2014b) and SDM (Worth et al. 2011). They use Support Vector Machines (SVM) in a comprehensive prediction to integrate the outputs from different methods into one predictor, the output of which is signified as Gibbs free energy (∆∆G), in which negative values denote the destabilizing impact the mutation has on the structure of the protein. In addition, a PREDICT-SNP (a consensus classifier) integrated server was used for predicting the point mutation impact on hSOD1 protein function (Bendl et al. 2014a). PREDICT-SNP processes the prediction by combining the seven most efficient prediction algorithms (SIFT (Ng and Henikoff 2003), SNAP (Bromberg and Rost 2007), PhD-SNP (Capriotti et al. 2006), PolyPhen-1, PolyPhen-2 (Adzhubei et al. 2013), PANTHER (Thomas et al. 2003), SNPs&GO (Calabrese et al. 2009), PMUT (Ferrer-Costa et al. 2005), and MAPP (Stone and Sidow 2005)) to present an optimal prediction along with the confidence predictions score. Furthermore, we investigated the effect of alteration in amino acid on protein folding free energy by the SAAFEC-SEQ server (Li et al. 2021). As a simple method that is based upon evolutionary information, DDGun provides a prediction of the ∆∆G for single and multiple variations from available sequence and structure information (DDGun3D). It is based only on sequence-based properties and DDGun3D as well as structure-based characteristics (Montanucci et al. 2019). DeepDDG is a neural network-based method that predicts alterations in the proteins’ stability caused by point mutations (Cao et al. 2019).
Molecular dynamics (MD) simulations
Structural comparison between WT-hSOD1, V148G, and I149T mutants (apo/holo forms) was made based on simulation of molecular dynamics (MD). In this study, we used monomeric hSOD1 to evaluate stability and dynamics because each monomer's dynamics or folding kinetics are independent of dimerization and the dimer interface (Nordlund and Oliveberg 2008). As reported in several studies, monomeric hSOD1 was used to investigate the impacts of mutations on the structure, folding kinetics, stability, and dynamics of the protein (Ferraroni, et al. 1999; Leinartaite et al. 2010; Nordlund et al. 2009; Nordlund and Oliveberg 2006; Schmidlin et al. 2009). Therefore, apo/holo mutations at (V148G and I149T) were entered using PyMOL into the hSOD1 protein structure (DeLano 2002), in sub-unit A alone and sub-unit B unchanged to function as a control for studying and comparing the impact of the mutation on the hSOD1 molecule dynamics. It uses this technique to preserve WT SOD1 chain dynamics under the same simulation conditions. GROMACS v4.6.5 package with GROMOS96 54A7 force field (Huang et al. 2011; Schmid et al. 2011) was applied for performing the MD simulations of mutants (apo/holo hSOD1) structures. The models were placed at the center of a dodecahedral box and solvated with a simple point charge (SPC) water model and an appropriate amount of Na+ and Cl˗ ions was used to create the topology file and neutralize the system. The steepest descent algorithm was applied to minimize energy in the system. In addition, in the minimized system, position restraints were used to equilibrate constant number, volume, and temperature (NVT) and also constant number, pressure and temperature (NPT) for a short MD simulation (100 ps) to stabilize them at 300 K until the convergence of potential energy. Periodic boundary conditions are used to confirm whether the system components are present within the simulation box. The temperature is kept at 300 K and the pressure is maintained at 1 atm. A leap-frog algorithm with a time step of 2 fs was applied for integration of Newton’s equation in the MD simulation and data was gathered every 10 ps. Then, MD simulations of the equilibrated system were carried out for 150 ns, and all the atoms were freely allowed to modify their positions. Subsequently, the following GROMACS distribution programs were applied to analyze the trajectories: gmx rms, gmx rmsf, gmx gyrate, gmx h-bond, gmx sasa, and gmx dssp. These MD analyses yielded parameter values for root‐mean‐square deviation (RMSD), root‐mean‐square fluctuation (RMSF), the radius of gyration (Rg), hydrogen bonds (H-bond), solvent accessible surface area (SASA), and secondary structure (SS) do_dssp.
Essential dynamics
We used essential dynamics or principal component analysis (PCA) to shed light on the collective movement of holo/apo-WT-SOD1 and mutants (Amadei et al. 1993). Consequently, we obtained the covariance matrix related to atomic coordinates from the trajectory and retrieved collective motions from diagonalizing matrix, as eigenvectors and eigenvalues, which provided each motion’s amplitude. The atoms’ motion in each eigenvector indicated the direction of protein motion. On this basis, to quantify WT and mutants’ motion, across the trajectory, the highest amplitude of motions was plotted, as represented in the first two eigenvectors. GROMACS v4.6.5 package for the holo/apo-WT-SOD1 and mutants was applied to the essential dynamics. Using the Bio3D package, the inter-residue mean smallest distances map was plotted between the WT and mutants (Grant et al. 2006).
Experimental methods
Expression and purification of holo/apo-WT-hSOD1 and variants
The Quick-change site-directed mutagenesis is performed for constructing single-point mutations including I149T and V148G. The recombinant pET28a vector containing the human SOD1 gene (pET28a-hSOD) was used as the template. Point mutations are created by designing two mutagenesis primers for V148G: forward primer (5′- G GCT TGT GGT GGA ATT GGG ATC GCC CAA TAA AC -3′) with mutation site (highlight), and reverse primer (5′—TG GGC GAT CCC AAT TCC ACC ACA AGC CAA AC—3′) with mutation site (highlight) and also two primers were used for I149T mutation: forward primer (5′ TG GCT TGT GGT GTA ACT GGG ATC GCC CAA TAA AC 3′) with mutation site (highlight), and reverse primer (5′ TTG GGC GAT CCC AGT TAC ACC ACA AGC CAA AC 3′) with mutation site (highlight) were designed to amplify pET28a-hSOD as a template. The Quick-Change site-directed mutagenesis reactions were performed using HL Taq DNA polymerase. In the following cycles, 95 °C for 5 min for one cycle (initial denaturation), followed by 20 cycles at 95 °C for 45 s (denaturation), 60 °C for 45 s (annealing), and 72 °C for 8 min (extension). The product of PCR was analyzed by 1% agarose gel electrophoresis and then digested with Dpn I to destroy methylated parental plasmid DNA. In this study, E. coli DH5α cells were used as the host strain to maintain and amplify the recombinant vector and E. coli BL21 (DE3) cells were utilized for recombinant protein expression. The pET28a-hSOD were transformed into E. coli DH5α cells by the heat shock (calcium chloride) procedure. After selection colonies containing mutant pET28a-hSOD1, the recombinant vectors were purified using a plasmid miniprep extraction kit. Sequencing was performed using an automatic sequencer and after confirming them, the recombinant vectors were transformed to the expression host E. coli BL21 (DE3) competent cells by heat shock. Primarily, the colony was cultured in 5 ml of Luria–Bertani (LB) medium supplemented with 50 µg/ mL kanamycin for 16–18 h at 37 °C under 160 rpm agitations. After inoculation into a 100 mL culture and reaching absorbance of about 0.6–0.8 OD at 600 nm, at 37 °C, 220 rpm, the bacterial culture was induced with 0.8 mM isopropyl-1-thio-d-galactopyranoside (IPTG), 200 µM CuSO4 and 60 μM ZnSO4 (just for holo-SOD1) for 18–22 h at 22 °C and then cells were harvested by centrifugation at 6000 g for 15 min. The supernatant was loaded onto Ni–NTA agarose affinity chromatography (ABT, USA). After purification, one part of SOD1 dialysis by serial dialysis against CuSO4 and ZnSO4 at 4 °C for numerous days was described previously (Beyer et al. 1987). For preparing the metal-free (apo) state of the SOD1, stepwise dialysis was not performed. Protein concentration was measured by the Bradford method, and the purity of SOD1 was checked by SDS-PAGE.
Specific activity of apo/holo WT-SOD1 and mutants
The specific activity of apo/holo WT-SOD1 and mutants was assessed by inhibiting pyrogallol autoxidation using the Marklund method, as previously reported (Baziyar et al. 2022a; Marklund and Marklund 1974). One unit of SOD1 activity was expressed as the amount needed for inhibiting 50% pyrogallol autoxidation.
Intrinsic and ANS fluorescence assay of apo/ holo SOD1 forms
Intrinsic fluorescence spectra were recorded using an FP-8300 spectrofluorometer JASCO fluorescence spectrophotometer at 25 °C. The concentrations of apo/holo WT-SOD1, V148G, and I149T variants were 20 µg/ml in phosphate buffer (20 mM and pH 7.4). The excitation wavelength was fixed at 295 nm, and emission spectra were monitored between 300 and 450 nm (slit width 5/10 nm each). To monitor exposed hydrophobic patches apo and holo forms, ANS (8-anilino-1-naphthalene-sulfonic acid) fluorescence assay was carried out using FP-8300 spectrofluorometer JASCO fluorescence with an excitation wavelength of 350 nm and emission spectra between 370 and 700 nm (slit widths, ex 5 nm/em 10 nm). The concentration of WT-SOD1, V148G, and I149T variants in apo/holo forms was 20 µg/ml in 20 mM phosphate buffer (pH 7.4). ANS concentration was 30 µM and the molar ratio of protein to ANS (1:30) was used.
Results and discussion
Computational section
Effects of the substitution mutation on structural SOD1 protein
In the current study, we describe a combined experimental and bioinformatics approaches (in silico and MD simulation) to evaluate the possible impacts of ALS-associated mutations of holo/apo forms of WT/ V148G / I149T SOD1 variants located at the dimer interface for better understanding the sequence and structural consequences of substitution mutations in hSOD1 protein. The impact of mutation on the stability of the SOD1 dimer is considered as the difference between mutant and WT-SOD1 in the stimulated conformational free energy, and the dissociation propensity as the difference between the SOD1 dimer and two monomers’ conformational free energies. The implications of the mutations showed that structural changes and local interactions could affect the protein's hydrophobicity, flexibility, stability, and folding capability and alteration of secondary structures at the dimer interface (Ray et al. 2005). Loss of metal ions and reduction of the intramolecular disulfide bond destabilizes the native dimer and produces an apo monomer as the entry point for aggregation (Rakhit et al. 2004). For WT-SOD1, residues 1–9 and 141–152 are predicted to be the most prone region for aggregation, and mutations occurring near or located at the dimer interface increase the aggregation propensity of the terminal residue. Aggregation happens by a pathway including metal loss and dimer dissociation into apo-SOD1 monomers (Khare, et al. 2005). Therefore, a qualitative analysis of the aggregation hot spots in SOD1 points to the FALS mutations’ potential for inducing SOD1 aggregation via two mechanisms. Our analysis here and previously published results suggests that mutations in the dimer interface can lead to direct enhancement of the hot spot regions’ propensity for aggregation, while other mutations are likely to cause destabilization of the dimer leading to exposure of the aggregation hot spots (Khare et al. 2006). Accordingly, it can be concluded that FALS mutations in SOD1 can lead to induction of aggregation via one or a compilation of the following mechanisms: (i) decreased stability of dimer, (ii) decreased stability of monomer, (iii) increasing dimer dissociation propensity, and (iv) changed catalytic properties. Overall, these studies provide a general framework for the mutation's effect on FALS-associated SOD1 aggregation, leading to reduced stability of the apo-SOD1 dimer and monomer and/or increased dimer dissociation compared to WT, subsequently leading to protein aggregation.
Predicting the functional and structural effects of dimer interface mutations
Interactions between amino acids and the environment cause the determination of the protein structure, leading to a stable three-dimensional structure. In the protein folding process, a stable three-dimensional protein structure is consistent with the least of the Gibbs free energy (ΔG) of the protein–environment system. ΔG of protein folding includes two entropic effects (hydrophobic impact and protein conformation) and the energy of interactions within the protein (such as hydrogen bonds, hydrophobic and electrostatic interactions, etc.) (Stirling and Hieter 2017). Non-synonymous mutations may cause an increase or decrease in protein function and change protein stability, thus preventing structural changes required for protein function (Luigi, et al. 2016). Concerning the difference in the Gibbs free energy of protein unfolding (ΔG), the impacts of non-synonymous mutations on the stability of the protein are measured. ΔG is a non-linear function that includes several factors, such as pH values, temperature, organic solvents, salt concentration, urea, and other chemical factors (Quan et al. 2016). Experimentally, the difference between mutant and wild-type proteins in Gibbs free energy of unfolding is calculated (ΔΔGu) (Sanavia et al. 2020). Therefore, the difference in unfolding free energy between mutant and wild-type proteins ΔΔGu = ΔGumutant- ΔGuwild-type is calculated. The symbol ΔΔG represents changes in protein stability; if it reduces (ΔΔGu < 0) or increases (ΔΔGu > 0), it will be calculated as such. However, the reverse is also likely (ΔΔGf = ΔGfmutant − ΔGfwild-type) (Mishra 2022). Therefore, it is critical to determine which description is assumed. In this evaluation, we used the symbol ΔΔG to refer to ΔΔGu. Subsequently, in this study, we predicted measurable stability changes and estimated structural destabilization of hSOD1-related using various algorithms that calculate the difference in the protein’s structural stability caused by substitution mutation. To further study the possible impacts of ALS-associated mutations I149T and V148G variants located at the dimer interface, we used various integrated bioinformatics servers to predict how single-point mutations influence the stability of the protein. Here, an integrated prediction server known as i-Stable was used to predict changes in the stability of proteins obtained from single amino acid residue alternations using an SVM (support vector machine). The role of mutation on the stability and structure of hSOD1 was examined by five tools of prediction of ΔΔG calculation with web servers, such as I-mutant2 PDB, i-Stable, I-mutant2.0 SEQ, AUTO-Muter RF, AUTO-MUTE SVM, and DUET. As an SVM model, Mutant2.0 estimates the value of ddG protein and the direction of stability alteration (Chen, et al. 2020). The sequence (I-Mutant-SEO) and structure (I-Mutant_PDB) information can be used to construct i-Sable. AUTO-MUTE computes any disorder in the environment due to a single amino acid substitution that creates random forest (RF) (AUTO-MUTE_RF) and supports vector machine (AUTO-MUTE-SVM) (Chen et al. 2013). In addition, DUET was employed as a web server for an approach of integrated computation to study missense mutation in proteins. There are two complementary approaches in DUET (SDM and mCSM) to make a comprehensive prediction based on the results of segregated approaches into one predictor based on SVM (Pires et al. 2014a). SAAFEC-SEQ (Li et al. 2021) (web server) depends on a gradient-boosting decision-tree machine learning approach that utilizes physico-chemical properties, evolutionary information, and sequence features to predict the value of ΔΔG. SDM (Worth et al. 2011) (web server) is a statistical potential energy function that utilizes environment-specific amino acid substitution frequencies inside homologous protein families to determine a score of stability as a proxy of the free energy difference between mutant and wild-type proteins. mCSM (Pires et al. 2014b) (web server) takes the structural signatures based on graphs to encode the distance patterns between atoms, which was utilized for representing the protein residue environment and also predicting and studying the effects of one-point mutation on the stability protein. DDGun3D and DDGun (Montanucci et al. 2019) (stand-alone tool) represent untrained methods that mix evolutionary information and statistical potentials to estimate the ΔΔG. In comparison with the sequence-based DDGun, DDGun3D contains structural information that is scored using Bastolla-Vendruscolo statistical potential (Bastolla et al. 2001) and assigns weights to the linear combination via the accessibility of the mutated amino acid. The two approaches contain antisymmetric features and offer an easy add-on feature to predict several changes. The results of integrated server calculations for the V148G and I149T mutations showed destabilizing effects on the structure of WT-hSOD1, leading to a better understanding of the effect of the mutation on hSOD1 (Table 1). The use of multiple algorithms provides greater reliability. Previous reports have established the role of a missense mutation in protein stability alterations (Compiani and Capriotti 2013; Ó’Fágáin 2017; Tian et al. 2015).
Furthermore, we used Predict SNP computational algorithms to find how amino acid substitutions affect the protein function. Results obtained from computational algorithms and servers can predict the nature of substitution, i.e., whether it is neutral or deleterious because single-nucleotide polymorphisms (SNPs) and genetic variations in the human population are common (Wu and Jiang 2013). SNPs cause synonymous and non-synonymous alters in the human genome. Non-synonymous point mutations cause structural changes that can significantly affect proteins' folding energy and stability. Realizing the impacts of non-synonymous point mutations on the stability of proteins is considered vital and key biological trouble. Numerous neurological illnesses and genetic disorders are related to the misfolding of proteins, which are caused by mutations in genes (Yates and Sternberg 2013). Therefore, a better understanding of how non-synonymous mutations affect the stability of proteins is necessary to correlate their role in various diseases. To differentiate intolerant and tolerant coding mutations, Sorting Intolerant From Tolerant (SIFT) was utilized. This is based on several algorithm information that predicts deleterious or tolerated substitutions for all positions of the query sequence. The servers determine the probability of an amino acid to be tolerated at a given position. Substitutions with probabilities below a tolerance level of 0.05 are considered intolerant or deleterious and substitutions with a tolerance level ≥ 0.05 are considered tolerated (Ng and Henikoff 2003). The probable effects of an amino acid substitution on the function and structure of a protein can be predicted using PolyPhen2 (Polymorphism Phenotyping). The software uses comparative evolutionary and structural considerations. The prediction depends on many features, such as phylogenetic and structural information and sequence, which determine the substitution. Each substitution of amino acid contains a qualitative prediction (“potentially damaging”, probably damaging, benign, and unknown). The software output ranges from 0.0 (tolerated) to 1.0 (deleterious) (Thomas et al. 2003). Another software for checking the effect of deleterious mutations on the function and structure of protein is a multivariate analysis of protein polymorphism (MAPP) (Stone and Sidow 2005). The software predicts the functional effects of amino acid substitutions using the measurement of physicochemical deviation in a column of a protein sequence alignment. The higher the obtained deviation, the higher the probability of affecting the normal function of the protein. The evolutionary conservation of amino acids is used by PANTHER (Protein Analysis Through Evolutionary Relationships) (Thomas et al. 2003) to determine pathogenic coding variants. It employs evolutionary alignment-related proteins to determine the term of the current state of a given amino acid preserved in its ancestors. The probability of functional changes is higher with a longer preservation time. Predictor of Human Deleterious Single-Nucleotide Polymorphism (PhD-SNP) (Capriotti et al. 2006) is a classifier trained to predict if nsSNP is pertinent to disease using a machine learning technique. By beginning with a protein sequence, the support vector machine (SVM) can determine if nsSNPs are neutral or deleterious. Screening for non-acceptable polymorphisms (SNAP) (Bromberg and Rost 2007) is a neural-network-based classifier that predicts the functional effect of single amino acid substitutions. The software depends on sequence information and functional and structural annotations to determine if an nsSNP can disrupt the function of a protein. As a consensus classifier, Predict SNP (Bendl et al. 2014b) mixes the outcome of six ways of prediction (MAPP, nsSNP Analyzer, Panther, PolyPhen-2, PhD-SNP, PolyPhen-1, SNAP, and SIFT) to examine the effects of nsSNPs on the function of the protein. A sequence-dependent prediction method, PROVEAN (Choi, et al. 2012) can predict the functional role of protein variations. The tool depends on an alignment-based score that determines the variation in sequence similarity of a protein prior and following introducing an amino acid variation. Hence, we predicted the impact of the variants on hSOD1 protein structure and function with ten tools presented in Table 2. The data of Predict SNP analysis for the V148G and I149T variants demonstrated that eight algorithms assessed these variants as destructive, and the predicted confidence score was found 0.87. Using multiple parameters ensures optimal reliability. Overall, the results from our prediction on the protein structure indicated that the substitution effect changed the protein folding, stability, and function (Silva et al. 2019; Berdyński et al. 2022).
Molecular dynamics (MD) simulations
MD is a calculational technique that dissolves Newton’s equations of movement and predicts an atomic system’s time-dependent move. The simulations coming out of this technique are utilized to reproduce the protein’s actual behavior, which is used for evaluating the study of biomolecular processes that include structural changes and protein folding and as well as predicting the impacts of mutations on the structure of the protein (Hollingsworth and Dror 2018). For a better realization of the effects amino acid substitutions have on the structure of the protein in the present study, we conducted simulations of the WT-SOD1 (PDB ID: 2C9V) and ALS-linked variants, i.e., apo/holo SOD1 mutants’ forms (V148G/I149T). Hence, we conducted molecular dynamics simulations for 150 ns to obtain equilibrated systems for WT and mutant proteins (apo/holo forms).
Root mean square deviation (RMSD)
The RMSD was used to determine the difference between a protein’s backbone Cα atoms and its initial structural conformation to its final position. The deviation occurring pending the protein simulation can be applied to find its stability to its structural conformation. RMSD is, in fact, the most helpful approach to comparing the protein structures and can be obtained as follows:
where rfinal (i) denotes an atom’s final coordinates i, rinitial(i) represents the atom’s initial coordinate, and N shows the number of atoms (Jiang et al. 2019).
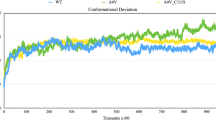
The backbone RMSD of WT, V148G, and I149T mutants (apo/holo forms) of the proteins was calculated over 150 ns simulations (Fig. 1). At the beginning of the simulation, an initial increase was observed, and until 75 ns, all the structures were floating in a stable state. A sudden increase was found in the holo V148G mutant toward the end of the simulation, which was caused by the disruption of vital loops, and dimer interface, signifying structural instability (Fig. 1A). The average RMSD of backbone and Cα atoms of WT-SOD1 was 0.23 ± 0.032 nm, while for V148G and I149T mutated holo-SOD1, the values were 0.27 ± 0.055 and 0.22 ± 0.019 nm, respectively. Therefore, the results of RMSD showed that the point mutation in SOD1 had altered the overall protein backbone. However, the RMSD of holo WT-SOD1 and mutants fluctuated less during the simulation. The RMSD related to backbone atoms of apo-WT-SOD1 floated in a stable state up to 100 ns. An abrupt rise in RMSD values between 100 and 120 ns was observed in the simulation of apo-WT-SOD1, and then it was accompanied by a decrease in fluctuations (Fig. 1B). The more significant fluctuations in the RMSD of apo-SOD1 mutants (V148G/I149T) compared to holo SOD1 were observed, which were attributed to metal ion removal and protein aggregation. The average RMSD values of WT, V148G, and I149T mutants of apo-SOD1 were 0.23 ± 0.046, 0.31 ± 0.032, and 0.28 ± 0.047 nm, respectively. Generally, the computational results showed that mutation and metal deficiency lead to the loss of protein structural stability and may cause aggregation. Furthermore, the probability distribution of RMSD values for the WT-SOD1 and the V148G and I149T mutants in the apo/holo forms is shown in Fig. 1C, D. The results of the current study are matched with those of previous reports (Namadyan 2022; Srinivasan and Rajasekaran 2017).

RMSD as a function of time. Respectively (A and B), RMSD for backbone atoms of the (holo/apo) WT-hSOD1 and the mutants V148G and I149T are shown as a function of time. (C and D) Probability density function (PDF) of the (holo/apo) WT-hSOD1, V148G, and I149T mutant’s conformations over the period of simulation time
Root mean square fluctuations (RMSF)
Root Mean Square Fluctuation (RMSF) helps examine positional flexibility in the structures of protein, enabling the diagnosis of flexible and rigid regions. For more accurate analysis, the residual protein flexibility was calculated for the Cα atoms through RMSF, which is a measurement of deviation between atom i position and some reference positions in protein molecules (Jiang et al. 2019). During MD simulation, we measured the RMSF alterations using the following equation:
in which T represents the time and riref denotes atom i's reference position.
We examined the flexibility of 153 residues within WT-hSOD1 and mutants, as displayed in Fig. 2. Generally, mutation has altered the mutated proteins’ flexibility compared to the WT-hSOD1. We found reduced flexibility in the residues forming beta-sheets but increased flexibility in those forming loops in mutants that are functionally important relative to WT-SOD1 (holo/apo forms) because the binding loops are metal-binding (loop IV) and electrostatic loops (loop VII) maintain the structural stability of the protein (Banci et al. 2009; Ding and Dokholyan 2008). Specifically, the average flexibility values related to two functionally important loop residues, i.e., loop IV (49–83) and loop VII (121–142), and the total average calculated RMSF values in the holo/apo-WT-SOD1, and mutants are presented in Table 3. Since loop IV in SOD1 is close to the interface between two monomers, the fluctuations are also likely to have a crucial impact on maintaining the SOD1 dimer structure. The RMSF of residues of apo-SOD1 (Fig. 2B) indicated larger values than holo, showing that metal ions loss from their active sites might be the driving force for apo-SOD1’s higher propensity for aggregation compared to holo-SOD1(Fig. 2A). The RMSF related to loops IV and VII in apo-SOD1 had more significant fluctuations than in holo-SOD1, revealing apo-SOD1’s high propensity for aggregation in comparison with holo-SOD1, showing a strong deformation or disorder within these loops, pointing to the conclusion that the mutation altered protein flexibility, which is likely to cause the protein to lose structural stability, and the elimination of metal ions from the active sites leads to greater loosening and dynamics of the loops, which is believed to increase the propensity to form aggregates formation (Arnesano et al. 2004). Moreover, according to previous reports, loop VII creates an optimal electrostatic field for superoxide anion radical uptake. The deformation due to the mutation leads to a disturbance in the orientation of the metal ligands for the reaction and thus decreases its catalytic activity (Arnesano et al. 2004).

RMSF as a function of time. Respectively (A and B), RMSF for the (holo/apo) WT-hSOD1, V148G and I149T mutants are shown as a function of time
Radius of gyration (Rg)
Rg is the mass‐weighted root mean square distance of a set of atoms from their common center of mass, such that the Rg values are directly proportional to the volume of the protein. Therefore, this analysis yields the structure’s overall dimensions. A protein’s Rg provides information about the protein structure’s relative compactness that is associated with the stability of the protein with the sum of intra-molecular interactions in the structure of the protein. A low radius of gyration indicates that the atoms in a structure are close to their center of mass, and a high radius means the atoms are far away from this center (Jiang et al. 2019). Below is the equation we will use for calculating the radius of gyration:
where: ri denotes the atom position at index i, mi is the mass of the atom at index i, rCM is the mass center, N represents the number of atoms being counted, and r2RG is the square of the radius of gyration.
Figure 3 illustrates the conformational structures’ (A and B) compactness and the probability distribution (C and D) of Rg values for the holo/apo-WT, V148G and I149T mutants hSOD1. From the beginning of the simulation time until 75 ns, all the structures float in a stable state, and after that, a sudden increase in the slope for the V148G mutant and a decrease in the slope for the I149T mutant was observed (Fig. 3A). Also, an initial increase up to 40 ns was observed for the I149T mutant, and then until the end of the simulation time, it was accompanied by a decrease in fluctuations in comparison to the WT and theV148G mutant. As shown in Fig. 3B, increased/decreased fluctuations were observed for the apo-WT and V148G mutant proteins, and for the I149T mutant, a decrease in fluctuations was observed with an increase in protein compaction. Specifically, the probability distributions of the structures for the holo WT, V148G and I149T mutants were 1.44, 1.46 and 1.43 nm, respectively (Fig. 3C). The probability distributions of the structures for the apo-WT, V148G and I149T mutants were 1.44, 1.42, and 1.4 nm, respectively (Fig. 3D). The lower Rg value in apo-SOD1 compared to holo-SOD1 points to the protein’s increased compaction, probably because of the lack of metal ions that maintain the protein’s high stability in its fully functional state (Sheng et al. 2012). In holo-SOD1, the Rg value’s probability distribution was higher compared to apo-SOD1. Therefore, holo-SOD1 with metal had a lower propensity for aggregation than the apo form. Finally, our outcomes showed that the mutant SOD1 (apo/holo) structures have a decreased number of intra-molecular interactions and had lower stability than the WT structures. Generally, our outcomes suggested that the apo/holo mutants of the SOD1 structure have a reduced number of intra-molecular interactions and lower stability compared to the WT structures. The results indicated that different mutations affect SOD1 structure to different extents, as previously reported (Baziyar et al. 2022b).

A, B Radius of gyration as a function of time. The radius of gyration of the (holo/apo) WT-SOD1, V148G and I149T mutants during the MD trajectory is shown. C, D Probability distribution of the (Rg) of (apo/holo) WT-SOD1, V148G and I149T mutants
Hydrogen bond analysis
Hydrogen-bonding and hydrophobic interactions are the most important instigators of protein folding; however, the extent to which the folded states of proteins are stabilized by the hydrogen bonds is not known and is contentious. H bond dignifies the strengthening of the bond and stabilization of any chemical structure and higher favoring intramolecular hydrogen bonding. The H bonds usually are altered around the mutation site when mutations influence the proteins (Hubbard and Haider 2010). Therefore, the formation of charge between residues is considerably significant in maintaining the proteins’ structural conformation. The number of H bonds for both mutants was calculated to examine the effect the mutation produces hSOD1 apo/holo forms’ conformational compactness (Fig. 4A, B). The probability distribution of the number of hydrogen bonds within the apo and holo SOD1 was calculated; we found that there was a higher probability of finding more hydrogen bonds (WT, V148G, and I149T were 103, 107, and 105, respectively) in apo-SOD1 than in holo-SOD1 (Fig. 4D). While the probability distributions of the number of hydrogen bonds for the holo WT, V148G, and I149T were 100, 97, and 104, respectively (Fig. 4C). As a consequence, the H bonds number of apo mutants increased compared to the WT, showing increased protein conformational changes. The results of the previous studies revealed that mutants of SOD1 have a propensity for protein aggregation into oligomers and amyloid fibrils through creating H bonds within the molecules and hydrophobic interactions (Yamazaki et al. 2022). Overall, our results indicated that the structures of both apo mutants have more intramolecular interactions and are less stable than holo-SOD1 structures. Comparatively, in apo-SOD1, the number of H bonds is reduced, but the value of Rg is increased, and vice versa in holo-SOD1. These results are in line with the results related to the Rg parameter.

A, B Hydrogen bonds (H bond) function of time. The hydrogen bonds of the holo/apo-WT-SOD1, V148G and I149T mutants during the MD trajectory are shown. C, D Probability distribution of the H-bond of apo/holo WT-SOD1, V148G and I149T mutants
Solvent accessible surface area (SASA)
As a measure of the exposed surface in the structure of proteins, SASA can be accessed by the solvent molecules. SASA is generally regarded as a means of evaluating the protein-solvent interactions, which then can help obtain the protein’s features and functions. On this basis, SASA analysis reveals the degree of exposure of the protein to its environment during the time. The protein’s increased SASA represents buried residues’ protein unfolding and exposure. Recently, studies have reported the areas in which the surface properties are very dependent on the protein’s structural conformations, and the alterations in its functional properties are likely to be induced by a slight change in the conformations (Jiang et al. 2019). The protein’s SASA was calculated as follows:
in which, A, R, Li, I, and Zi represent the surface area, the radius of the atom, the length of the arc drawn on a given section the section i's perpendicular distance from the sphere center, respectively (Jiang et al. 2019).
The obtained SASA values in all the simulations (Fig. 5) showed a stable behavior in all simulations. A decrease in fluctuations was observed for the holo I149T mutant during the simulation time (Fig. 5A). This designated that mutation from Ile149 to Thr leads to the SOD1 forming a compact state. In both cases, the ΔSASA value reduced, but there was a higher reduction in apo-SOD1 (Fig. 5B). This alteration in the dynamics causes structure destabilization, leading to the structure unfolding and dimer dissociation and, consequently, aggregation. These findings indicate that apo-SOD1 has a higher propensity for aggregation than holo-SOD1 as loss of metal ions leads to destabilization of the native dimer and generation of an apo monomer, which is the entry point to aggregation based on the studies (Khare et al. 2006). Further, we computed SASA distribution in SOD1 for the apo/holo WT and the mutants’ system (Fig. 5C, D). This analysis showed that the mean values for holo/apo-WT were (87.7 ± 2.06) and (86.5 ± 1.7), respectively, and for mutants, were as follows: holo V148G (87.7 ± 2.1), holo I149T (86.9 ± 2.2), apo-V148G (84.9 ± 2.6) and apo-I149T (84.6 ± 2.3). The fluctuations of Rg (Fig. 3) and H-bonds (Fig. 4) observed in the simulations confirm this behavior and are consistent with other results.

A, B Solvent accessible surface area (SASA) function of time. The SASA of the holo/apo-WT-SOD1, V148G and I149T mutants during the MD trajectory are shown. C, D Probability distribution of the SASA of apo/holo WT-SOD1, V148G and I149T mutants
Disparity in the secondary structure of holo/apo-WT-SOD1 and mutants
Secondary structure is a vital component in studying protein structural characteristics. In the aggregation process of the proteins involved in neurodegenerative disorders, changing the content of secondary structures plays a decisive role and is of significant importance (Kumar et al. 2021). Therefore, in this study, we evaluated the effect of mutation on SOD1 secondary structure propensities using the DSSP program. Considerable changes were observed in the secondary structure contents of holo/apo-WT-SOD1 and mutants during the simulation (Fig. 6). Comparatively, the secondary structure content’s propensity was analyzed in the apo/holo SOD1 protein, showing a noticeable change following increased β-sheet, bend and turn contents of apo-SOD1 in comparison to that of the holo-SOD1, as presented in Table 4. Accordingly, the coil's tendency is decreased in apo-SOD1, pointing to the significant role of the Zn ion in retaining the secondary structural feature of the SOD1 protein. The percentage of the 310-Helix content of holo-SOD1 did not reveal any alterations in all the conditions; nevertheless, minor alterations in the 310-Helix content of apo-SOD1 were observed. Furthermore, the α-helix contents were decreased in apo-SOD1 in comparison to holo-SOD1. The α-helix can be found in loops IV & VII of SOD1 and is believed to be a functionally significant loop. A decline in the α-helical content led to the extension of loops IV & VII, destabilizing the protein, and, consequently, making the protein aggregation-prone. These results are consistent with previous reports (Jahan and Nayeem 2020).

Secondary structural propensity of residues in apo/holo WT and mutants SOD1 obtained over the entire simulation time
Configurational entropies of apo/holo WT-SOD1and mutants
A protein’s increased configurational entropy indicates its increased conformational fluctuation/disorder. Such conformational disorder in apo/holo SOD1 can be various along different unfolding pathways in water. We also found that loops IV & VII of apo-SOD1 have different unfolding behaviors compared to holo SOD1 in the specified conditions. The differential unfolding behaviors shown by apo/holo SOD1 in different conditions caused the gain or loss of entropy, as analyzed in this part. The approach put forward by Schlitter is used to measure a protein’s configurational entropy (Schlitter 1993), which is efficient for the direct calculation of large macromolecules’ absolute entropies from their Cartesian coordinates and based on approximation of one-dimensional quantum mechanical harmonic oscillator. The absolute entropy (Sabs) is estimated using the following equation:
In which, KB, T, e, ħ, M, and σ represent Boltzmann's constant, absolute temperature, Euler's number, Planck's constant divided by 2π, 3N dimensional diagonal matrix that contains N atomic masses of the solute, and covariance matrix related to atom positional fluctuations, respectively. The inequity in the above equation arises when the entropy obtained from Schlitter’s method is an upper bound to the system’s true absolute entropy (Sabs). The covariance matrix elements (σij) were calculated as follows:
in which xi represents the macromolecule atoms’ Cartesian coordinates after eliminating the center-of-mass translation and rotation motion w.r.t. the reference structure of the macromolecule. In the aforementioned process, the computed entropy is the configurational entropy within the molecule. For obtaining the covariance matrix, the time was averaged over the simulated trajectories using monomer SOD1’s initial structure as the reference structure. You can find further details related to the method in the literature (Schlitter 1993; Andricioaei and Karplus 2001). The method of triangularization also referred to as the Cholesky decomposition method (Andricioaei and Karplus 2001), is applied to specify the determinant of the symmetric positive definite matrix, 1 + \(KBTe2/h2\) M½σ M½. One of the advantages associated with this method is concerned with calculation of the entropy contribution of a certain subset of atoms that form a segment of macromolecules like proteins, which is not possible in other available approaches. The cumulative configurational entropies (S) of apo/holo WT-SOD1 and mutants in water at 298 K were computed in the simulation trajectories. Figure 7A and B displays the entropy contributions obtained from total protein. For normalization of the entropy values, we divided them by the number of atoms in order not to come up with a different number of backbone and side-chain atoms. Apo-SOD1 in the water had a higher contribution to the protein entropy compared to the holo-SOD1 meaning that the increase in the entropy of apo-SOD1 in water was greater compared to holo-SOD1, which is likely to cause larger conformational changes in apo-SOD1, indicating that the great conformational alterations in the protein stem from the disturbed SOD1 sidechain. It is believed that Loops IV and VII are functionally significant loops because they are responsible for the SOD1 protein’s stability (Sheng et al. 2012). In this system, the entropy of the whole protein atoms increased in the apo-SOD1 forms (Fig. 7B), indicating protein destabilization where the destabilization in apo is greater than in holo-SOD1. Therefore, the results show that metal defects can affect the stability and configuration of the structure.

Cumulative configuration entropies (per atom) of the protein total backbone (A, B) of apo/holo WT-SOD1, V148G and I149T mutants in water, respectively
Essential dynamics
To analyze MD simulations PCA was used, which is called essential dynamics (ED) (Amadei et al. 1993). Using ED linear, the high-dimensional and complex data within a molecular trajectory is turned into a low-dimensional space where large-scale protein movements happen (Sang et al. 2017), decreasing the number of dimensions required for describing the dynamics of the protein. Via this statistical technique, the observed movements are systematically filtered from the largest scale to the smallest in a molecular trajectory through a covariance matrix developed with the Cartesian coordinates, which represent atomic positions (David and Jacobs 2014). ED enables the separation of the vital motions of a protein from the rest of the movements. The essential motions, i.e., those related to the largest scales, are normally biologically relevant; they include such motions as opening, closing, and flexing. But the remaining ones describe small and biologically irrelevant local fluctuations. The former is often confined to the first two PCA modes (principal components) (Stein et al. 2006). The projections related to the WT-SOD1 MD trajectories and variants into the subspace spanned by PC1 and PC2 are displayed in (Fig. 8). Based on the projection of the two main ingredients of WT-SOD1, i.e., PC1 and PC2 (holo/apo forms) and mutants (Fig. 8A and B), it can be observed that eigenvectors revealed an obvious difference between the WT and mutants in terms of their collective motion. The holo-SOD1 variants were distributed over a higher area in conformational space, but the apo-SOD1 variants were distributed in a smaller area. Altered cluster shapes were also seen in all variants’ conformational space compared to the WT-SOD1. Specifically, the eigenvectors pointed to an obvious difference between the holo/apo-WT and the mutants in collective motion. The ED analysis, accordingly, revealed changes in all variants’ overall essential dynamics. In addition, the principal component analysis confirmed the role of the change in collected motion’ projection in the protein’s impaired function (Berendsen and Hayward 2000). Therefore, based on the analysis, holo-SOD1’s motion over the first two eigenvectors was dissimilar in comparison to apo-SOD1, proving WT and apo-SOD1 mutants’ dysfunction. Additionally, we shed more light on the conformational dynamics of the Mean smallest distances motion between trajectories of the holo/apo-WT-SOD1 and mutants. Protein structure prediction (PSP) has recently undergone significant progress through the prediction of inter-residue distances by deep learning models and applying the predictions in conformational search (Rahman et al. 2022). Protein inter-residue contact maps present a translation and rotation invariant topological representation of a protein and are applicable as an intermediary step in the prediction of protein structure (Kukic et al. 2014). To illuminate the impact of mutation and (holo/apo forms) SOD1 on the global conformation of the structure of SOD1, time-averaged distance maps were determined for WT and mutants (Fig. 9). The main differences between holo/apo-WT and mutants are observed to examine the mean smallest distance between protein residues. As shown in the dotted boxes in (Fig. 9A), there are corresponding reduced interactions in holo-SOD1 (WT and mutants); while in (Fig. 9B), apo-WT and considerably apo-V148G and apo-I149T show a tight network of interactions including residues 121–142 in loop VII. Specifically, residues 121–142 are longer located near other residues, which is indicative of destabilization or loss of critical contacts, leading to more compaction in the structure of protein. The structural consequences of these observations are the change in stabilizing interactions and the reduction of the tendency to form critical contacts, as well as the weakening of contact between adjacent residues, which ultimately leads to the creation of new contacts in the protein structure. Generally, the changes in the distance between protein residues are more pronounced in apo-SOD1 than in holo-SOD1.

PCA for the holo/apo-WT-SOD1 and its variants. Projections for the first two principal components extracted from the MD trajectories. A Comparison between the PCA projections for the holo-WT-SOD1 and its variants V148G and I149T. B Comparison between the PCA projections for the apo-WT-SOD1 and its variants V148G and I149T

Mean smallest distance matrices for holo-WT-SOD1 and mutants (A) and apo-WT-SOD1 and mutants (B). The time-averaged minimum distance of each residue to every other residue is plotted in heat maps with white indicating the smallest distance and black indicating the largest distance ranging from 0 to 2.5 nm. This map reveals differences in the global conformational space
Experimental section
Enzymatic activity assay
hSOD1 is a highly stable enzyme and dimerization is an important factor in its stability. Despite hSOD1’s higher stability, its structure is destabilized by the point mutations, as a result of the loss of protein stability and misfolding and the dissociation of dimerization, finally leading to hSOD1 accumulation. The enzymatic activity of holo WT-hSOD1, V148G, and I149T mutants were (7031 ± 345), (3564 ± 295), and (3765 ± 278) U/mg, respectively, whereas for apo-WT-hSOD1, V148G, and I149T mutants were (2736 ± 368), (1378 ± 325), and (1158 ± 248) U/mg, respectively (Fig. 10). The results of our study revealed reduced enzymatic activity for apo-SOD1 and mutants compared to holo WT-hSOD1. Although some hydrogen bonds from the side to the main chain and intramolecular interactions are important for the folding and stability of hSOD1, interestingly, mutation and metal deficiency can lead to an aberrant increase of hydrophobic and hydrogen bonds and these changes may lead to reduced enzymatic activity in hSOD1 (Tiwari et al. 2009). Dimer interface mutants Val148Gly and Ile149Thr tend to be the most destabilizing and reduce the affinity for dimer formation, hence may be to influence the protein stability and structure. The particular sensitivity of the SOD1 dimer interface to mutations-induced disruption, as displayed here, can be rationalized by further examination of the structural and dynamic features associated with the interface. The SOD1 interface is not evolutionarily conserved, has a relatively small size (10% of monomer surface area) and is highly hydrophobic (80% nonpolar residues) (Rumfeldt et al. 2008). Its small size and highly hydrophobic nature indicate the low extent of monomer–monomer interaction and its selectivity such that mutation may easily perturb association. Structural studies reported significant changes in the interface (Hough et al. 2004). Additionally, the stabilizing interactions formed by metals’ boundness to SOD1 limit flexibility in loops IV and VII; however, without metal, these loops acquire conformational freedom, and are believed to create energetic frustration in the protein’s apo form. The regions with enhanced dynamics, particularly in loop IV, which constitutes a part of the dimer interface, are likely to easily be perturbation by mutations, compromising the stability of the interface. Furthermore, based on the results of RMSF, we discovered that in the two mentioned important loops, higher fluctuations were observed in apo-SOD1 in comparison to holo-SOD1 for the whole simulation, suggesting a great defect or disturbance in these loops as a consequence of greater flexibility compared to native loops, which is consistent with loss of loop activity, finally leading to the loss of the entire hSOD1 protein’s catalytic function. The fluctuations may have a significant effect on keeping hSOD1’s dimeric structure due to the reason that loop IV in hSOD1 is near the interface between the two monomers. In addition, other findings revealed that loop VII creates an optimal electrostatic field for the gathering of the superoxide anion radicals, and the deformation caused by this mutation leads to the disrupted direction of metal ligands for the deformation reaction that may, as a consequence, reduces its enzyme activity (Banci et al. 2009; Arnesano et al. 2004). These indicate the loss of metal, the substitution of amino acids of different sizes, and their chemical nature, which changes the protein’s hydrophobic free energy.

Specific activity measurements of holo/apo-WT-hSOD1 compared to mutants. The data were expressed as the mean ± SD (n = 3)
Fluorescence spectroscopy analysis
The proteins’ intrinsic (Trp) fluorescence presents good details on their structures and dynamics. The intrinsic fluorescence spectra of apo/holo WT-SOD1 and mutants are demonstrated in Fig. 11a and b, respectively. Based on the findings, the mutations have influenced the conformation of SOD1 to different extents, as previously shown (Baziyar et al. 2022a). Therefore, the changes in the circumambient environment, the only tryptophan (W32) in the intrinsic fluorescence of both holo mutants, were observed compared to holo WT-hSOD1. Concerning holo I149T, an apparent blue shift deviation accompanies the higher tryptophan fluorescence, suggesting that the environment circumambient tryptophan residue’s polarity reduced, but the holo V148G mutant displayed a lower intensity for fluorescence (Fig. 11A), showing the protein’s conformational changes and the change of the environment of the only tryptophan into more hydrophilic one is accompanied by the red shift of the fluorescence signal. As shown in these figures (comparing 11A with 11B), the spectra of apo-WT/ V148G /I149T proteins indicate that the enzyme metalation has a significant impact on SOD1 variants’ structures. It is demonstrated that both V148G/I149T mutations caused a reduction in apo-SOD1 fluorescence that might be due to soluble aggregates of these mutants. In other words, metal deficiency had a direct effect on the three-dimensional structure of the studied SOD1 types, and it seems that the three-dimensional structure of the enzyme (both apo/holo forms) undergoes structural changes due to mutations in positions 148 and 149. Therefore, these findings are matched with the results from molecular dynamic analysis. To understand the mutations’ contribution to alterations in the hydrophobicity of hSOD1, ANS is applied to monitor hydrophobic surface accessibility in β-sheeted structures when the protein aggregation is formed and also to diagnose the protein population’s compact, partially folded intermediate. The increase in ANS fluorescence and blueshift revealed that ANS is bound to exposure to contagious hydrophobic districts. The feebler conformational condition may cause the ANS boundness to intermediate surfaces of hSOD1 mutants. Changes in protein surface hydrophobicity contribute to some stages of SOD1 aggregation, and possibly, this parameter may occur through SOD1 mutation as well as metallization/demetallization (Roberts et al. 2007). In preliminary evaluations, as observed in Fig. 11C, considerable increases in ANS intensities were witnessed for both mutants (V148G/I149T) in comparison with the holo WT, showing an increase in hydrophobic surfaces in the protein. Therefore, as holo proteins, with high ANS fluorescence, the hydrophobic patches/hydrophobic interactions may be sufficiently exposed to ANS, leading to increased fluorescence emission. This may indirectly confirm that conformational perturbation (partial/complete unfolding) is essentially a prerequisite for protein aggregation. To investigate the hydrophobic surfaces under the metal deficiency condition (apo-SOD1), as it was already mentioned, ANS fluorescence was examined (Fig. 11D). Decreased ANS fluorescence intensity of apo-mutants compared to WT indicates reduced access of ANS to hydrophobic patches due to disrupted dimer interface and dissociation of dimerization and formation of monomers meaning that the growth of pre-fibrillar species and amyloid aggregations to each other led to coverage of the available hydrophobic surface, causing the eventual burial of hydrophobic patches and consequently reducing the emission of the ANS fluorescence. This decrease in ANS fluorescence intensity was more obvious in the apo-I149T mutant compared to the apo-V148G mutant, because pre-fibrillar species and amyloid aggregation in this mutant may be increased and thus decrease the hydrophobicity of the surface. These results are matched with the findings reported in the previous studies (Famil Samavati et al. 2020; Li et al. 2010).

Structural characteristics of hSOD1 by intrinsic and ANS fluorescence emission. (A, B) Intrinsic fluorescence emission of holo/apo-WT-hSOD1, V148G and I149T mutants in phosphate buffer (20 mM, pH 7.4). The excitation wavelength was 295 nm. (C, D) The ANS fluorescence emission of holo/apo-WT-hSOD1, V148G and I149T mutants in phosphate buffer (20 mM, pH 7.4). The concentration of hSOD1 was 0.02 mg/ml, at 25 °C. The concentration of the ANS in the enzyme solutions was 30 µM, and the molar ratio of protein to ANS was 1:30. The excitation wavelength was 350 nm
Conclusion
Our purpose in this study was to illuminate the destabilizing impacts of substitution mutations on the overall structure of holo/apo hSOD1. The mutations investigated were positioned within the dimer interface, inducing the protein structural instability. To investigate non-synonymous substitution mutations and to better understand the sequence and structural significance of the holo/apo hSOD1 protein, a combination of bioinformatics approaches and experimental methods were used. At first, the change in Δ∆G value of missense mutations was predicted using several algorithms. The simulation results showed that apo-SOD1 mutants significantly changed the structure of the protein compared to the holo-SOD1 mutants, which may lead to misfolding and aggregation. Subsequently, the formation of intramolecular H bonds was significantly increased in apo-SOD1 compared to holo-SOD1, thus increasing protein compaction. This increase in aberrant H bonds leading an increase in the compactness of the structure, which was consistent with the results of the Rg and SASA. Therefore, apo-SOD1 was aggregation-prone more than holo SOD1. As well as, the results of the experimental section showed that metal deficiency and hSOD1 mutation change the protein folding pattern, which leads to structural changes and ultimately causes protein aggregation and affects the enzyme activity, and subsequently leads to hSOD1-linked fALS. Loss of protein folding and changes in hydrophobicity and H-bonds could lead to changes at the dimer interface, which significantly reduces the stability of hSOD1. Overall, more severe structural changes were observed in apo-SOD1 compared to holo-SOD1. The native sequences in the formation of the stable tertiary structure of the hSOD1 protein are necessary to maintain the biological activity, the importance of which is well shown in this study. Dissociation of Cu and/or Zn ions from SOD1 causes protein aggregation/oligomerization in vitro, ultimately disturbing the dimer-monomer equilibrium, which causes destabilizes the dimer and increases the dimer dissociation propensity to apo monomer. Nevertheless, the pathological contribution of such metal dissociation to the SOD1 misfolding remains ambiguous. Although the factors and mechanisms that lead to protein aggregation are still not well understood; however, our findings can provide valuable insights into the theoretical and experimental mechanisms linked to holo/apo-WT-SOD1, V148G, and I149T mutants and their subsequent effect on neurodegenerative disorders in humans.
Data availability
The data that support the findings of this study are available on request.
References
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protocol Hum Genet 76(1): 7.20. 1–7.20. 41
Amadei A, Linssen AB, Berendsen HJ (1993) Essential dynamics of proteins. Proteins: Struct Funct Bioinf 17(4): 412–425
Andricioaei I, Karplus M (2001) On the calculation of entropy from covariance matrices of the atomic fluctuations. J Chem Phys 115(14):6289–6292
Arnesano F et al (2004) The unusually stable quaternary structure of human Cu, Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem 279(46):47998–48003
Banci L et al (2009) Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc Natl Acad Sci 106(17):6980–6985
Bastolla, U., et al., How to guarantee optimal stability for most representative structures in the protein data bank. Proteins: Structure, Function, and Bioinformatics, 2001. 44(2): p. 79–96.
Baziyar P, Seyedalipour B, Hosseinkhani S (2022a) Zinc binding loop mutations of hSOD1 promote amyloid fibrils under physiological conditions: Implications for initiation of amyotrophic lateral sclerosis. Biochimie 199:170–181
Baziyar P et al (2022b) Development of in Silico Analysis and Molecular Dynamics Simulation on L67P and D76Y Mutants of the Human Superoxide Dismutase 1 (hSOD1) Related to Amyotrophic Lateral Sclerosis. Iran J Biotechnol 20(4):26–37
Bendl J et al (2014b) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10(1):e1003440
Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED Zendulka J, Brezovsky J, Damborsky J (2014a) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10:e1003440
Berdyński M et al (2022) SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci Rep 12(1):1–11
Berendsen HJ, Hayward S (2000) Collective protein dynamics in relation to function. Curr Opin Struct Biol 10(2):165–169
Berman HM et al (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Bernard E et al (2020) Clinical and molecular landscape of ALS patients with SOD1 mutations: Novel pathogenic variants and novel phenotypes. A single ALS center study. Int J Mol Sci 21(18):6807
Beyer WF Jr et al (1987) Examination of the role of arginine-143 in the human copper and zinc superoxide dismutase by site-specific mutagenesis. J Biol Chem 262(23):11182–11187
Bhatia NK et al (2015) Curcumin binds to the pre-fibrillar aggregates of Cu/Zn superoxide dismutase (SOD1) and alters its amyloidogenic pathway resulting in reduced cytotoxicity. Biochim Biophys Acta (BBA) Protein Proteom 1854(5): 426–436
Bromberg Y, Rost B (2007) SNAP: predict effect of non-synonymous polymorphisms on function. Nucl Acids Res 35(11):3823–3835
Calabrese R et al (2009) Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat 30(8):1237–1244
Cao H et al (2019) DeepDDG: predicting the stability change of protein point mutations using neural networks. J Chem Inf Model 59(4):1508–1514
Capriotti E, Calabrese R, Casadio R (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22(22):2729–2734
Capriotti E, Fariselli P, Casadio R (2005) I-Mutant2. 0: predicting stability changes upon mutation from the protein sequence or structure. Nucl Acids Res 33(suppl_2): W306–W310
Cardoso RM et al (2002) Insights into Lou Gehrig’s disease from the structure and instability of the A4V mutant of human Cu, Zn superoxide dismutase. J Mol Biol 324(2):247–256
Chen C-W, Lin J, Chu Y-W (2013) iStable: off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinf BioMed Central
Chen C-W et al (2020) iStable 2.0: predicting protein thermal stability changes by integrating various characteristic modules. Comput Struct Biotechnol J 18:622–630
Choi, Y., et al., Predicting the functional effect of amino acid substitutions and indels. 2012.
Compiani M, Capriotti E (2013) Computational and theoretical methods for protein folding. Biochemistry 52(48):8601–8624
Da Silva ANR et al (2019) SOD1 in amyotrophic lateral sclerosis development–in silico analysis and molecular dynamics of A4F and A4V variants. J Cell Biochem 120(10):17822–17830
David CC, Jacobs DJ (2014) Principal component analysis: a method for determining the essential dynamics of proteins. Protein dynamics. Springer, pp 193–226
De Lano WL (2002) Pymol: an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr 40(1): 82–92
Deng H-X et al (1993) Amyotrophic Lateral Ssclerosis and Structural Defects in Cu, Zn superoxide dismutase. Science 261(5124):1047–1051
Ding F, Dokholyan NV (2008) Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc Natl Acad Sci 105(50):19696–19701
Famil Samavati S et al (2020) Reduced thermodynamic stability as prerequisite for aggregation of SOD1 mutants: a path through the reduction in intramolecular disulfide bonds. J Iran Chem Soc 17(8):2053–2071
Famil Samavati S et al (2022) Study on the effects of various incubation conditions on aggregation of SOD1 variants: disulfide bond reduction and demetallation synergistically promote generation of non-amyloid amorphous aggregates from SOD1 mutants. J Iran Chem Soc 19(5):1755–1771
Fay JM et al (2016) A phosphomimetic mutation stabilizes SOD1 and rescues cell viability in the context of an ALS-associated mutation. Structure 24(11):1898–1906
Ferraroni M et al (1999) The crystal structure of the monomeric human SOD mutant F50E/G51E/E133Q at atomic resolution. The enzyme mechanism revisited. J Mol Biol 288(3): 413–426
Ferrer-Costa C et al (2005) PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21(14):3176–3178
Fong GC et al (2006) Clinical phenotypes of a large Chinese multigenerational kindred with autosomal dominant familial ALS due to Ile149Thr SOD1 gene mutation. Amyotroph Lateral Scler 7(3):142–149
Frieden C (2007) Protein aggregation processes: In search of the mechanism. Protein Sci 16(11):2334–2344
Ghosh DK, Kumar A, Ranjan A (2020) T54R mutation destabilizes the dimer of superoxide dismutase 1 T54R by inducing steric clashes at the dimer interface. RSC Adv 10(18):10776–10788
Grant BJ et al (2006) Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 22(21):2695–2696
Hollingsworth SA, Dror RO (2018) Molecular dynamics simulation for all. Neuron 99(6):1129–1143
Hough MA et al (2004) Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci 101(16):5976–5981
Huang W, Lin Z, van Gunsteren WF (2011) Validation of the GROMOS 54A7 force field with respect to β-peptide folding. J Chem Theory Comput 7(5):1237–1243
Hubbard RE, Haider MK (2010) Hydrogen bonds in proteins: role and strength. eLS
Ivanova MI et al (2014) Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci 111(1):197–201
Jackson C et al (2019) Radicava (edaravone) for amyotrophic lateral sclerosis: US experience at 1 year after launch. Amyotrophic Lateral Sclerosis Frontotemporal Degen 20(7–8):605–610
Jahan I, Nayeem SM (2020) Conformational dynamics of superoxide dismutase (SOD1) in osmolytes: a molecular dynamics simulation study. RSC Adv 10(46):27598–27614
Jaiswal MK (2019) Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev 39(2):733–748
Jamadagni SN, Godawat R, Garde S (2011) Hydrophobicity of proteins and interfaces: Insights from density fluctuations. Annu Rev Chem Biomol Eng 2:147–171
Jiang Z et al (2019) Effects of an electric field on the conformational transition of the protein: a molecular dynamics simulation study. Polymers 11(2):282
Khare SD, Caplow M, Dokholyan NV (2006) FALS mutations in Cu, Zn superoxide dismutase destabilize the dimer and increase dimer dissociation propensity: a large-scale thermodynamic analysis. Amyloid 13(4):226–235
Khare SD et al (2005) Sequence and structural determinants of Cu, Zn superoxide dismutase aggregation. PROTEINS: Struct Funct Bioinf 61(3):617–632
Krebs BB, De Mesquita JF (2016) Amyotrophic lateral sclerosis type 20-In Silico analysis and molecular dynamics simulation of hnRNPA1. PLoS ONE 11(7):e0158939
Krivickas LS (2003) Amyotrophic lateral sclerosis and other motor neuron diseases. Phys Med Rehabil Clin 14(2):327–345
Kukic P et al (2014) Toward an accurate prediction of inter-residue distances in proteins using 2D recursive neural networks. BMC Bioinf 15(1):1–15
Kumar R et al (2014) Synthesis and antimicrobial studies of pyrimidine pyrazole heterocycles. Adv Chem 2014:1–12
Kumar S et al (2021) Explicit-solvent molecular dynamics simulations revealed conformational regain and aggregation inhibition of I113T SOD1 by Himalayan bioactive molecules. J Mol Liq 339:116798
Leinartaite L et al (2010) Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J Am Chem Soc 132(38):13495–13504
Li H-T et al (2010) Roles of zinc and copper in modulating the oxidative refolding of bovine copper, zinc superoxide dismutase. Acta Biochim Biophys Sin 42(3):183–194
Li G, Panday SK, Alexov E (2021) SAAFEC-SEQ: a sequence-based method for predicting the effect of single point mutations on protein thermodynamic stability. Int J Mol Sci 22(2):606
Luigi MP et al (2016) Large scale analysis of protein stability in OMIM disease related human protein variants
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47(3):469–474
Masso M, Vaisman II (2014) AUTO-MUTE 2.0: a portable framework with enhanced capabilities for predicting protein functional consequences upon mutation. Adv Bioinf 2014
Mishra SK (2022) Protein stability changes upon point mutations identified with a Gaussian network model simulating protein unfolding behavior
Molnar KS et al (2009) A common property of amyotrophic lateral sclerosis-associated variants: destabilization of the copper/zinc superoxide dismutase electrostatic loop. J Biol Chem 284(45):30965–30973
Montanucci L et al (2019) DDGun: an untrained method for the prediction of protein stability changes upon single and multiple point variations. BMC Bioinf 20(14):1–10
Namadyan N et al (2022) Biochemical and biophysical properties of the novel ALS-linked hSOD1 mutants: an experimental study accompanied by in silico analysis. J Iran Chem Soc p 1–14
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucl Acids Res 31(13):3812–3814
Nordlund A, Oliveberg M (2006) Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: parallels to precursors in amyloid disease. Proc Natl Acad Sci 103(27):10218–10223
Nordlund A, Oliveberg M (2008) SOD1-associated ALS: a promising system for elucidating the origin of protein-misfolding disease. HFSP J 2(6):354–364
Nordlund A et al (2009) Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proc Natl Acad Sci 106(24):9667–9672
Ó’Fágáin C (2017) Protein stability: enhancement and measurement. Protein Chromatography. Springer, pp 101–129
Pires DE, Ascher DB, Blundell TL (2014a) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucl Acids Res 42(W1):W314–W319
Pires DE, Ascher DB, Blundell TL (2014b) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30(3):335–342
Quan L, Lv Q, Zhang Y (2016) STRUM: structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics 32(19):2936–2946
Rahman J et al (2022) Enhancing protein inter-residue real distance prediction by scrutinising deep learning models. Sci Rep 12(1):1–13
Rakhit R et al (2004) Monomeric Cu, Zn-superoxide Dismutase Is a Common Misfolding Intermediate in the Oxidation Models of Sporadic and Familial Amyotrophic Lateral Sclerosis*[boxs]. J Biol Chem 279(15):15499–15504
Ray SS et al (2005) Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. Proc Natl Acad Sci 102(10):3639–3644
Roberts BR et al (2007) Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol 373(4):877–890
Rose PW et al (2016) The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucl Acid Res p gkw1000
Rumfeldt JA et al (2008) Conformational stability and folding mechanisms of dimeric proteins. Prog Biophys Mol Biol 98(1):61–84
Sanavia T et al (2020) Limitations and challenges in protein stability prediction upon genome variations: towards future applications in precision medicine. Comput Struct Biotechnol J 18:1968–1979
Sang P et al (2017) In silico screening, molecular docking, and molecular dynamics studies of SNP-derived human P5CR mutants. J Biomol Struct Dyn 35(11):2441–2453
Schlitter J (1993) Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chem Phys Lett 215(6):617–621
Schmid N et al (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur Biophys J 40(7):843–856
Schmidlin T, Kennedy BK, Daggett V (2009) Structural changes to monomeric CuZn superoxide dismutase caused by the familial amyotrophic lateral sclerosis-associated mutation A4V. Biophys J 97(6):1709–1718
Sheng Y et al (2012) SOD1 aggregation and ALS: role of metallation states and disulfide status. Curr Top Med Chem 12(22):2560–2572
Srinivasan E, Rajasekaran R (2017) Computational investigation of the human SOD1 mutant, Cys146Arg, that directs familial amyotrophic lateral sclerosis. Mol BioSyst 13(8):1495–1503
Stein SAM et al (2006) Principal components analysis: a review of its application on molecular dynamics data. Annu Rep Comput Chem 2:233–261
Stirling PC, Hieter P (2017) Canonical DNA repair pathways influence R-loop-driven genome instability. J Mol Biol 429(21):3132–3138
Stone EA, Sidow A (2005) Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res 15(7):978–986
Svensson A-KE et al (2010) Metal-free ALS variants of dimeric human Cu, Zn-superoxide dismutase have enhanced populations of monomeric species. PLoS ONE 5(4):e10064
Tang L et al (2019) Better survival in female SOD1-mutant patients with ALS: a study of SOD1-related natural history. Transl Neurodegen 8(1):1–10
Thomas PD et al (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13(9):2129–2141
Tian R, Basu MK, Capriotti E (2015) Computational methods and resources for the interpretation of genomic variants in cancer. BMC Genom 16(8):1–19
Tiwari A et al (2009) Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J Biol Chem 284(40):27746–27758
Worth CL, Preissner R, Blundell TL (2011) SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucl Acids Res 39(suppl_2): W215–W222
Wright GS, Antonyuk SV, Hasnain SS (2019) The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q Rev Biophys 52
Wu, J. and R. Jiang, Prediction of deleterious nonsynonymous single-nucleotide polymorphism for human diseases. The Scientific World Journal, 2013. 2013.
Yamazaki K et al (2022) SOD1 gains pro-oxidant activity upon aberrant oligomerization: change in enzymatic activity by intramolecular disulfide bond cleavage. Sci Rep 12(1):1–9
Yates CM, Sternberg MJ (2013) The effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on protein–protein interactions. J Mol Biol 425(21):3949–3963
Zarei S et al (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 6
Author information
Authors and Affiliations
Contributions
HDZ and HMH contributed to methodology, perform experiments, validation, data curation, formal analysis, writing—review. BS contributed to conceptualization, supervision, methodology, validation, data curation, formal analysis, writing—review and editing. PB contributed to methodology, validation, formal analysis, data curation, writing—original draft preparation. SH contributed to conceptualization, validation, data curation, writing—review and editing. MA contributed to methodology, perform experiments, formal analysis.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zaji, H.D., Seyedalipour, B., Hanun, H.M. et al. Computational insight into in silico analysis and molecular dynamics simulation of the dimer interface residues of ALS-linked hSOD1 forms in apo/holo states: a combined experimental and bioinformatic perspective. 3 Biotech 13, 92 (2023). https://doi.org/10.1007/s13205-023-03514-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-023-03514-1