
Abstract
Single Nucleotide Polymorphisms (SNPs) are key in understanding complex diseases. Nonsynonymous single-nucleotide polymorphisms (nsSNPs) occur in protein-coding regions, potentially altering amino acid sequences, protein structure and function. Computational methods are vital for distinguishing deleterious nsSNPs from neutral ones. We investigated the role of NLRP3 gene in neuroinflammation associated with Alzheimer’s disease (AD) pathogenesis. A total of 893 missense (nsSNPs) were obtained from the dbSNP database and subjected to rigorous filtering using bioinformatics tools like SIFT, Align GVGD, PolyPhen-2, and PANTHER to identify potentially damaging variants. Of these, 18 nsSNPs were consistently predicted to have deleterious effects across all tools. Notably, 16 of these variants exhibited reduced protein stability, while only 4 were predicted to be buried within the protein structure. Among the identified nsSNPs, rs180177442 (R262L and R262P), rs201875324 (T659I), and rs139814109 (T897M) were classified as high-risk variants due to their significant deleterious impact, probable damaging effects, and association with decreased protein stability. Molecular docking and simulation analyses were conducted utilizing Memantine, a standard drug utilized in AD treatment, to investigate potential interactions with the altered protein structures. Additional clinical and genetic investigations are necessary to elucidate the underlying mechanisms that link NLRP3 polymorphisms with the initiation of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) stands as a prevalent neurodegenerative condition, particularly affecting the elderly, aged sixty years and above [1]. The intricate mechanisms underlying its pathophysiology and etiology remain incompletely elucidated [2]. In addition to neuronal loss, various contributing factors have been identified in AD onset [3]. Notably, neuroinflammation [3] and dementia [4] emerge consequent to the dysregulation of the nucleotide-binding oligomerization domain (NOD)-Like Receptor Pyrin Domain-Containing 3 (NLRP3) protein. NLRP3, a tripartite protein belonging to the NALP family, is renowned for its involvement in apoptosis and inflammation [5]. Structurally, NLRP3 comprises an amino-terminal pyrin domain, a central nucleotide-binding and oligomerization domain (NOD; also referred to as the NACHT domain), and a C-terminal leucine-rich repeat domain [6]. Research indicates that inflammation induced by viral or bacterial infections plays a significant role in the pro-inflammatory activation of immune cells resident in the central nervous system (CNS), such as astrocytes and microglia, leading to neuronal death [7, 8]. Ever since the discovery of NLRP3 as a prominent inflammasome linked to various age-related inflammatory conditions like gout, multiple sclerosis, diabetes, ulcerative colitis, and AD [9, 10], NLRP3 has garnered significant attention as a potential therapeutic target. This attention stems from its influence on activated astrocytes and microglia, which are known to exhibit enrichment in genes associated with AD [11].
The human NLRP3 gene spans approximately 32.9 kilobases (kb) in length, positioned on chromosome 1q44 and comprising nine exons. Variations in single-nucleotide polymorphisms within the NLRP3 gene can potentially alter its functionality, thereby augmenting the activation of inflammasomes and the production of IL-1β [12, 13]. Notably, next-generation sequencing (NGS) technologies provide extensive insights into sequence variability, facilitating comprehensive analysis of genetic alterations. Exploring the impact of extensive sequence variation data within biological systems can be challenging and resource-intensive [14]. Conversely, in silico analysis methods present an accessible, cost-effective, and efficient approach for evaluating the potential consequences of amino acid alterations. Leveraging protein structure and sequence information, these techniques offer predictive insights into phenotypic changes resulting from genomic variations. Specifically, non-synonymous single-nucleotide polymorphisms (nsSNPs) introduce modifications to protein sequences, potentially influencing their functionality. Therefore, conducting initial in-silico analyses can augment our capacity to identify nsSNPs for experimental investigations. These analyses involve assessing the potential influence of an amino acid variant on protein structure and functionality [15, 16].
Numerous genotyping analyses have been undertaken previously to identify diverse single nucleotide polymorphisms (SNPs) within NLRP3, which have been associated with various inflammatory and autoimmune disorders. These disorders include multiple sclerosis, rheumatoid arthritis, psoriatic arthritis, celiac disease, myasthenia gravis, and ulcerative colitis, among others [17, 18]. Several studies have indicated a correlation between NLRP3 polymorphic sites and the susceptibility to certain autoimmune disorders. However, conflicting evidence exists, with some studies proposing a protective effect of NLRP3 gene polymorphisms at these sites against disease onset [19, 20]. Nevertheless, there is a dearth of research investigating the structural and functional implications of these SNPs. In this study, we utilized in silico methodologies to pinpoint pathogenic SNPs within the NLRP3 gene, aiming to prioritize SNPs for subsequent genetic mapping investigations. Various computational techniques and tools were employed to fulfill the objectives of this study. Leveraging the in-silico approach offers significant advantages in identifying candidate SNPs, as it is both accessible and cost-effective, thereby facilitating future genetic studies.
Materials and Methods
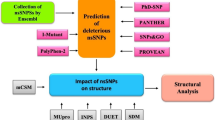
To provide a comprehensive analysis of nsSNPs in the NLRP3 gene and their implications for Alzheimer’s disease, we employed a series of bioinformatics tools and methodologies. The following tools were used in the present study to ensure a thorough evaluation of the genetic variants and their potential impacts on protein function and disease association (Table 1).
Sequence Retrieval and Mining of nsSNPs in the NLRP3 Gene
The UniProt database (UniProt ID: Q96P20) offers essential information regarding the human NLRP3 fasta sequence. Given the pivotal role of NLRP3 inflammasome activation in Alzheimer’s disease (AD) pathogenesis, exploring missense single nucleotide variations (nsSNVs) within the NLRP3 gene is imperative. The NLRP3/caspase-1/IL-1β axis represents a crucial pathological pathway associated with AD, emphasizing the significance of investigating genetic anomalies [21]. These nsSNVs can profoundly influence both the structure and function of the protein, potentially impacting disease progression. To initiate a comprehensive molecular analysis, we retrieved nsSNPs from the SNP database (https://www.ncbi.nlm.nih.gov/snp/), laying the groundwork for further investigation [22].
Identification and Prediction of Effects of SNPs
Several computational tools were sequentially employed to assess the functional and structural impacts of deleterious SNPs within the NLRP3 gene as outlined by Wanarase et al., [22].
SIFT (Sorting Intolerant from Tolerant) tool, available at https://sift.bii.a-star.edu.sg/www/SIFT_dbSNP.html, evaluates the phenotypic effects of amino acid substitutions by considering protein evolution and function. Scores range from 0 to 1, where values below 0.05 indicate damaging substitutions [23]. The Align GVGD web server (http://agvgd.hci.utah.edu/) predicts the deleteriousness of missense substitutions based on protein multiple sequence alignments and biophysical characteristics of amino acids. Variants are classified on a spectrum, ranging from C0 to C65, with C65 being the most likely to impact protein function [22]. PolyPhen-2 (Polymorphism Phenotyping V2), accessible at http://genetics.bwh.harvard.edu/pph2/, predicts the structural and functional consequences of amino acid substitutions using physical and comparative considerations. Variants with a PSIC score above 0.85 are considered probably damaging [22, 24]. The PANTHER cSNP classification system (http://pantherdb.org/tools/csnpScoreForm.jsp) assesses the evolutionary conservation and molecular functions of proteins, providing position-specific evolutionary conservation (PSEC) scores. Input queries include the protein sequence, amino acid variants, and the human organism [22].
Prediction of Solvent Accessibility
Understanding interaction interfaces and active sites within folded proteins is crucial for unraveling molecular behavior. Changes in these regions, such as amino acid substitutions, can disrupt binding affinity, especially in enzymes where catalytic activity is essential. NetSurf-2.0 is a valuable tool for analyzing surface composition, solvent accessibility, structural disorder, and other factors affecting amino acid residues. Utilizing protein sequences in FASTA format, NetSurf-2.0 (http://www.cbs.dtu.dk/services/NetSurfP/) employs deep neural networks trained on solved protein structures for accurate estimations [25, 26].
Homology Modeling and Functional Analysis
For structural assessment, SWISS-MODEL employs a hierarchical approach encompassing protein modeling, structural refinement, and functional annotation. By utilizing appropriate templates sourced from the Protein Data Bank (PDB), the server generates the top five models. The TIMP1 FASTA sequence serves as input for this comprehensive analysis [27].
The SWISS-MODEL web server (https://swissmodel.expasy.org/) leverages the updated UniProtKB proteome to conduct homology modeling of protein structures. Using the FASTA sequence as the input query, SWISS-MODEL provides critical metrics including the Ramachandran plot, Z Score, comparison plots, global and local quality scores, and molprobity score, offering a thorough evaluation of the modeled protein [28].
To assess the geometry of each residue and the overall structure, PROCHECK analysis (https://servicesn.mbi.ucla.edu/PROCHECK/) was employed [29]. Additionally, the ERRAT server (https://servicesn.mbi.ucla.edu/ERRAT/) was utilized to validate the overall model quality by evaluating the statistical relationship of non-bonded interactions between various types of atoms based on typical atomic interactions. Predicted models in PDB format serve as the basis for input queries in both PROCHECK and ERRAT analyses.
Prediction of Disease Association
Performing disease association predictions and structural analyses of nsSNPs is crucial for identifying genetic risk factors, and understanding the molecular mechanisms of diseases. In the present study, we have employed SNP& GO (https://snps.biofold.org/snps-and-go/snps-and-go.html), a SVM-based classifier that predicts the disease-associated variations using the PANTHER, GO and PhD-SNp terms corresponding to the query protein. The output is predicted as neutral variation (Probability <0.5) or as disease associated variation (Probability>0.5) [30].
PhD-SNP (https://snps.biofold.org/phd-snp/phd-snp.html) is a SVM classifier designed to predict if a nonsynonymous single-point mutation is disease-associated or neutral using protein sequence data. This tool employs BLAST software to compare mutations against the UniRef90 database, considering various mutation characteristics. The output is a score between 0 and 1, with scores below 0.5 indicating a likely disease association [31].
Assessing nsSNP Impact on Protein Aggregation
Protein aggregation is a hallmark of Alzheimer’s disease, making it essential to study the impact of nsSNPs on this process. The Solubility based on Disorder and Aggregation (SODA) tool (http://protein.bio.unipd.it/soda/) is designed to evaluate how mutations affect protein aggregation, disorder, and secondary structure. By inputting protein sequences or PDB files, SODA employs methods like PASTA 2.0, ESpritz-NMR, and Fells to predict variations, including insertions, deletions, substitutions, and duplications. It provides a final score indicating the solubility difference between the wild-type and mutant proteins, offering insights critical for Alzheimer’s research [32].
Analyzing Non-Covalent Interactions
Mutations can alter the non-covalent interactions within a protein, impacting its stability, function, and overall structure. Understanding these changes is critical for comprehending the molecular mechanisms underlying diseases like Alzheimer’s. The Arpeggio server is a valuable tool for analyzing these interactions by calculating the number of interatomic contacts in a protein structure. This server supports PDB format files and identifies about 15 types of interatomic interactions. Using OpenBabel and SMARTS queries, Arpeggio assigns atom types and identifies nearest-neighbor atoms within a 5 Å radius, generating a Structural Interaction Fingerprint (SIFt) for each pairwise contact [33].
Construction of Protein-Protein Interaction Network (PPIN)
The STRING (https://string-db.org/) database was used to predict the functional association of NLRP3 with other proteins. The NLRP3 protein was mapped against Homo sapiens (NCBI taxonomy ID: 9606) and a PPIN was constructed at the highest confidence (confidence score: 0.900) by including known interactions (from curated databases and experimentally determined), predicted interaction (gene neighbourhood, gene fusions and gene co-occurrence), and others (text mining, co-expression, and protein homology) [34]. The predicted PPIN was further subjected to clustering to identify functional proteins majorly interacting with NLRP3.
Protein-Protein Docking
The NEK7 was identified as a major functional protein interacting with NLRP3. In order to perform protein-protein docking the 3D structure of NEK7 (PSB ID: 5DE3) was retrieved from the PDB database. The modelled structure of wild-type NLRP3 and the cleaned and prepared structure of NEK7 were subjected to global protein-protein docking using the HDOCK server [35]. Following docking the docking scores were calculated by a knowledge-based iterative scoring function wherein a more negative docking score indicated a more possible binding model. The tool states that a docking score of −200 or better and a confidence score above 0.7 suggest empirically defined docking between the two molecules. The docked structures of the wild-type and the mutant type were further visualised for the molecular interactions among the proteins.
Molecular Docking of a Memantine with the NLRP3 Protein and Its Mutant Forms
Docking analysis was conducted using the PyRx tool (https://pyrx.sourceforge.io/) to evaluate the binding affinity of the selected ligand with both the NLRP3 wild type and R262L mutated protein structures. The mutated protein structure and the wild type were individually assessed against Memantine (PubChem CID ID: 4054) using AutoDock Vina 2.0 integrated into PyRx.
In this analysis, the modeled proteins served as the macromolecules, while Memantine was designated as the flexible entity for docking. A grid box was generated with the center dimensions of X:184.614, Y:201.012, and Z:134.0352 for all the structures. This investigation aimed to elucidate Memantine’s binding mechanisms and potential interactions with the NLRP3 protein and its mutant variant, offering valuable insights into Memantine’s efficacy. The docking results were quantified based on binding affinity, representing the energy associated with ligand binding [26]. Throughout the docking process, the ligands maintained nine degrees of flexibility, with the binding pose exhibiting zero Root Mean Square Deviation (RMSD), deemed optimal for maximal binding efficacy. Additionally, interactions were visualized using BIOVIA Discovery Studio and the protein structures (wild and mutant) were superimposed to understand the structural alignment (https://discover.3ds.com/discovery-studio-visualizer-download) [36].
Molecular Dynamic Simulation
The R262L mutation stands out as it may significantly impact the NLRP3. The Molecular Dynamics Simulation (MDS) using GROMACS software was employed to evaluate the stability of the R262L and wild-type NLRP3 models. Subsequently, MDS was conducted using the GROMACS package with the GROMOS96 54a7 force field. Solvation was achieved using the SPCE water model, and a salt concentration of 0.15 M with NA+ and Cl- ions was maintained, while the complex’s pH was set at 7. The equilibration process consisted of two phases: first, a 10-ns v-rescale algorithm heating period gradually increased the temperature to approximately 300 K in the NVT ensemble. This was followed by a 5-ns restrained phase to allow solvent settling, succeeded by a 10-ns NPT equilibration phase with gradually removed restraints. Once equilibrated, the systems underwent a 100 ns preparation period for graphing and MMPBSA analysis for the last 50 ns, with pressure maintained at 1 bar and temperature at 300 K [36].
MDS assessed variations in Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), and hydrogen bond count of the complex, providing a comprehensive evaluation of its stability.
Results
Retrieval of SNPs
The dbSNP database was used to retrieve the SNPs for the human NLRP3 gene. It contained 13825 SNPs in total, out of which 7 inframe deletions, 2 inframe indel, 6 inframe insertions, 2 initiator codon variants, 12178 introns, 893 missense (nsSNPs), 225 non-coding transcript variants and 512 synonymous. For this study, only nsSNPs of NLRP3 were selected, which contributed to only 6.45% of all SNPs known in the human NLRP3 gene (Fig. 1).

Prediction of SNPs associated with the human NLRP3 gene. The figure shows the presence of mutations in the NLRP3 genes retrieved from the dbSNP database. A total of 13825 SNPs were retrieved of which, 0.05% were inframe deletions, 0.014% were inframe indels, 0.04% were inframe insertions, 0.014% were initiator codon variants, 88% were introns, 6.45% were missense, 1.62% were non-coding transcripts and 3.7% were synonymous
Identification and Prediction of Effects of SNPs
In the initial screening of 893 non-synonymous single nucleotide polymorphisms (nsSNPs), SIFT analysis categorized 86 as deleterious and 116 as tolerated, with the remaining nsSNPs remaining unclassified. To refine these results, Align GVGD, PolyPhen-2, and PANTHER methodologies were employed to filter the SIFT predictions and assess the potential consequences of the nsSNPs.
Align GVGD analysis revealed insights into the potential functional impact of the nsSNPs. Among the 86 deleterious nsSNPs identified by SIFT, 42 (48.84%) were classified as having the highest likelihood of functional impact (Class 65), indicating a higher probability of inducing functional changes. Additionally, 40 nsSNPs (46.51%) were classified into other classes (C0, C15, C25, C35, and C55), suggesting a lower likelihood of functional impact. The remaining mutations were excluded from further analysis due to their association with unknown amino acids. Subsequently, only the 42 nsSNPs classified as having the highest likelihood of functional impact (Class C65) were subjected to further scrutiny (Table 2). PolyPhen-2 analysis identified 4 nsSNPs as possibly damaging, 26 as probably damaging, and 12 as benign. PANTHER analysis indicated that 6 nsSNPs were possibly damaging, 20 were probably damaging, and 16 were probably benign (Table 2).
Through a comprehensive analysis, 18 non-synonymous single nucleotide polymorphisms (nsSNPs) were identified as significant candidates. These nsSNPs were predicted to be deleterious by SIFT, categorized as Class C65 in Align-GVGD analysis, and deemed probably damaging according to predictions from PolyPhen-2 and PANTHER. The selection of these 18 nsSNPs was made meticulously, considering the robustness of the analyses and the consistency of outcomes across various prediction techniques (Table 3).
Prediction of Changes in Protein Stability
The 18 chosen nsSNPs underwent analysis using the MUpro and I-Mutant web servers to assess the impact of point mutations on protein stability, as indicated by the change in free energy value. For MUpro analysis, the range of DDG values for the nsSNPs varies from −1.92 kcal/mol to −0.1738 kcal/mol. Among these mutations, all are predicted to decrease stability (Table 4). In the case of I-Mutant analysis, some mutations are predicted to increase stability while others are predicted to decrease stability. Specifically, 2 mutations are predicted to increase stability, while 16 mutations are predicted to decrease stability (Table 4). Additionally, there are 16 mutations that are predicted to decrease stability by both MUpro and I-Mutant tools, suggesting a consensus in their assessment of these mutations and were subjected to further screening.
Protein stability is a crucial determinant of protein function. Stable proteins are more likely to maintain their correct folding and functional conformation, which is essential for their normal biological activity. Conversely, decreased stability can lead to misfolding, aggregation, and dysfunctional interactions. These effects are particularly relevant in the study of disease-associated proteins, where even minor perturbations in stability can have significant pathological consequences. In our study, we focused on nsSNPs that decrease the stability of the NLRP3 protein rather than those that increase it. NLRP3 is a central component of the inflammasome complex, which plays a critical role in the inflammatory response. Decreased stability in the context of NLRP3 does not necessarily equate to a loss of function. Instead, it can result in aberrant activation and heightened inflammatory responses as they may form aggregates leading to excessive and chronic inflammation.
Prediction of Solvent Accessibility
The analysis conducted through NetSurfP aimed to predict the solvent accessibility of various mutations within the NLRP3 gene (Fig. 2, Table 5). These predictions provide valuable insights into the potential functional implications of each mutation, shedding light on their respective roles within the protein structure.

Solvent accessibility and stability prediction by NetSurfP
In Table 4, the classification of each mutation based on solvent accessibility is outlined, along with corresponding RSA (Relative Surface Accessibility) and ASA (Absolute Surface Accessibility) values. Notably, the mutations exhibit a mixture of buried and exposed classifications, suggesting diverse interactions with the protein environment. Among the mutations classified as “Buried,” including D305H, T954M, T350M, L307P, G303D, D305G, C261W, F525C, Y443H, C998S, G303C, and L362Q, the RSA values range from 0.015 to 0.223. These mutations are likely situated within the core of the protein, with limited exposure to the surrounding solvent environment. The corresponding ASA values further support this inference, ranging from 2.152 to 32.141 (Table 5). Conversely, mutations classified as “Exposed,” such as T897M, R262P, R262L, and T659I, exhibit higher RSA values ranging from 0.28 to 0.339. These mutations are positioned on the protein surface, allowing for greater interaction with solvent molecules. As a result, their ASA values are comparatively higher, ranging from 38.841 to 64.733 (Table 5).
Validation of Protein Models for Structural and Functional Analysis
The three-dimensional structural analysis of the human NLRP3 protein was conducted to investigate the impact of deleterious nsSNPs on both wild-type and mutant-type structures. Notably, mutations such as T897M, R262P, R262L, and T659I were found to induce destabilizing effects in the NLRP3 protein. The SWISS-MODEL platform was utilized to generate three-dimensional models of both the wild-type and mutant-type NLRP3 proteins, employing the 7pzc.1.A template from the UniProtKB proteome as the most suitable alignment. Through this process, detailed structural insights into the conformational changes induced by the mutations were obtained. Analysis of the Ramachandran plot revealed that approximately 86% of the residues in each structure occupied the most favoured region, indicating the overall high quality and reliability of the modelled protein structures. This analysis provides valuable information regarding the structural stability and integrity of the NLRP3 protein under the influence of deleterious nsSNPs (Table 6, Fig. 3).

Modeled structures of (a) Wild type (b) R262L (c) R262P (d) T659I (e) T897M mutants of NLRP3
Prediction of Disease Association and Aggregation Propensity
The nsSNPs can alter the amino acid sequence of proteins, potentially affecting their structure and function. Understanding whether these changes are associated with diseases helps in identifying genetic risk factors and mechanisms underlying various conditions. In the present study, the disease association was predicted with SNP&GO and PhD-SNP servers wherein a probability score greater than 0.5 were predicted to be disease-associated variation (Table 7). The rs180177442 (R262P, R262L) was predicted to be disease-associated across both servers.
The aggregation propensity for the wild-type NLRP3 protein is −5.798, serving as a baseline for evaluating the effects of various mutations. The SODA tool predicts protein aggregation propensity, with more negative values indicating a higher tendency to aggregate. The R262L mutation exhibits a significantly negative aggregation value of −19.634, indicating a much higher propensity to aggregate and reduced solubility compared to the wild type (Table 7). Consequently, the R262L mutation was prioritized for further analysis.
Analyzing Non-Covalent Interactions
The non-covalent interaction analysis of wild-type and R262L mutated NLRP3 reveals subtle but significant differences that could explain the mutant’s behavior. The R262L mutant shows a slight decrease in hydrogen bonds and weak hydrogen interactions compared to the wild type, indicating minor alterations in stability and local interactions. The decrease in ionic interactions further suggests potential disruptions in electrostatic stability. Interestingly, the R262L mutant has an increase in aromatic contacts, hydrophobic contacts, carbonyl interactions, polar contacts, and weak polar contacts, which suggests a compensatory mechanism to maintain structural integrity (Table 8). The increase in hydrophobic contacts, in particular, aligns with previous reports indicating that changes in hydrophobic interactions significantly affect protein stability. These changes contribute to the mutant’s higher aggregation propensity and lower solubility, reinforcing its association with disease.
Protein-Protein Interaction Network for NLRP3
The STRING analysis for NLRP3 reveals its crucial role in innate immunity and inflammation, particularly through its involvement in the NLRP3 inflammasome. As a sensor, NLRP3 interacts with several key proteins such as PYCARD, NEK7, AIM2, and CASP1, facilitating the formation and activation of the inflammasome complex. This complex subsequently activates caspase-1, leading to the maturation and secretion of pro-inflammatory cytokines IL-1β and IL-18, which are central to the inflammatory response. The interactions with proteins like MAVS and TXNIP further underscore the role of NLRP3 in mediating inflammatory pathways and responding to pathogenic threats (Fig. 4).

Protein-protein interaction network for NLRP3. The results show (a) the PPIN generated with STRING at the highest confidence of 0.900 and the highlighted regions (b, c) represent the major clusters in the network
Protein-Protein Docking
STRING identified two major clusters, with NLRP3 showing significant interaction with NEK7. NEK7 is essential for the activation of the NLRP3 inflammasome, a multi-protein complex that plays a pivotal role in the inflammatory response associated with Alzheimer’s disease. NEK7 interacts with NLRP3, facilitating its activation, which leads to the production of pro-inflammatory cytokines. This inflammation is a key feature of Alzheimer’s disease pathology, contributing to neuronal damage and disease progression. Due to its critical role in inflammasome activation, NEK7 was docked with wild type and R262L mutated model of NLRP3 (Fig. 5).

Protein-protein docking of wild and mutated model of NLRP3 with NEK7
The docking results between NLRP3 and NEK7 reveal significant differences between the wild-type and R262L mutant. The docking score (DS) for the wild-type is −281.76 with a confidence score (CS) of 0.933, indicating a strong binding model. For the R262L mutant, the DS is −295.11 with a CS of 0.948, suggesting an even stronger interaction. Visualization of the interactions shows that the wild-type NLRP3 forms contacts with 32 residues in NEK7, establishing 2 salt bridges, 5 hydrogen bonds, and 229 non-bonded contacts. The hydrogen bonds are precise, with distances ranging from 2.23 to 3.13 Å. In contrast, the R262L mutant forms contacts with 30 residues in NLRP3 and 298 residues in NEK7, with 4 salt bridges, 6 hydrogen bonds, and 210 non-bonded contacts (Fig. 5). The hydrogen bonds in the R262L mutant interaction are generally slightly shorter, indicating stronger interactions, with distances between 1.78 and 3.28 Å. This increase in hydrogen bonds and salt bridges in the R262L mutant suggests a more robust and possibly more pathogenic interaction with NEK7, highlighting the mutant’s potential to exacerbate inflammatory responses, which is critical in the context of Alzheimer’s disease. These results underscore the R262L mutant’s enhanced propensity to aggregate and interact more strongly with NEK7, reflecting its potential for increased inflammatory activity.
Molecular Docking
Following molecular docking with memantine, disparities in binding affinity and interaction residues were noted between the wild-type and mutated NLRP3 models. Relative to the wild type, the R262L mutant displayed a slightly elevated binding affinity (−6.6 kcal/mol versus −6.5 kcal/mol) (Table 9). Upon examination of interaction residues, TRP A:416 consistently engaged with memantine in both wild-type and mutant models (Fig. 6), underscoring its pivotal role in drug binding. However, the mutants exhibited additional or altered interactions compared to the wild type. Notably, the R262L mutant formed a conventional hydrogen bond with SER A:345 (Fig. 6). These variances in interaction residues and bond types suggest structural modifications induced by mutations, potentially influencing drug-binding efficacy.

Molecular visualization of molecular docking of NLRP3 with Memantine. a 2D visualization of Wild-type (b) 3D visualization of Wild-type (c) 2D visualization of R262L mutant (d) 3D visualization of R262L mutant. Purple color indicates hydrogen bond donors and green color indicates hydrogen bond acceptors
The wild type and the mutated models were superimposed to have a better understanding on the structural alignment. The structures had a 100% overlapping (Topology score: 1.00), RMSD values of 0.06, with a 1.00 fragment score and 21.17 Z-score (Fig. 7).

Alignment of wild-type NLRP3 with R262L mutant. a Cartoon representation (b) Backbone representation of NLRP3 with R262L mutant (Green: Wild type NLRP3; Brown: R262L mutant), c Pairwise sequence alignment of NLRP3 with R262L mutant
Molecular Dynamic Simulation
Molecular dynamics simulations were carried out separately for 100 ns on two systems that are the wild NLRP3 and the R262L mutated model. The stability of the of the complexes was investigated with the RMSD values. The Root Mean Square Deviation (RMSD) graph provides a quantitative measure of the structural stability of the wild-type NLRP3 protein versus the R262L mutant over a 100 ns molecular dynamics simulation. RMSD values (Fig. 8), plotted over time, reflect the average distance between backbone atoms of the protein at a given time point and the initial structure, thus serving as an indicator of conformational changes and stability.

RMSD plots for wild type and R262Lmutated model of NLRP3
In this graph, the wild-type NLRP3, represented by the orange line, maintains relatively low RMSD values throughout the simulation, fluctuating around 0.4 to 0.5 nm. This indicates that the wild-type protein remains close to its original conformation with only minor deviations, demonstrating a stable structure over time. Such stability is crucial for maintaining the protein’s functional integrity and proper interaction with other molecules, such as NEK7, in the inflammasome complex. Conversely, the R262L mutant, shown by the black line, exhibits consistently higher RMSD values, fluctuating around 0.5 to 0.7 nm. This suggests that the mutant protein undergoes greater conformational changes and deviates more significantly from its initial structure. The increased RMSD values indicate that the R262L mutation induces structural instability, potentially leading to alterations in the protein’s functional domains and interaction surfaces (Fig. 8).
The Root Mean Square Fluctuation (RMSF) graph illustrates the flexibility of individual residues in the wild-type and R262L mutant NLRP3 proteins over the course of the simulation. RMSF measures the average deviation of each residue from its mean position, providing insight into the dynamic behavior and flexibility of specific regions within the protein. In this graph (Fig. 9), the wild-type NLRP3, represented by the orange line, shows relatively consistent fluctuations across most residues, with peaks indicating regions of higher flexibility. The R262L mutant, depicted by the black line, displays generally higher RMSF values compared to the wild type, particularly in the regions around residues 200, 400, and 900. This indicates that these regions in the mutant protein are more flexible and experience greater deviations from their mean positions. The increased flexibility in the R262L mutant can be attributed to the structural instability introduced by the mutation, which disrupts the local environment and weakens stabilizing interactions.

RMSF plots for wild type and R262L mutated model of NLRP3
The Radius of Gyration (Rg) graph provides insight into the overall compactness and structural integrity of the wild-type NLRP3 and the R262L mutant proteins over the course of the simulation. Rg measures the distribution of a protein’s mass around its center of mass, with lower values indicating a more compact structure. In this graph (Fig. 10), the wild-type NLRP3, represented by the orange line, displays relatively consistent Rg values around 4.0 nm throughout the simulation, with slight fluctuations. This stability in Rg values indicates that the wild-type protein maintains a relatively compact and stable structure, which is crucial for its proper function and interactions with other proteins. Conversely, the R262L mutant, depicted by the black line, shows slightly higher Rg values, fluctuating around 4.1 nm. These higher Rg values suggest that the mutant protein adopts a less compact structure compared to the wild type. The fluctuations in the Rg values for the R262L mutant also appear more pronounced, indicating greater variability in the protein’s overall shape and compactness during the simulation.

Rg plots for wild type and R262L mutated model of NLRP3
The Solvent Accessible Surface Area (SASA) graph measures the surface area of the protein that is accessible to solvent molecules, providing insight into the protein’s exposure to the surrounding environment. SASA is an important parameter that reflects the folding state, stability, and potential interaction sites of a protein. Higher SASA values indicate more surface area is exposed to solvent, suggesting a more unfolded or extended conformation, while lower values indicate a more compact and folded structure.
The wild-type NLRP3, represented by the orange line, shows a gradual decrease in SASA values over the simulation time, stabilizing around 800 nm². This decrease indicates that the wild-type protein becomes more compact as the simulation progresses, reducing its solvent-exposed surface area (Fig. 11). Such behavior is typical of a stable protein that folds correctly and maintains a compact conformation, essential for its proper biological function. The R262L mutant, depicted by the black line, also shows a decrease in SASA values, but with more fluctuations and generally slightly higher values than the wild type. The R262L mutant stabilizes around 830 nm², which is higher than the wild type, suggesting that the mutant protein retains more exposed surface area throughout the simulation. The increased SASA values and greater fluctuations indicate that the R262L mutant adopts a less compact and more dynamic conformation compared to the wild type. The higher SASA values for the R262L mutant suggest that this mutation causes the protein to adopt a more open structure, potentially exposing more hydrophobic regions to the solvent. This exposure can lead to increased interactions with solvent molecules, promoting aggregation and reducing solubility, as observed in the experimental data. The increased solvent exposure also implies that the mutant protein may be less stable, with a greater tendency to unfold or misfold.

Solvent accessible surface area plot for wild type and R262L mutated model of NLRP3
Discussion
NLRP3 is recognized for its involvement in sensing various molecular patterns, leading to the assembly of the inflammasome complex, activation of caspase-1, and subsequent release of the pro-inflammatory cytokine IL-1β, thereby orchestrating innate immune responses. The NLRP3 inflammasome is implicated in AD, as its activation within microglial cells by amyloid-beta (Aβ) peptides initiate neuroinflammatory processes [37]. It is widely acknowledged that polymorphisms play a significant role in modulating gene expression, potentially influencing inter-individual variations in disease susceptibility and severity [38]. SNPs are the most common type of genetic variation, occurring at a frequency of more than 1% at specific genomic positions [39]. These variations can occur within coding regions, non-coding regions, or intergenic regions of the human genome. Among SNPs, the nsSNPs stand out as they entail a single amino acid substitution within a gene’s coding region, potentially leading to significant alterations in the structural and functional characteristics of the resultant protein [40]. Consequently, nsSNPs have garnered considerable attention in recent research endeavours. Therefore, investigating the relationship between genetic mutations and their phenotypic consequences holds promise in elucidating the underlying mechanisms contributing to AD.
Numerous research endeavours have aimed to anticipate the functional implications of nsSNPs, particularly discerning whether they bear disease relevance or remain neutral, by scrutinizing various attributes of these polymorphisms. Certain attributes rely solely on sequence information, such as the specific residues encountered at the SNP locus. Single-base alterations occurring within protein-coding regions of DNA can induce modifications in amino acids, thereby potentially impacting protein structure and function [39].
Focusing on mitigating neuroinflammation as a therapeutic approach for progressive neurological disorders represents a logical strategy for neuroprotection. However, to effectively implement such strategies, it is imperative to pinpoint the cellular and molecular pathways pivotal in disease-related neuroinflammation [41]. Research conducted by Martinez et al. underscores the necessity of NLRP3 in the manifestation of inflammatory symptoms and neurodegeneration in a mouse model of Parkinson’s disease. This study identified several SNPs concentrated in the highly conserved NLRP3 NACHT domain. Among these variants, one synonymous SNP, rs7525979, was found to be significantly associated with a reduced risk of Parkinson’s disease development [42]. In our study, our focus centered on nsSNPs within the human NLRP3 gene. Analysis conducted through the dbSNP database revealed a total of 13,825 SNPs associated with the NLRP3 gene, with nsSNPs (missense) and synonymous SNPs accounting for 6.4% and 3.7% of the total mutations, respectively. Intriguingly, a substantial proportion (approximately 88%) of these mutations were found within intronic regions, representing non-coding segments of the gene. Further scrutiny was directed towards the 893 nsSNPs identified in our investigation.
Characterizing these nsSNPs based on their impact on protein structure was imperative, prompting the utilization of various bioinformatics tools. Our methodology incorporated diverse parameters describing pathogenicity, providing molecular insights into the effects of these mutations. Given the complexity of predicting the pathogenic effects of SNPs, employing a single method proved insufficient. Therefore, we employed a combination of multiple methods to cross-validate and enhance the robustness of our predictions. Specifically, our study utilized a suite of in silico prediction algorithms, including SIFT, Align GVGD, PolyPhen-2, and PANTHER.
The mutations including rs121908153 (D305H), rs139814109 (T840M, T897M, T954M), rs151344629 (T350M), rs180177431 (L307P), rs180177433 (T438I), rs180177441 (G303D), rs180177442 (R262P, R262L), rs180177447 (D305G), rs180177475 (C261W), rs180177478 (F525C), rs180177499 (Y443H), rs199517145 (C998S), rs201504984 (G303C), rs201593863 (L362Q), and rs201875324 (T659I), were predicted to be deleterious and probably damaging by all prediction software. Except for the mutation T840M and T438I, all the other mutations were predicted to have a destabilizing effect on the protein stability. A decline in protein stability can precipitate structural alterations that impede the protein’s capacity to execute its intended functions efficiently. This destabilization may induce structural modifications, such as misfolding or unfolding, compromising the protein’s functional efficacy. Notably, among the four nsSNPs analyzed—rs180177442 (R262L and R262P), rs201875324 (T659I), and rs139814109 (T897M)—the prediction indicated that they were exposed in the protein. The term “buried” signifies that the amino acid resides deep within the protein’s core, shielded from the surrounding solvent. Conversely, an “exposed” residue is positioned on the protein’s surface, directly interacting with the solvent environment. Buried residues serve a pivotal role in upholding the structural stability and integrity of proteins [43].
Mutations affecting buried residues can disrupt the intricate network of interactions within the protein core, resulting in destabilization of its structure. Buried residues often coincide with critical functional sites within proteins, such as catalytic sites or binding interfaces. Mutations in these residues can interfere with essential sites, thus altering the protein’s function. Given their pivotal role in protein folding and stability, buried residues are typically more evolutionarily conserved. Consequently, mutations in buried residues are more likely to exert a significant impact on both protein structure and function [44]. To gain insights into their three-dimensional structures, these mutated proteins were modeled using 7pzc.1 as the template. Subsequently, molecular docking studies were conducted with Memantine to elucidate potential interactions.
Using SNP&GO and PhD-SNP servers, the nsSNPs R262L and R262P were identified as disease-associated, with R262L showing a higher aggregation propensity (−19.634) and reduced solubility. The non-covalent interaction analysis highlighted that the R262L mutant had slight decreases in hydrogen bonds and ionic interactions but increases in hydrophobic and polar contacts, suggesting enhanced aggregation and disease association. STRING analysis revealed NLRP3’s crucial role in inflammation through interactions with proteins like NEK7, which is vital for inflammasome activation. Docking studies showed stronger interactions between R262L NLRP3 and NEK7 compared to the wild type, with the mutant forming more hydrogen bonds and salt bridges, indicating a potentially more pathogenic interaction. These findings underscore the R262L mutation’s enhanced inflammatory response potential, relevant to Alzheimer’s disease progression.
Memantine is a medication prescribed for alleviating symptoms associated with AD and dementia. Classified as an NMDA receptor antagonist, it operates by mitigating aberrant brain activity. While it does not offer a cure for AD, memantine demonstrates efficacy in enhancing cognitive functions, and memory retention and decelerating the advancement of the disease [45]. The molecular docking analysis of memantine with wild type and mutated NLRP3 models revealed variations in binding affinity and interaction residues. Despite minor fluctuations in binding affinity among mutants compared to the wild type, interaction residues differed significantly. These findings suggest structural alterations induced by mutations, potentially influencing drug-protein interactions. Specifically, the R262L mutation stands out as it may significantly impact the NLRP3-memantine binding interface, highlighting its importance for further investigation in understanding drug efficacy and potential therapeutic implications. The R262L mutant model was subjected to molecular dynamics simulations to understand the structural stability and flexibility of the wild and mutated structures over time.
The higher RMSD values for the R262L mutant are indicative of its decreased structural integrity. This instability can lead to improper folding or increased flexibility, which may expose hydrophobic regions and promote aggregation. The structural changes observed in the R262L mutant are consistent with its predicted higher aggregation propensity and reduced solubility, factors that contribute to its pathogenicity. The higher fluctuations observed in the R262L mutant suggest that the mutation induces localized structural instability, making certain regions of the protein more dynamic and less stable. Furthermore, the increased structural fluctuations in the R262L mutant could disrupt its ability to interact effectively with NEK7 and other inflammasome components. Moreover, the regions with increased RMSF in the R262L mutant may correlate with areas critical for the protein’s role in inflammasome activation and the inflammatory response. The heightened fluctuations in these regions could disrupt the precise molecular interactions required for proper inflammasome assembly and function, potentially leading to aberrant inflammatory signaling. This is particularly relevant in the context of Alzheimer’s disease, where dysregulated inflammation is a key pathological feature. The increased Rg values for the R262L mutant reflect a structural impact caused by the mutation, leading to a more expanded and less stable conformation. This expansion could result from the disruption of key interactions within the protein’s core, leading to a loosening of its tertiary structure. The higher Rg values and increased fluctuations suggest that the R262L mutant is less able to maintain a tightly packed conformation and further may potentially contribute to aggregation due to the exposure of hydrophobic residues. The changes in SASA values for the R262L mutant highlight its structural impact on NLRP3. The mutation appears to disrupt the protein’s ability to maintain a tightly packed conformation, leading to an increase in solvent-exposed surface area.
The R262L mutant (rs108177442) (Alleles: G > A/G > C/G > T) identified in the present study is reported to be present in 1 in 264690 people (Allele frequency: 0.000004) as per the data available in dbSNP (accessed on 22nd July 2024). The R262L coding variant is associated with Familial cold autoinflammatory syndrome 1 (ClinVar Accession: RCV000084231.1) and is considered to be likely-pathogenic in cases of Chronic infantile neurological, cutaneous and articular syndrome (ClinVar Accession: RCV001028056.1) and CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders ClinVar Accession: RCV000788842.1).
Previously, Zhang et al. examined a Chinese population and found that variants rs10754558 and rs10925019 within NLRP3 are linked to ulcerative colitis development [46]. Conversely, Varghese et al. conducted a study among Caucasians and concluded that there is no association between NLRP3 polymorphisms and inflammatory bowel disease [47]. Yang et al. proposed that the rs4353135 polymorphism may contribute to the pathophysiology of juvenile idiopathic arthritis in a Taiwanese population [48]. Additionally, Dehghan et al. emphasized the potential critical role of NLRP3 SNPs in regulating inflammation underlying cardiovascular disease [49]. Several limitations need to be acknowledged in our study. While we have identified high-risk nsSNPs within the conserved domain that exhibit a destabilizing effect on the protein, their functional associations were not characterized in this investigation. Further clinical and genetic studies are warranted to explore the mechanisms underlying the correlation between NLRP3 polymorphisms and the onset of AD.
Conclusion
Detection of deleterious variants is a prominent area of research, with bioinformatics tools offering insights into the impact of nsSNPs or missense mutations on protein stability and structure. Our analysis of 893 nsSNPs revealed 18 deleterious variants associated with the NLRP3 gene. Notably, rs180177442 (R262L and R262P), rs201875324 (T659I), and rs139814109 (T897M) were classified as high-risk variants due to their significant deleterious effects and association with reduced protein stability. These findings suggest potential targets for proteomic studies, diagnostics, and therapeutic interventions in AD. Further investigation through in vivo experiments is warranted to elucidate the implications of these nsSNPs in AD pathology.
References
Serrano-Pozo, A., & Growdon, J. H. (2019). Is Alzheimer’s disease risk modifiable? Journal of Alzheimer’s Disease: JAD, 67(3), 795–819. https://doi.org/10.3233/jad181028.
Silva, M. V. F., Loures, C., de, M. G., Alves, L. C. V., de Souza, L. C., Borges, K. B. G., & Carvalho, M. D. G. (2019). Alzheimer’s disease: risk factors and potentially protective measures. Journal of Biomedical Science, 26(1), 33 https://doi.org/10.1186/s12929-019-0524-y.
Mangalmurti, A., & Lukens, J. R. (2022). How neurons die in Alzheimer’s disease: Implications for neuroinflammation. Current Opinion in Neurobiology, 75(102575), 102575 https://doi.org/10.1016/j.conb.2022.102575.
James, B. D., & Bennett, D. A. (2019). Causes and patterns of dementia: An update in the era of redefining Alzheimer’s disease. Annual Review of Public Health, 40(1), 65–84. https://doi.org/10.1146/annurev-publhealth-040218-043758.
de Brito Toscano, E. C., Rocha, N. P., Lopes, B. N. A., Suemoto, C. K., & Teixeira, A. L. (2021). Neuroinflammation in Alzheimer’s disease: Focus on NLRP1 and NLRP3 inflammasomes. Current Protein & Peptide Science, 22(8), 584–598. https://doi.org/10.2174/1389203722666210916141436.
Gao, X. (2022). Mechanism of NLRP3 inflammasome activation and its role in Alzheimers disease. Exploration of Immunology, 2(3), 229–244.
Geyer, S., Jacobs, M., & Hsu, N.-J. (2019). Immunity against bacterial infection of the central nervous system: An astrocyte perspective. Frontiers in Molecular Neuroscience, 12, 57 https://doi.org/10.3389/fnmol.2019.00057.
Furr, S. R., & Marriott, I. (2012). Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Frontiers in Microbiology, 3, 201 https://doi.org/10.3389/fmicb.2012.00201.
Fusco, R., Siracusa, R., Genovese, T., Cuzzocrea, S., & Di Paola, R. (2020). Focus on the role of NLRP3 inflammasome in diseases. International Journal of Molecular Sciences, 21(12), 4223 https://doi.org/10.3390/ijms21124223.
Mangan, M. S. J., Olhava, E. J., Roush, W. R., Seidel, H. M., Glick, G. D., & Latz, E. (2018). Targeting the NLRP3 inflammasome in inflammatory diseases. Nature Reviews. Drug Discovery, 17(9), 688 https://doi.org/10.1038/nrd.2018.149.
Bai, H., & Zhang, Q. (2021). Activation of NLRP3 inflammasome and onset of Alzheimer’s disease. Frontiers in Immunology, 12, 701282 https://doi.org/10.3389/fimmu.2021.701282.
Hanaei, S., Sadr, M., Rezaei, A., Shahkarami, S., Ebrahimi Daryani, N., Bidoki, A. Z., & Rezaei, N. (2018). Association of NLRP3 single nucleotide polymorphisms with ulcerative colitis: A case-control study. Clinics and Research in Hepatology and Gastroenterology, 42(3), 269–275. https://doi.org/10.1016/j.clinre.2017.09.003.
Zhang, Q., Fan, H. W., Zhang, J. Z., Wang, Y. M., & Xing, H. J. (2015). NLRP3 rs35829419 polymorphism is associated with increased susceptibility to multiple diseases in humans. Genetics and Molecular Research: GMR, 14(4), 13968–13980. https://doi.org/10.4238/2015.October.29.17.
Satam H, Joshi K, Mangrolia U, Waghoo S, Zaidi G, Rawool S, Thakare R. P, Banday S, Mishra A. K, Das G, & Malonia S. K. (2023). Next-generation sequencing technology: Current trends and advancements. Biology, 12(7), 997. https://doi.org/10.3390/biology12070997.
Yazar, M., & Özbek, P. (2021). In silico tools and approaches for the prediction of functional and structural effects of single-nucleotide polymorphisms on proteins: An expert review. Omics: A Journal of Integrative Biology, 25(1), 23–37. https://doi.org/10.1089/omi.2020.0141.
Sukumar, S., Krishnan, A., & Banerjee, S. (2021). An overview of bioinformatics resources for SNP analysis. In Advances in bioinformatics (pp. 113–135). Singapore: Springer.
Wu, Z., Wu, S., & Liang, T. (2021). Association of NLRP3 rs35829419 and rs10754558 polymorphisms with risks of autoimmune diseases: A systematic review and meta-analysis. Frontiers in Genetics, 12, 690860 https://doi.org/10.3389/fgene.2021.690860.
Cheng, L., Liang, X., Qian, L., Luo, C., & Li, D. (2021). NLRP3 gene polymorphisms and expression in rheumatoid arthritis. Experimental and Therapeutic Medicine, 22(4), 1110 https://doi.org/10.3892/etm.2021.10544.
Addobbati, C., da Cruz, H. L. A., Adelino, J. E., Melo Tavares Ramos, A. L., Fragoso, T. S., Domingues, A., & Sandrin-Garcia, P. (2018). Polymorphisms and expression of inflammasome genes are associated with the development and severity of rheumatoid arthritis in Brazilian patients. Inflammation Research, 67(3), 255–264. https://doi.org/10.1007/s00011-017-1119-2.
Heidari, Z., Salimi, S., Rokni, M., Rezaei, M., Khalafi, N., Shahroudi, M. J., & Saravani, M. (2021). Association of IL-1β, NLRP3, and COX-2 gene polymorphisms with autoimmune thyroid disease risk and clinical features in the Iranian population. BioMed Research International, 2021, 7729238 https://doi.org/10.1155/2021/7729238.
Wanarase, S. R., Chavan, S. V., Sharma, S., & D, S. (2023). Evaluation of SNPs from human IGFBP6 associated with gene expression: an in-silico study. Journal of Biomolecular Structure & Dynamics, 41(23), 13937–13949. https://doi.org/10.1080/07391102.2023.2192793.
Liu, Q., Zhang, M.-M., Guo, M.-X., Zhang, Q.-P., Li, N.-Z., Cheng, J., & Yi, L.-T. (2022). Inhibition of microglial NLRP3 with MCC950 attenuates microglial morphology and NLRP3/caspase-1/IL-1β signaling in stress-induced mice. Journal of Neuroimmune Pharmacology: The Official Journal of the Society on NeuroImmune Pharmacology, 17(3–4), 503–514. https://doi.org/10.1007/s11481-021-10037-0.
Chai, C.-Y., Maran, S., Thew, H.-Y., Tan, Y.-C., Rahman, N. M. A. N. A., Cheng, W.-H., & Yap, W.-S. (2022). Predicting deleterious non-synonymous single nucleotide polymorphisms (nsSNPs) of HRAS gene and in silico evaluation of their structural and functional consequences towards diagnosis and prognosis of cancer. Biology, 11(11), 1604 https://doi.org/10.3390/biology11111604.
Wang, Z., Huang, C., Lv, H., Zhang, M., & Li, X. (2020). In silico analysis and high-risk pathogenic phenotype predictions of non-synonymous single nucleotide polymorphisms in human Crystallin beta A4 gene associated with congenital cataract. PloS One, 15(1), e0227859 https://doi.org/10.1371/journal.pone.0227859.
Jayasurya, B.R., Swathy, J. S., Susha, D., & Sharma, S. (2023). Molecular docking and investigation of Boswellia serrata phytocompounds as cancer therapeutics to target growth factor receptors: An in silico approach. International Journal of Applied Pharmaceutics, 15(4),173–183. https://doi.org/10.22159/ijap.2023v15i4.47833.
Galgale, S., Zainab, R., Kumar A., P., Nithya, Susha & Sharma, S. (2023). Molecular docking and dynamic simulation-based screening identifies inhibitors of targeted SARS-CoV-2 3clpro and human ace2. International Journal of Applied Pharmaceutics, 15(6), 297–308. https://doi.org/10.22159/ijap.2023v15i6.48782.
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M., & Ng, P. C. (2016). SIFT missense predictions for genomes. Nature protocols, 11(1), 1–9. https://doi.org/10.1038/nprot.2015.123.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013 Jan; Chapter 7: Unit 7.20. https://doi.org/10.1002/0471142905.hg0720s76.
Khan, S. M., Faisal, A.-R. M., Nila, T. A., Binti, N. N., Hosen, M. I., & Shekhar, H. U. (2021). A computational in silico approach to predict high-risk coding and non-coding SNPs of human PLCG1 gene. PloS one, 16(11), e0260054 https://doi.org/10.1371/journal.pone.0260054.
Manfredi, M., Savojardo, C., Martelli, P. L., & Casadio, R. (2022). E-SNPs&GO: embedding of protein sequence and function improves the annotation of human pathogenic variants. Bioinformatics (Oxford, England), 38(23), 5168–5174. https://doi.org/10.1093/bioinformatics/btac678.
Mustafa, H. A., Albkrye, A. M. S., AbdAlla, B. M., Khair, M. A. M., Abdelwahid, N., & Elnasri, H. A. (2020). Computational determination of human PPARG gene: SNPs and prediction of their effect on protein functions of diabetic patients. Clinical and Translational Medicine, 9(1), 7 https://doi.org/10.1186/s40169-020-0258-1.
Paladin, L., Piovesan, D., & Tosatto, S. C. E. (2017). SODA: prediction of protein solubility from disorder and aggregation propensity. Nucleic Acids Research, 45(W1), W236–W240. https://doi.org/10.1093/nar/gkx412.
Jubb, H. C., Higueruelo, A. P., Ochoa-Montaño, B., Pitt, W. R., Ascher, D. B., & Blundell, T. L. (2017). Arpeggio: A web server for calculating and visualising interatomic interactions in protein structures. Journal of Molecular Biology, 429(3), 365–371. https://doi.org/10.1016/j.jmb.2016.12.004.
Szklarczyk, D., Franceschini, A., Wyder, S., Forslund, K., Heller, D., Huerta-Cepas, J., & von Mering, C. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research, 43, D447–D452. https://doi.org/10.1093/nar/gku1003.
Yan, Y., Tao, H., He, J., & Huang, S.-Y. (2020). The HDOCK server for integrated protein-protein docking. Nature Protocols, 15(5), 1829–1852. https://doi.org/10.1038/s41596-020-0312-x.
Bhowmik, R., Nath, R., Sharma, S., Roy, R., & Biswas, R. (2022). High-throughput screening and dynamic studies of selected compounds against SARS-CoV-2. International Journal of Applied Pharmaceutics, 14(1), 251–260. https://doi.org/10.22159/ijap.2022v14i1.43105.
Zhang, Y., Zhao, Y., Zhang, J., & Yang, G. (2020). Mechanisms of NLRP3 inflammasome activation: Its role in the treatment of Alzheimer’s disease. Neurochemical Research, 45(11), 2560–2572. https://doi.org/10.1007/s11064-020-03121-z.
Tan, M.-S., Yu, J.-T., Jiang, T., Zhu, X.-C., Wang, H.-F., Zhang, W., & Tan, L. (2013). NLRP3 polymorphisms are associated with late-onset Alzheimer’s disease in Han Chinese. Journal of Neuroimmunology, 265(1–2), 91–95. https://doi.org/10.1016/j.jneuroim.2013.10.002.
Chen, R., Davydov, E. V., Sirota, M., & Butte, A. J. (2010). Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PloS one, 5(10), e13574 https://doi.org/10.1371/journal.pone.0013574.
Vihinen, M. (2022). When a synonymous variant is nonsynonymous. Genes, 13(8), 1485 https://doi.org/10.3390/genes13081485.
Dhapola, R., Hota, S. S., Sarma, P., Bhattacharyya, A., Medhi, B., & Reddy, D. H. (2021). Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology, 29(6), 1669–1681. https://doi.org/10.1007/s10787-021-00889-6.
Martinez, E. M., Young, A. L., Patankar, Y. R., Berwin, B. L., Wang, L., von Herrmann, K. M., & Havrda, M. C. (2017). Editor’s highlight: Nlrp3 is required for inflammatory changes and nigral cell loss resulting from chronic intragastric rotenone exposure in mice. Toxicological Sciences: An Official Journal of the Society of Toxicology, 159(1), 64–75. https://doi.org/10.1093/toxsci/kfx117.
Dinesh, S. & Sharma, S. (2023). Prediction of high-risk Nssnps associated with wisp3 gene expression: an in silico study. International Journal of Applied Pharmaceutics, 15, 161–170.
Bhor, S., Tonny, S. H., Dinesh, S., & Sharma, S. (2024). Computational screening of damaging nsSNPs in human SOD1 genes associated with amyotrophic lateral sclerosis identifies destabilising effects of G38R and G42D mutations through in silico evaluation. In Silico Pharmacology, 12(1), 20 https://doi.org/10.1007/s40203-024-00191-7.
Marotta, G., Basagni, F., Rosini, M., & Minarini, A. (2020). Memantine derivatives as multitarget agents in Alzheimer’s disease. Molecules (Basel, Switzerland), 25(17), 4005 https://doi.org/10.3390/molecules25174005.
Zhang, H.-X., Wang, Z.-T., Lu, X.-X., Wang, Y.-G., Zhong, J., & Liu, J. (2014). NLRP3 gene is associated with ulcerative colitis (UC), but not Crohn’s disease (CD), in Chinese Han population. Inflammation Research, 63(12), 979–985. https://doi.org/10.1007/s00011-014-0774-9.
Varghese, G. P., Uporova, L., Halfvarson, J., Sirsjö, A., & Fransén, K. (2015). Polymorphism in the NLRP3 inflammasome-associated EIF2AK2 gene and inflammatory bowel disease. Molecular Medicine Reports, 11(6), 4579–4584. https://doi.org/10.3892/mmr.2015.3236.
Yang, C.-A., Huang, S.-T., & Chiang, B.-L. (2014). Association of NLRP3 and CARD8 genetic polymorphisms with juvenile idiopathic arthritis in a Taiwanese population. Scandinavian Journal of Rheumatology, 43(2), 146–152. https://doi.org/10.3109/03009742.2013.834962.
Dehghan, A., Yang, Q., Peters, A., Basu, S., Bis, J. C., Rudnicka, A. R., & Folsom, A. R. (2009). Association of novel genetic Loci with circulating fibrinogen levels: a genome-wide association study in 6 population-based cohorts: A genome-wide association study in 6 population-based cohorts. Circulation. Cardiovascular Genetics, 2(2), 125–133. https://doi.org/10.1161/CIRCGENETICS.108.825224.
Acknowledgements
H.B. extends her appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project Number PNURSP2024R146, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author contributions
Mitesh Patel, AA and BB contributed to study concept and design, collected/analyzed data, project administration and drafted the original manuscript. Alya Redhwan, CC, Mohd Adnan, Hadeel R Bakhsh, Nawaf Alshammari, Malvi Surti, Mansi Parashar and Mirav Patel contributed to methodology, data curation, investigation, visualization, analysis, review and editing; Mitesh Patel, DD, Alya Redhwan, EE, Mohd Adnan,FF, Dinesh MS, and Sameer S. contributed to critical revision of the manuscript, validation, formal analysis and study supervision. All authors read and approved the final manuscript.
Funding
This work is supported by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number PNURSP2024R146, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Redhwan, A., Adnan, M., Bakhsh, H.R. et al. Computational Identification and Functional Analysis of Potentially Pathogenic nsSNPs in the NLRP3 Gene Linked to Alzheimer’s Disease. Cell Biochem Biophys (2024). https://doi.org/10.1007/s12013-024-01465-9
Accepted:
Published:
DOI: https://doi.org/10.1007/s12013-024-01465-9