
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating motor neuron disease for which there is currently no robust therapy. Recent progress in understanding ALS disease mechanisms and genetics in combination with innovations in gene modulation strategies creates promising new options for the development of ALS therapies. In recent years, six gene modulation therapies have been tested in ALS patients. These target gain-of-function pathology of the most common ALS genes, SOD1, C9ORF72, FUS, and ATXN2, using adeno-associated virus (AAV)-mediated microRNAs and antisense oligonucleotides (ASOs). Here, we review the latest clinical and preclinical advances in gene modulation approaches for ALS, including gene silencing, gene correction, and gene augmentation. These techniques have the potential to positively impact the direction of future research trials and transform ALS treatments for this grave disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS), the most common motor neuron disease in adults, is characterized by degeneration of upper and lower motor neurons in the brain and spinal cord, causing progressive muscle weakness and ultimately resulting in respiratory failure. Most ALS patient die within 3–5 years of symptom onset [1]. ALS pathology is not caused by a single process, but by complex combinations of molecular mechanisms, which include neuroinflammation [2], defective RNA metabolism [3], oxidative stress [4], mitochondrial dysfunction [4], cytoskeletal disturbances, altered exon splicing, defective nucleo-cytoplasmic and axonal transport [5], toxic protein aggregation, perturbations in autophagy [6], and glutamate excitotoxicity [7]. These can be triggered by genetic [8], toxic [9], and/or environmental [10] factors. It must be emphasized that many views of ALS pathophysiology are predicated on studies of models in vitro and in animals; confirmation in human tissues has been challenging, although recent analyses of human ALS motor neurons in vitro are proving very instructive. Presently, approximately 30 genes have been robustly documented to trigger ALS when mutated [11, 12]. These monogenetic mutations, which have a high effect size, cause ALS in roughly 15% of patients. Additionally, there are more than 100 gene mutations found with a low-to-moderate effect size that likely contribute to the risk of developing ALS or modifying its phenotype [11, 13]. The majority of ALS cases (90–95%) are sporadic originating from de novo mutations or unknown causes, while approximately 5–10% of patients have a family history of mutations with an autosomal dominant inheritance pattern [14]. Due to the heterogeneity of disease mechanisms, identification of disease-modifying treatments or cures has been challenging. After decades of research and more than 120 clinical trials [15], only two therapeutic approaches have been FDA approved. Both riluzole [16] and edaravone [17] slow disease progression only modestly. Recently, advances in gene therapy have rendered it a promising field for the treatment of ALS due to its potential to correct underlying pathogenic mechanisms by attenuating gain-of-function toxicity and/or addressing haploinsufficiency at the genetic level. This review will present an overview of the current gene modulation therapies for the treatment of ALS, focusing on two modalities: viral delivery and antisense oligonucleotide (ASO) treatment.
Pathological Mechanisms of Major ALS Genes
SOD1
The first ALS-associated gene, superoxide dismutase-1 (SOD1), was identified in 1993 [18]. Mutations in the SOD1 gene account for 10–14% of familial and 1–2% of sporadic ALS [19]. ALS-causing SOD1 mutations typically display an autosomal dominant inheritance pattern. The canonical role of SOD1 is to protect cells from toxic reactive oxygen species by catalyzing the dismutation of superoxide anion to oxygen and hydrogen peroxide [20]. To date, 217 SOD1 variants have been identified in ALS patients [21], the majority of which are missense mutations. Disease severity and duration depends significantly on the variant, with some causing a relatively slow progression and others a much faster progression [22]. While a portion of SOD1 variants decrease the dismutation enzyme activity of SOD1, there is no clear correlation between dismutase activity, disease onset, and progression in adult humans [23].
Although the exact mechanisms of SOD1-induced motor neuron death are unclear, there is ample evidence that toxic gain of functions (GOF), generated by mutant SOD1, are at play. Mutations can induce conformational and functional changes in SOD1 protein that can cause toxicity via interaction with other proteins through various mechanisms, such upregulation of reactive oxygen species, endoplasmic reticulum stress, excitotoxicity, mitochondrial dysfunction, prion-like propagation, axonal transport disruption, and non-cell autonomous toxicity of neuroglia [24]. It is striking that forced expression of high levels of mutant but not wild-type SOD1 protein cause motor neuron disease in mice [25]. While the resulting ALS model reproduces many aspects of human ALS, an important caveat is that the high levels of SOD1 expression in some models (e.g., SOD1G93A transgenic ALS mice) do not represent the 1:1 ratio of mutant to WT protein in human ALS arising from these mutations. (On the other hand, the SOD1G85R mouse with normal levels of mutant SOD1 protein (0.2–1.0 fold of wild type) does develop motor neuron death, albeit after several months [26]). It is correctly argued that caution must be exercised in interpreting conclusions about molecular pathophysiology in any models based on excessive levels of mutant transgene expression.
While there is overwhelming evidence that SOD1-ALS is caused by GOF pathology, there is also evidence that LOF may play a modifying role. Although Sod1−/− mice do not exhibit motor neuron loss, they have neuromuscular, neuronal, and extra-neuronal phenotypes [27]. Two recent reports underscore this point. In 2019, two cases were published describing a 2-year-old child [28] and a 6-year-old child [29], both with homozygosity for a nucleotide duplication (c.335dupG) in SOD1 that resulted in a complete absence of SOD1 enzymatic activity in cells. The 2-year-old patient showed predominantly impairment of upper motor neurons, whereas other organ systems were unaffected [28]. The 6-year-old showed a loss of motor abilities, tetraspasticity, and mild cerebellar atrophy [29]. Although the truncated SOD1 protein may have contributed to motor disease, the symptoms observed in these patients were much more severe than in patients homozygous for full-length SOD1 mutations that preserved some SOD activity [30]. This suggests that the total loss of SOD1 enzymatic activity contributed to motor-neuron dysfunction, which could have implications for therapies aiming at complete knock down of SOD1.
TDP-43
Transactive response (TAR) DNA-binding protein 43 (TDP-43), encoded by the TARDBP gene, was identified in 2006 as an ALS-associated gene [31, 32]. Pathogenic missense mutations in TARDBP account for 3.3% of familial ALS cases and 0.5% of sporadic ALS cases [33] and typically display an autosomal dominant inheritance pattern. TDP-43 is a DNA/RNA-binding protein, normally located in the nucleus, which regulates various steps of RNA metabolism, including transcription, translation, surveillance of splicing, microRNA biogenesis, and RNA transport [34, 35]. Intriguingly, > 95% of ALS patients do not harbor mutations in the TARDBP gene, yet demonstrate widespread abnormalities involving the TDP-43 protein, which in ALS is aberrantly cleaved, hyperphosphorylated, and mislocated to the cytoplasmic where it forms neuronal inclusions [31]. The pathogenicity of TDP-43 cytoplasmic aggregates may be linked to both loss of nuclear functions and gain of toxic functions, leading to dysregulation of RNA metabolism and splicing defects, impaired mitochondrial function and axonal transport, defects in proteostasis, stress granules, and amyloid-like aggregate formation that may spread from cell to cell in a prion-like manner [36]. Importantly, overexpression of both aggregation-prone mutant TDP-43 and non-aggregating WT TDP-43 is toxic [37, 38]. This complex pattern renders TARDBP a difficult gene target to treat with gene therapy.
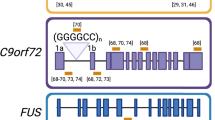
FUS
In 2009, pathogenic variants of the fused in sarcoma (FUS) gene, also called “translocated in liposarcoma,” were identified to be ALS-causing [39, 40]. FUS mutations account for 3% of familial cases and 0.4% of sporadic cases [33] and typically display an autosomal dominant inheritance pattern. Most pathogenic FUS variants are associated with early-onset ALS symptoms and juvenile patients [41]. FUS is a ubiquitously expressed RNA-binding protein predominantly located in the nucleus that is involved in DNA repair and multiple aspects of RNA metabolism, such as transcription, translation, pre-mRNA splicing, mRNA transport and stability, and processing of microRNAs and other non-coding RNAs [42]. To date, 126 different pathogenic FUS variants have been identified in ALS patients; the majority are missense mutations [43]. The functional consequences of these pathogenic mutations are not precisely defined, but there is strong evidence that supports a toxic GOF mechanism in ALS-FUS. Most mutations are located in exon 15, which encodes the C-terminal region of the protein containing the nuclear localization signal (NLS) domain [43]. These mutations disrupt the nuclear localization signal and result in mislocalization of FUS to the cytoplasm [44, 45]. Nuclear-to-cytoplasmic mislocalization of FUS is commonly found in neurons and glia of post-mortem brains of FUS-ALS patients [44]. It is still debated whether this has a direct or indirect impact on motor neuron toxicity. LOF does not seem to be sufficient to cause motor neuron death [46]; however, there is evidence that it might contribute to the pathogenesis of ALS [47].
C9ORF72
In 2011, a hexanucleotide repeat expansion (HRE) in intron 1 of the Chromosome 9 Open Reading Frame 72 (C9ORF72) gene was revealed to be the most common cause of both inherited (40%) and sporadic (5–6%) ALS [48,49,50] as well as the most common genetic cause of frontotemporal dementia (FTD) [51]. While the size of the repeat is usually less than 24 in healthy individuals, the number of repeats in the HRE mutation can reach a magnitude of thousands in ALS patients [52, 53]. The C9ORF72 gene has three transcript variants: V1, V2, and V3 [48]. V1 translates into a 222 amino acid protein while V2 and V3 both translate into the most predominant C9ORF72 481 amino acid protein [48]. V1 and V3 prior to mRNA splicing harbor the HRE. The C9ORF72 protein interacts with endosomes and is required for normal vesicle trafficking and lysosomal biogenesis in motor neurons [54, 55]. The HRE gives rise to three pathological hallmarks of C9ORF72-ALS. First, it impairs transcription, leading to C9ORF72 haploinsufficiency that compromises neuronal viability [55, 56]. Second, sense and antisense transcriptions of the C9ORF72 HRE produce G4C2 or C4G2 transcripts that accumulate in the cell nuclei and sequester RNA-binding proteins, resulting in RNA foci [48, 57, 58]. Third, both the sense and antisense transcripts of the HRE can serve as templates for repeat-associated non-AUG (RAN) translation of toxic poly-dipeptides, producing glycine-arginine (GR), glycine-proline (GP), and glycine-alanine (GA) poly-dipeptides in the sense direction and proline-alanine (PA), proline-arginine (PR), and GP in the anti-sense direction [58, 59]. These aggregation-prone dipeptides have been found in the brains and spinal cords of C9ORF72-ALS and FTD patients [59,60,61] and have proven to be toxic in cell culture [62,63,64] and in different animal models [65,66,67]. Similarly to SOD1, C9ORF72 LOF does not seem to cause ALS by itself; rather, there appears to be a direct form of cooperative pathogenesis between gain- and loss-of-function mechanisms, in which C9ORF72 haploinsufficiency impairs clearance of poly-dipeptides, making motor neurons hypersensitive to dipeptide pathology [55].
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) have proven to be greatly effective in the treatment of a wide range of genetic disorders, coming a long way from their first practical application formulated by Grineva in 1967 [68]. Grineva proposed a method of attaching active chemical groups to oligonucleotides to direct these groups toward a target nucleic acid, which she called “of complementary-addressed modification.” It took another 32 years before the first ASO therapy was approved by the FDA, in 1998. Since then, a total of 8 FDA-approved ASOs are on the market [69]. ASOs are single-stranded synthetic nucleic acids, generally 12–30 nucleotides in length, that bind sequence specifically to target RNA and can modulate protein expression through various mechanisms. They are generally categorized as having mechanisms promoting RNA cleavage and degradation or steric blocking. How ASOs modulate RNA depends on the function of the targeted RNA, where the ASO is designed to bind, and the chemistry and design [70]. The broad range of target options that ASO therapies offer renders them an intriguing treatment modality for common mutations in known ALS genes.
SOD1
The papers describing the first ASOs designed to lower SOD1 levels were published in 1994. These ASOs were developed to study the role of SOD1 loss in motor neuron death [71, 72]. A decade later, Miller and colleagues [73] published a study of the first ASO that was designed to be a therapeutic strategy for SOD1-ALS. Presymptomatic SOD1G93A rats carrying a human SOD1 mutation variant, which is expressed 8–16-fold above endogenous SOD1, received intracerebroventricular (ICV) injections of an ASO against human SOD1 called ISIS 333611. This ASO reduced WT and mutant SOD1 mRNA through RNase H activity and extended survival from 122 to 132 days [73]. These results led to a phase I clinical trial (NCT01041222] in which ISIS 333611 was tested in SOD1-ALS patients in a clinical trial by Ionis to assess its safety, tolerability, and pharmacokinetics. A single intrathecal infusion of ISIS 333611 was well tolerated with no serious adverse events, but at the low doses used, there were no reductions of SOD1 protein in the cerebrospinal fluid (CSF) of treated patients [74]. Ionis halted the testing of this ASO in favor of a newer generation of ASOs, which were initially tested in SOD1G93A rats and SOD1G93A mice. Median disease onset in treated SOD1G93A mice was delayed by 43 days, and survival was increased from 168 to 205 days [75]. Treated SOD1G93A rats lived 64 days longer [75] (Table 1 and Fig. 1), a marked increase in survival compared to ISIS 333611-treated rats [73]. Moreover, after treatment was initiated, there was an increase in the amplitudes of compound muscle action potentials, which had begun to fall prior to treatment. After these successful results, the new generation ASO, Tofersen (BIIB067), was tested in a phase I/II study (NCT02623699; sponsored by Biogen) in SOD1-ALS patients. Patients received 5 lumbar intrathecal injections over 12 weeks, with different treatment groups being given a 20, 40, 60, or 100 mg dose per injection. The primary outcome of this trial was safety and pharmacokinetics. All patients, including the placebo group, reported adverse effects, although most were related to the intrathecal lumbar punction, such as headache and post-lumbar puncture syndrome. Nevertheless, in general, the trial demonstrated safety at all doses. Furthermore, CSF SOD1 protein levels were dose-dependently reduced by up to 36% in the 100 mg treated group. Additionally, levels of CSF neurofilaments, biomarkers for ALS neurodegeneration, were decreased in the 100 mg treated group. In a small cohort of patients treated at 100 mg, there was a decline in disease progression, measured by the revised ALS functional rating scale (ALSFRS-R). This decline was apparent in both slow- and fast-progressing groups, but was more pronounced in the fast-progressing group [76]. After these successful results, a phase III, randomized, double-blind clinical trial named VALOR (NCT02623699) was initiated in 2019. This 28-week trial was designed to evaluate the efficacy, safety, tolerability, and pharmacodynamic effects of 100 mg Tofersen on SOD1-ALS patients. The study population consisted of 56% fast-progressing patients and 46% slow-progressing patients. It was reported at the American Neurological Association 2021 Annual Meeting that the primary endpoint as measured by ALSFRS-R was not met after 28 weeks; treatment did not significantly slow disease progression (p value of 0.97) [77, 78]. Nevertheless, both SOD1 levels and neurofilament levels were strongly decreased after 28 weeks in the treated groups. Furthermore, the VALOR trial has an open-label extension in which patients who were treated from the outset (week 0 onwards) demonstrated better results than those patients who did not receive Tofersen until later (only starting Tofersen after receiving a placebo treatment for 28 weeks). The first group demonstrated lower declines in motor and respiratory function, muscle strength, and quality of life after the first 28 weeks [77]. It remains unclear why the VALOR trial failed, given that there was a decrement in both CSF SOD1 and neurofilament levels. This may have reflected the relatively short length of the study. It is also possible that by the time treatment was started, the complex downstream pathological events were self-sustaining independently of levels of mutant SOD1 protein; that is to say, effective intervention may only be possible earlier in the disease course. For example, the fast-progressing group started treatment at an average of 8.3 months after diagnosis, more than half-way through the expect survival for the SOD1A5V rapidly progressing cohort. A new clinical phase III trial (ATLAS, NCT04856982) is currently ongoing, in which presymptomatic adult carriers of a SOD1 mutation with elevated neurofilament (NF) will be treated with 100 mg Tofersen via intrathecal injection on days 1, 15, and 29, and every 28 days thereafter for up to 2 years [79]. This trial will likely address the challenge optimal timing of trial initiation early in the therapeutic window.

Age at treatment plotted against extension of survival in treated SOD1G93A mice (Table 1)
C9ORF72
Soon after the discovery that the hexanucleotide expansion (HRE) in the C9ORF72 gene is the most frequent genetic cause of ALS, multiple ASO-based treatments targeting C9ORF72 mRNA were published [80,81,82,83,84,85]. ASOs can penetrate the nucleus and so have the potential to target both newly transcribed pre-mRNAs as well as those sequestered in RNA foci. Since only the lesser expressed V1 and V3 C9ORF72 transcript variants carry the HRE, ASOs can be designed to target only those transcripts by targeting exon 1 or intron 1, without drastically lowering overall C9ORF72 expression. But, regardless of whether they target transcripts harboring transcripts selectively or all transcripts, ASOs were shown to blunt toxic, acquired adverse consequences of the HRE, including the deposition of RNA foci [80,81,82,83,84,85] and toxic polydipeptides translated via non-AUG start codons (RAN translation) from the expansion [82, 84, 85]. This was documented using induced pluripotent stem cells (iPSCs) [80, 81, 86], C9-patient-derived fibroblasts [81, 83, 86,87,88], C9 mouse cortical primary neurons [82, 86], and mouse models [84,85,86, 88]. Notably, one study showed a reduction in the number of inclusions immunopositive for pTDP-43 and ataxin-2 after treatment with an ASO targeting C9ORF72, which validates that C9 HRE pathologies are associated with TDP-43 pathology [85].
In 2018, Ionis and Biogen began the first clinical trial testing ASOs against C9ORF72 (NCT03626012). This trial tested an ASO designated BIIB078, or IONIS-C9Rx. The primary objective of this phase I study was to evaluate the safety and tolerability of BIIB078 in C9-ALS patients. The secondary objective of this study was to evaluate the pharmacokinetic profile of BIIB078 and its effects on clinical function measured by slow vital capacity, hand-held dynamometry, the Iowa Oral Pressure Instrument, and the ALSFRS-R. There were 6 treatment cohorts assessed in the dose-escalating trial, and BIIB078 was administered intrathecally. In 2022, it was announced that although BIIB078 at 60 mg was generally well tolerated, it did not meet any secondary efficacy endpoints and did not demonstrate clinical benefit compared to the placebo group. Participants in the BIIB078 90 mg dose cohort trended toward a greater decline than those in the placebo group across secondary endpoints. Based on these results, the BIIB078 clinical development program, including its ongoing open-label extension study, was discontinued [89].
Wave Life Sciences started a phase Ib/IIa trial called FOCUS-C9 (NCT04931862) to test their ASO, WVE-004. WVE-004 is a stereopure ASO targeting C9ORF72 mRNA variants carrying the HRE. Preclinical testing of WVE-004 in C9-BAC transgenic mice showed reductions of repeat-containing mRNA by 60–80% in spinal cord and 40–50% percent in cortex up to 6 months later. WVE-004 also diminished dipeptide repeat proteins in C9-BAC mice by approximately 90% in the spinal cord and cortex but did not affect overall C9orf72 protein levels [90]. The FOCUS-C9 trial will assess safety and tolerability of WVE-004 in C9-ALS/FTD patients. Each participant is to receive a single intrathecal injection of one of four doses of WVE-004 or a placebo, with the potential to proceed to a multi-dosing phase after 10 weeks. The primary outcome is the percentage of patients with adverse events 24 weeks after injection. The trial will also measure WVE-004 levels and GP polydipeptides in CSF at the beginning and end of the study. The study is still ongoing, and no interim analysis was published before the writing of this review. The FOCUS-C9 trial is scheduled to be completed at the beginning of 2023.
In a parallel program, a team at UMass Chan Medical School developed an ASO targeting intron 1 adjacent to the HRE, degrading specifically repeat-containing C9ORF72 mRNA variants by RNase H activity. Treatment with ASO5, which was designed by Jonathan Watts, resulted in a marked reduction of RNA foci and toxic polydipeptides, while conserving overall C9ORF72 levels in patient-derived fibroblasts and C9-BAC transgenic mice [88]. Modification of a subset of the phosphodiester internucleoside linkages significantly improved the ASO tolerability without impairing potency [86, 88]. Treatment with the modified ASO5-2 in mice had no effect on total C9 transcript levels but reduced mutant-bearing HRE transcripts by ~80%. GP polydipeptides levels were lowered by 90%, which was sustained for 20 weeks after a single-dose ICV injection. In sheep, the lead ASO5-2 was well tolerated for 1 month following intrathecal administration. Intrathecal delivery of ASO5-2 to cynomolgus monkeys treated with 0, 1.5, or 6 mg of ASO5-2 at days 1, 14, 28, 57, and 85 produced no behavioral or neurological deficits out to 90 days; necropsies at 90 days showed no pathological findings attributable to ASO5-2. Encouraged by these results, we treated one C9-ALS patient with ASO5-2 (Afinersen, IND141673). This patient, who has 2400 HRE repeats, had developed motor symptoms. His CSF revealed elevated GP polydipeptide levels. He was treated with escalating doses of ASO5-2, beginning at 0.5 mg/kg and progressing to 2.0 mg/kg. This was safely tolerated. After sequential doses of 2.0 mg/kg, the relative CSF GP polydipeptide levels was reduced by approximately 80%, During the roughly 1 year of treatment, the patient’s ALSFRS-R score was largely stable. It is not possible to document clinical efficacy in a single anecdotal case like this. It will be important to discern whether ASO5-2 or the Wave ASO WVE-004 shows sustained good safety profiles and to determine whether these ASOs demonstrate clinical benefit or, like the Biogen ASO BIIB07, encounter clinically adverse features.
FUS
The first ASO treatment for FUS was implemented by Neil Shneider and his team at Columbia University, deploying an ASO originally developed by Ionis Pharmaceuticals [91]. The lead ASO, ION363 (also labeled jacifusen in honor of the first treated case) is directed against intron 6 of FUS and silences FUS in a non-allele-specific manner. In the extensive preclinical studies leading to the ASO trial, Shneider and colleagues first developed two FUS mouse models, one with a mouse FUS P517L mutation, which is equivalent to the human FUS P525L mutation, and another with a mouse FUS Δ14 mutation, which is equivalent to the human G466VfsX14 C-terminal FUS truncation mutation that causes skipping of FUS exon 14. Newborn MN-P517L/Δ14 mice received a single ICV dose of 20 μg of ION363. After 4 months, untreated MN-P517L/Δ14 mice had lost 12% of motor neurons and demonstrated both denervation and microgliosis. By contrast, ION363-treated mice, in which FUS brain FUS levels were reduced, did not exhibit these markers of disease progression. After 6 months, as ION363 concentrations declined, FUS levels returned to normal in the treated mice, which now revealed an increase in muscle denervation and microgliosis. Nevertheless, no motor neuron degeneration was observed in ION363-treated MN-P517L/Δ14 mice at 6 months after treatment.
These encouraging results paved the way for a human study. A 25-year-old FUS-ALS patient with a rapidly progressive disease course triggered by the FUSP525L mutation was treated with ION363 under the FDA compassionate use protocol. The treatment began > 6 months after clinical onset, at which point the patient was already not ambulatory and required ventilatory support (ALSFRS-R score 17). She received ascending intrathecal doses of ION363, starting with 20 mg up to a maximum monthly dose of 120 mg, for a total of 12 infusions over 10 months. The treatment was well tolerated without serious adverse events. Before treatment, the patient lost ~5 points on the ALSFRS-R score per month. In contrast, during the course of therapy, the rate of decline in the ALSFRS-R score slowed substantially. The patient died nearly a year after initial treatment from worsening of ventilatory and bulbar dysfunction. Immunohistochemical studies demonstrated broad distribution of ION363 throughout the patient’s CNS. Both total and P525L-mutant FUS protein were reduced by 90% or more in the brain, and pathological FUS aggregation was decreased throughout the CNS. Soluble FUS and other insoluble aberrant RNA-binding proteins, including TDP43, were also strongly reduced, but total levels of these RNA-binding proteins were unchanged, suggesting their redistribution toward a more soluble and WT state [91].
Based on the promising preliminary evidence in support of ION363 lowering FUS levels and slowing disease progression, 10 more patients received ION363 under the compassionate use program [92]. In 2021, this led Ionis Pharmaceuticals to start a phase III clinical trial (NCT04768972), led by Shneider, to determine whether ION363 provides any clinical benefit of slowing disease progression in symptomatic FUS-ALS patients. Subjects will be treated with intrathecal injections every 4–12 weeks over a 61-week period, and every 12 weeks for 85 weeks in the open-label extension treatment period. The study is set to finish early 2024 [93].
Additional ASO Approaches
Studies of ALS pathophysiology have disclosed other targets amenable to ASO-based gene suppression therapy. One well-studied target is ataxin-2 (ATXN2), whose mutations were detected as the cause of progressive, familial spinocerebellar ataxia type 2 (SCA2). The offending lesion is an expansion of CAG tracts encoding polyglutamine within the ATXN2 gene. While most individuals possess 22–23 CAG repeats, those with SCA2 have more than 34 repeats. Remarkably, in individuals with intermediate length expansions of 27–33 repeats, there is an 11-fold increased risk of developing ALS. Seminal investigations in both yeast and Drosophila found that ATXN2 promotes aggregation and toxicity of TDP-43 protein, while reducing levels of ATXN2 suppresses TDP-43 toxicity [94]. Although > 95% of ALS patients demonstrate abnormal TDP-43 protein [31], silencing TDP-43 itself is likely not an option because of its critical cellular functions [36]. Decreasing expression of ATXN2 with an ASO, however, could benefit most people with ALS. In a preclinical study, newborn TDP-43Tg/Tg mice treated with an ATXN2 ASO (developed by Ionis to treat SCA2) demonstrated 77% reduction of Atxn2 in TDP-43Tg/Tg mice, while human TARDPB mRNA levels were unaffected, and improvement in both motor performance and median survival (35%) [95]. In 2020, these positive results led to a phase I study by Biogen (NCT04494256) in which ALS patients, with and without a CAG repeat expansion in ATXN2, will receive one of four doses of BIIB105 by intrathecal injection. The primary outcome is to identify potential adverse events, and pharmacokinetics of BIIB105 is a secondary outcome. The study is set to end in 2024 [96]. Other genetic targets of potential relevance ALS include the p75 neurotrophin receptor [97], acetylcholinesterase [98], microRNA155 [99], and bone morphogenetic protein 4 [100]; in each instance, the corresponding ASOs have significantly prolonged survival in the SOD1G93A transgenic ALS mouse model.
Other RNA-Based Therapeutics
Small Activating RNAs
Although mutations in the major ALS-causing genes, such as SOD1, FUS, and C9ORF72, are mainly associated with GOF pathology, there are various ALS-causing and ALS disease-modifying genes that demonstrate LOF pathology when mutated. Recently, a study was published by Zhang et al. [101] in which 690 ALS-associated genes were identified by a machine-learning approach called RefMap. Remarkably, most of those genes showed LOF pathology, suggesting that gene therapy approaches to restore these genes’ functions would be warranted. One promising approach seeks to mediate gene expression by using small activating RNAs (saRNAs) [102] on the non-mutated allele. saRNAs are a class of small double-stranded non-coding RNAs. They can induce gene expression at a transcriptional level by targeting promoter sequences [103] or gene antisense transcripts [104]. saRNAs can be delivered in vivo by lipid nanoparticles, dendrimers, lipopolyplexes, and aptamers [102]. An saRNA treatment for advanced liver cancer, MTL-CEBPA, has been tested in a phase I clinical trial. MTL-CEBPA was encapsulated by lipid nanoparticles and was well tolerated [105]. saRNA treatment has also been shown to upregulate gene expression in neurons. A preclinical study on RETT syndrome found successful Foxg1 gene upregulation in mice treated with an intraventricular injection of an saRNA targeting Foxg1 [106]. Transient induction of LOF ALS genes using the saRNA system could hold promise for pharmacological approaches to ALS treatment.
AIMers
Wave recently published an exciting method to edit single base mutations in RNA transcripts without causing the permanent changes to the genome that occur with DNA-targeting approaches. AIMers are short, chemically modified oligonucleotides that direct A–I editing of endogenous transcripts by endogenous adenosine deaminases acting on RNA (ADAR) enzymes. ADAR enzymes edit adenine bases to inosine, which the translational machinery reads as guanine. To test this method in vivo, cynomolgus monkeys received subcutaneous injection of AIMers targeting the ACTB gene, once a day for 5 days. Liver biopsies 2 days and 45 days post dose showed 48.1% and 39.1% editing, respectively. There were no signs of hepatoxicity at 2 days post dose, when AIMer levels were highest. RNAseq showed that the AIMers were highly specific and no bystander editing was detected in the ACTB transcript. To confirm that AIMer editing with endogenous ADAR enzymes can restore expression of proteins, these investigators designed AIMers to edit SERPINA1 mRNA that has a E342K missense mutation, which causes α1-antitrypsin deficiency. In vitro editing resulted in mean editing percentages of 68–75%, and protein expression was significantly increased [107]. This method has potential to edit ALS-causing single base mutations. Although ADAR2 has been found to be downregulated in motor neurons of ALS patient, ADAR1 levels remained unaffected [108]. AIMers can be designed to work with ADAR1 or ADAR2, but ADAR1 AIMers performed better [107], so low levels of ADAR2 in ALS motor neurons should not be an issue when utilizing this method to treat ALS. There are to date 35 G > A disease-causing mutations found in SOD1 [21], 14 for FUS [43], and 14 for TARDBP [109] that could hypothetically be corrected by AIMers.
Viral Vectors
Viral vectors offer advantages for CNS disease therapy as they obviate the need for repeated invasive treatments. In theory, viral vector therapy is “one and done.” Diverse viral vector platforms have been developed and are continuously being improved for gene therapy and other applications. Among the main viral vector platforms are lentivirus, herpes virus, adenovirus, and in particular adeno-associated virus (AAV). AAV is a parvovirus that was discovered as a contaminant in adenovirus production [110]. It is found in vertebrate species, including human and non-human primates but has not been observed to cause any human diseases [111]. It is ~25 nm in size and has a capsid and a single-stranded DNA genome of ~4.7 kb [112]. Recombinant AAVs do not express AAV protein-coding sequences, but encapsidate therapeutic gene expression cassettes in their place. AAV has a packaging capacity of ~4.7-kb single-stranded genome and half that for double-stranded self-complementary cargo [113]. Once the gene expression cassette has been delivered to the cell, AAV remains episomal. AAV sequences do not typically integrate into the host genome [114], although in some usages (such as CRISPR/Cas9 editing), there may be some incorporation of AAV fragments into the host genome [115]. The only sequences with a viral origin are the inverted terminal repeats (ITRs) that are necessary for guiding genome replication and packaging during vector production [111].
The first recombinant AAV expression was demonstrated in vivo in 1993 [116], and the first human trial using AAV began in 1995 [117]. The use of AAV offers several advantages for treatment of ALS. Evolutionary, some viruses have evolved methods to breach the blood–brain barrier (BBB) to invade the CNS, an aspect that could be exploited in therapies for ALS. The two preferred serotypes for CNS delivery are AAV serotype 9 (AAV9) and rhesus monkey10 (AAVrh10), due to their strong tropism preferences for the CNS, particularly after delivery directly to the cerebrospinal fluid (CSF) [118,119,120]. Importantly AAV can transduce nondividing and dividing cells. This renders it particularly appropriate for post-mitotic cells such as neurons. In actively dividing cells, the therapeutic cargo may be diluted as the cell numbers increase, leading to only a transient therapeutic effect [111]. AAV can be used for gene augmentation, gene knockdown, gene editing as well as exon skipping.
AAV-Mediated Gene Silencing
SOD1
Several gene therapy strategies delivered by AAV aimed at reducing SOD1 have been developed in recent years (Fig. 2). These strategies include the use of short hairpin (sh) RNA [121,122,123], miRNA [124,125,126,127,128,129], AAV-delivered antisense sequences [130], CRISPR Cas9 [131,132,133], and antibodies [134,135,136] targeting SOD1. The first approach published was an shRNA against SOD1 delivered by AAV9 [121]. Treatment of SOD1G93A mice and WT non-human primates demonstrated a robust reduction of SOD1 in the spinal cord. Intravenous injections of AAV9-sh-SOD1 in presymptomatic (P1 and P12) and early symptomatic (P85) SOD1G93A mice delayed disease onset, improved motor performance, and extended survival by 30–51.5 days, with better results in earlier treated animals [121]. In a next study, they treated neonatal SOD1G93A mice with intrathecal injections via the cisterna magna, which resulted in a stronger effect rescue of disease onset, motor performance, and survival (61.5 days) [122]. Intravenous treatment of AAV9-sh-SOD1 in slower-progressing symptomatic SOD1G37R mice resulted in an increased motor performance and survival (86.5 days) [121]. Despite these positive results, Novartis Gene Therapies, which was involved in these studies, halted the development of AAV9-sh-SOD1 in 2020 [137].

Summary of gene therapy strategies reducing gain-of-function pathology used for ALS. a ASO strategies to induce RNase H-mediated degradation of mRNA. b AAV-mediated mRNA silencing through shRNA or miRNA RNA interference. c AAV-mediated gene silencing through CRISPR-Cas9. d AAV-mediated delivery of antibodies targeting misfolded protein. RISC, RNA-induced silencing complex; PAM, protospacer adjacent motif; sgRNA, single-guide RNA. Created with BioRender.com
We have explored the potential of artificial microRNAs (amiRNAs) to suppress SOD1. Mueller, Sena-Esteves, and others on our team designed several silencing constructs and tested different AAV serotypes, promoters, treatment routes, age at treatment, and animal models to design a safe and effective therapy to lower SOD1 levels. AAV9-amiRSOD1 ICV injections in neonatal SOD1G93A mice increased median survival by 69 days and prevented hindlimb paralysis [125]. As most ALS patients are diagnosed in adulthood, a subsequent study treated presymptomatic adult SOD1G93A mice (by intravenous tail vain injections). Treatment with the U6 promoter-driven construct resulted in substantially delayed disease onset, an increased survival of 27 days, preservation of limb strength and motor skills, and stabilization of respiratory physiology [124]. Treatment of marmosets with AAVrh10-U6-miR-SOD1 via lumbar intrathecal injection reduced SOD1 in motor neurons by 93% in the lumbar, 65% in the thoracic, and 92% in the cervical spinal cord regions; similar levels of silencing were observed in the non-motor neuron tissue [124]. In a subsequent study using cynomolgus macaques, we found 45–65% SOD1 silencing within motor neurons in different parts of the spinal cord. The procedure was well tolerated during both the infusion and the post-procedure period, monitored up to 92 days after the procedure [126]. These promising preclinical results culminated in the first human study. Two SOD1-ALS cases each received intrathecal infusions of 4 × 1014 genomes of AAVRh10.miR-SOD1 using an FDA-approved protocol. The first case, a 22-year-old man harboring the SOD1A5V mutation, was treated ~6 months after symptom onset. At infusion, he received prednisone, but nonetheless developed an adverse CNS inflammatory response and elevation of liver enzymes ~5 weeks after the infusion; over the ensuing 15–20 weeks, these responded to increased and then tapering doses of prednisone. His ALSFRS-R score, which initially was 42, fell to 38 at 6 months after treatment. Weakness of his left arm and leg, which heralded disease onset, continued to progress, but other functions, such as right knee extension, showed improvement. At 14 months after the treatment, he recovered minimal finger flexion and extension in the left hand. Unfortunately, his respiratory function (gauged by his vital capacity) declined relentlessly, and he died of respiratory arrest 15.6 months after treatment. At autopsy, spinal cord SOD1 protein levels were reduced by ~90% as compared to both healthy controls and untreated individuals with the same mutation. Strikingly, there was notable preservation of motor neurons in the right lumbosacral spinal cord. Because of the inflammatory response in the first case, the second individual in this study, a 56-year-old man with the SOD1D91A/D91A mutation, received immunosuppression therapy. Prednisone, intermittent rituximab, and sirolimus were administered for 6 months beginning at the time of infusion. On this regimen, this person did not develop an inflammatory response. His disease course was generally stable, although his ALSFRS-R score reduced from 28 to 24 throughout the 90-week period reported [127]. Further studies of this approach will be undertaken by ApicBio, which is planning a phase I/II clinical to begin in late 2022 [138].
Another interesting approach to silence SOD1 used an AAVrh10 that expressed an exon-2-targeting antisense sequence to skip exon 2 to generate a premature stop codon. This strategy combines the properties of ASO and AAV vectors, preventing the need for re-administration. Combined intracerebroventricular and intravenous treatment increased the survival of SOD1G93A mice injected either at birth or at P50 by 121 and 63 days, respectively. It also prevented weight loss and the decline of neuromuscular function for both neonatally and adult injected mice [130].
Various groups have developed CRISPR/Cas9 strategies delivered by AAV to disrupt SOD1 expression. Intravenous injections of AAV9 expressing Staphylococcus aureus (Sa) Cas9 and a gRNA targeting SOD1 in neonatal SOD1G93A mice extended survival by 30 days and significantly improved motor function [132]. Another study using a similar approach found 71 days increased survival after treating neonatal SOD1G93A mice with intracerebroventricular (ICV) injections [131]. Since both groups used the same serotype, dose, and SaCas9, the difference in survival could be explained by the use of a different administration route or the effectiveness of the gRNAs used. Another approach to suppress SOD1 protein levels was published by the Gaj group in 2020 [139], which used cytosine base editors. Cytosine base editors convert C–G to T–A in the DNA using the CRISPR system without making double-stranded breaks, thereby eliminating the likelihood of insertions, deletions, DNA translocations, and DSB-induced toxicity. Base editors can be used to correct disease-causing mutations but in this study were used to generate nonsense mutations in SOD1. Because of the limited packaging capabilities of AAV and the large size of Streptococcus pyogenes (Sp) Cas9, the Gaj group developed a resourceful split-intein system which used two AAV9 vectors expressing the two halves of a SpCas9-based cytosine base editor fused to intein fragments that are reassembled in vivo via trans-splicing. SOD1G93A mice were treated intrathecally with the AAV9 base editor. Deep sequencing of the spinal cord showed that only 1.2% of reads showed the correct C > T edit. High percentages of bystander editing, editing of non-target bases, were found in treated HEK293T cells. Despite the low correct editing efficiency, treatment in adult SOD1G93A mice resulted in a 40% reduction in SOD1 inclusions, a 12-day extension of survival, and improvement of neuromuscular function [139].
While the above strategies targeted DNA and RNA, AAV can also be utilized to deliver a DNA construct encoding an antibody against SOD1 protein. Intrathecal delivery of AAV2/1 expressing a monoclonal antibody targeting specifically misfolded SOD1 to P45 SOD1G93A mice resulted in a survival increase of 16 days. Misfolded spinal SOD1 as well as neuronal stress and gliosis were reduced [134]. Another study treated neonatal and P120 SOD1G93A mice with an intravenous injection of an AAV9 expressing an antibody also targeting misfolded SOD1 and found an extension of survival of 27 days for neonatally treated mice and 14 days for P120 treated mice [136]. Of all strategies discussed in this paragraph, only these two studies that used antibodies to target misfolded SOD1, specifically targeted pathological SOD1. The other studies utilizing shRNA, miRNA, CRISPR, and ASO delivered by AAV did not distinguish between mutant and wild-type SOD1 in their in vivo studies, although some studies demonstrated in vitro the possibility of specifically targeting mutant SOD1. To provide a more universal therapy for SOD1 patients, the in vivo studies mostly targeted SOD1 regions in which no mutations had been described. This could, however, come with a cost, as SOD1 LOF plays a modifying role in ALS [27, 28].
TDP-43
As mentioned earlier, silencing all TDP-43 may be problematic because of its many cellular functions [36]. One strategy for lowering TDP-43 was published in 2019 [140], which did not show overt toxicity in vivo. A single-chain antibody targeting an aggregation-prone region of TDP-43 was delivered by an AAV9 vector and tested in symptomatic TDP-43G348C mice. These mice demonstrate cytoplasmic accumulation of TDP-43 and memory impairment. Treatment reduced TDP-43 proteinopathy, microgliosis, motor defects, and cognitive impairment. In addition to reducing cytoplasmic insoluble TDP-43, the antibody also significantly reduced nuclear TDP-43, which has many critical cellular functions [36]. Nonetheless, the authors noted that the mice remained healthy after treatment; further, treatment in HEK293T cells did not affect cell survival [140].
C9ORF72
UniQure published two studies in 2019 [141, 142] in which they used amiRNAs targeting C9ORF72 that were delivered by AAV5. They first used a bidirectional targeting approach in vitro, by expressing two concatenated microRNA hairpins targeting both sense and antisense C9ORF72. Such a bidirectional approach is in theory advantageous for a C9ORF72 treatment, as RNA foci and toxic polydipeptides are formed by both the sense and the antisense strands. Surprisingly, the two concatenated amiRNAs did not perform better than single amiRNAs in HEK293T cells [141]. In a subsequent study, the UniQure team treated iPSC-derived frontal neurons harboring the offending C9ORF72 hexanucleotide expansion with two types of amiRNAs: (1) amiRNAs that target all C9ORF72 transcripts and (2) amiRNAs targeting intron 1 just before the HRE. Both types of amiRNAs reduced intronic C9ORF72 by ~ 30%, but overall C9ORF72 levels were only spared in intron-1 targeting amiRNA-treated cells. They next treated C9ORF72 mutant BAC transgenic mice with amiRNAs targeting all C9ORF72 transcript variants and demonstrated significant silencing of repeat-containing C9ORF72 and total C9ORF72 transcripts, and a significant reduction of RNA foci after treatment [142].
An alternative way to treat the toxic GOF pathology of C9ORF72, RNA foci, and toxic polydipeptides is to target the double-stranded RNA-dependent protein kinase (PKR) pathway, instead of directly targeting C9ORF72. The PKR pathway can phosphorylate the translation initiation factor eIF2 on its α subunit (eIF2α), which impairs translational initiation of most proteins while provoking the translation of a selective set of mRNAs [143]. C9ORF72 RAN-positive repeat expansion RNAs can activate the PKR pathway, and inhibition of PKR can decrease in turn RAN protein levels. ICV treatment of neonatal C9-500 mice with a dominant-negative AAV2/9-PKR-K296R resulted in a 50% and 60% reduction of poly-GA and poly-GP, respectively [144], making this an intriguing gene therapy alternative for C9ORF72-ALS.
AAV-Mediated Gene Correction
C9ORF72
Unlike most ALS genes that can have many different disease-causing mutations, only the HRE in C9ORF72 has been documented to cause pathology. This makes the C9ORF72 gene a strong candidate for gene correction. We developed an AAV9-mediated CRISPR/Cas9 gene-editing approach to remove the GGGGCCn HRE using gRNAs targeting sequences flanking the HRE.
This gene-editing approach was tested in vivo in three different C9-BAC transgenic mouse models by striatal injections and show that HRE excision resulted in a strong reduction of C9ORF72-related GOF pathology. Due to the large size of SpCas9 and the small packaging capability of AAV, adult mice received striatal injections with both an AAV9 expressing the Cas9 and an AAV9 expressing the gRNAs. Staining for both vectors demonstrated that > 50% of cells in the striatum were transduced with both. There was a ~50% reduction of poly-GP and poly-GR and a >50% reduction of RNA foci, but no effect on overall C9ORF72 mRNA and protein levels after in vivo treatment. In C9-ALS patient, iPSCs generated an isogenic cell line, in which the HRE was edited out of the expanded allele in all cells. This resulted in a complete obliteration of poly-GP and a significant 50% increase of C9ORF72 mRNA and protein levels. One of the drawbacks of using CRISPR/Cas9 is the potential for insertions and deletions at the editing site. In this study, this risk is blunted by targeting the intron, wherein insertions and deletions are not likely to change amino acid composition of the C9ORF72 protein or change transcription levels. A similar approach was recently published in which C9-BAC transgenic mice expressing Cas9 were treated with AAV1/2 gRNAs targeting the flanking regions of the HRE, which resulted in a reduction of RNA foci [145].
AAV-Mediated Gene Delivery
Although few of the major ALS-causing mutations arise from loss of function of the mutated ALS genes, AAV-mediated gene delivery holds potential for the ALS field. The vast majority of ALS cases are sporadic, in whom the exact pathological mechanisms underlying motor neuron death are not well understood. One strategy to treat both familial and sporadic ALS is to activate neuroprotection mechanisms through AAV-mediated delivery of neurotrophic factors and neuromuscular junction modulators. Neurotrophic factors regulate several physiological processes, such as neuronal differentiation and survival, neurogenesis, synapse maintenance, and axonal outgrowth [146], which makes them a promising strategy to treat ALS. Despite encouraging results in preclinical models, many different neurotrophic factors and other growth factors have been tested in clinical trials without success. Reasons for their failure are numerous. Some neurotrophic factors could not penetrate the BBB; some have a short half-life, some did not achieve therapeutic target tissue levels, and tissue levels were variable over time [146, 147]. AAV-mediated delivery of neurotrophic and growth factors have the potential to address those issues.
Treatment with an AAV expressing insulin-like growth factor 1 (IGF1) in early symptomatic SOD1G93A mice using different serotypes (AAV1, 2, 4, 9) and diverse administration routes (deep cerebellar nuclei, ICV, intramuscular, intravenous injections) resulted in comparable outcomes in different studies. Survival in treated mice (predominantly the SOD1G93A transgenic mice) in many studies was modestly increased (9–15 days) with improved muscle strength [148,149,150,151]. Expression of IGF1 also resulted in a strong decrease of astroglial and microglial responses and rescue of motor neurons in the spinal cord of SOD1G93A mice [148, 150, 151]. One of the studies compared ICV treatment of SOD1G93A mice with an AAV4 expressing IGF1 and an AAV4 expressing vascular endothelial growth factor (VEGF) and found a slightly improved survival (12 vs. 20 days). When given in combination, no additional benefit was observed, suggesting that IGF1 and VEGF likely activate a common signaling pathway that is therapeutically beneficial [149]. Early studies of AAV-mediated glial-derived neurotrophic factor (GDNF) delivery showed promise, extending the lifespan of SOD1G93A mice by 17 days after four-limb intramuscular delivery [152]. A more recent study used a systemic approach in which SOD1G93A rats were treated with AAV9-GDNF intravenous injections. This report documented that upregulation of GDNF outside the muscular system caused adverse effects, such a decrease in working memory, activity levels, and weight. While treatment had no effect on survival, the rats demonstrated modest improvement in strength and a delay of forelimb paralysis [153]. A study in 2021 used intravenous injections to overexpress GDNF with muscle-specific promoter [154], delaying progression of disease, preserving motor function, and reducing glial activity without prolonging survival. This strategy prevented the previously noted adverse cognitive effects [153]. AAV-mediated delivery of other neurotrophic factors and growth factors, such as granulocyte-colony stimulating factor and hepatocyte growth factor, produced moderately beneficial effects on survival with some improvement in motor function [155,156,157].
Macrophage migration inhibitory factor (MIF) has proven to inhibit mutant SOD1 misfolding specifically. Intraspinal injection of AAV2/9 in neonatal SOD1G93A mice resulted in a > 50% reduction of misfolded SOD1 in the spinal cord and an increase of survival of 30 days [158]. Another intriguing strategy to treat SOD1-ALS was published by Tung and colleagues who investigated endogenous motor neuron-enriched miRNAs and found that miR-17 ~ 92 is essential for embryonic distal limb-innervating motor neuron survival. Furthermore, miR-17 ~ 92 was strongly decreased in patient iPSCs and SOD1G93A mice. Intrathecal injection of AAV9-mir17 ~ 92 into presymptomatic adult SOD1G93A mice yielded a 23-day survival increase [159]. It must be said that these varied interventions have been tested almost exclusively in SOD1 animal models; their relevance has yet to be demonstrated in other ALS models; ultimately, it is hoped that they will be therapeutic in both familial and sporadic ALS patients. The authors also argue that this treatment might hold potential for other types of ALS, as impaired mir17 ~ 92 production could be a general phenomenon in familial ALS [159].
AAV-Mediated Gene Expression Activation
Suppressor tRNA Platform
As mentioned above, there are various ALS-causing and ALS disease-modifying genes that demonstrate LOF pathology when mutated [101]. About 11% of human pathogenic mutations are nonsense mutations [160], which are single nucleotide mutations that convert a sense codon to one of three stop codons (e.g., TAG, TGA, or TAA) in mRNA, resulting in incorrect termination of translation and generation of non-functional, truncated proteins. To read through premature termination codons to increase expression, a suppressor transfer RNA (tRNA) can be used. Suppressor tRNAs are derived from natural tRNAs, but have the anticodon modified to base pair with one of the three stop codons in the mRNA and to aminoacylate. This continues translation elongation through the premature termination codon and rescues full-length protein expression [161]. Suppressor tRNAs are limited in size and so can be delivered and expressed through AAV. A recent study on the lysosomal storage disease mucopolysaccharidosis type I showed successful gene restoration in vitro and in vivo in the Idua gene after AAV-mediated delivery of suppressor tRNA. Intravenous injections resulted in gene activity restoration to a level of 27% in WT mice, which almost completely normalized lysosome abundance in the liver [162].
There are several advantages to AAV-mediated suppressor tRNA delivery over AAV-mediated gene delivery. It avoids issues of cDNA cargo size constraints on packaging cargo in AAV. Furthermore, in contrast to gene replacement therapy, suppressor tRNAs only induce expression in cells that normally express the gene. Moreover, suppressor tRNAs operate on endogenous transcripts and therefore restore mRNA and protein levels under physiological expression regulation. This avoids toxicity related to “Goldilocks genes” in which both the absence and overexpression are detrimental to the cell, such as with TDP-43, for which research indicates a role in LOF pathology [36], while overexpression of WT TDP-43 causes toxicity [37, 38].
Tethered mRNA Amplifier
Another strategy is to increase mRNA expression at the post-transcriptional level by mimicking an mRNA poly(A) tail using a tethered mRNA amplifier. Poly(A) tails promote the process of translation through PABPC proteins (poly(A)-binding protein, cytoplasmic) that bind to the tail. A recent study found an increase of MeCP2 mRNA expression by tethering a known translational stimulator, PABPC1, to the 3′UTR of MeCP2. PABPC1 was fused to the RNA binding protein dCAS13b complex with a gRNA targeting the 3′UTR. In vitro MeCP2 protein levels were increased 1.5-fold after treatment. Up to twofold increase of protein expression was achieved for other genes associated with haploinsufficiency disorders. The authors minimized the mRNA amplifier to a size that is suitable for AAV for future in vivo delivery [163]. This strategy can be used for haploinsufficiency diseases in which one allele is unaffected by mutations, such as TBK1-ALS [164]. Like the suppressor tRNA platform, this strategy is potentially useful for enhancing the expression of large genes that cannot be packaged by AAV because of size constraints.
Challenges
Therapeutic Window
Several animal studies have concluded that early treatment correlates with functional benefit, a point that is likely to be central to the design of ALS gene therapies (Table 2). Various parameters merit consideration when determining the therapeutic window for ALS treatment. First, not all affected cell types and tissues have the same window. Post-mitotic motor neurons cannot be replaced after degeneration and, moreover, have limited capacity to regenerate; this dictates that treatment should ideally start before degeneration becomes irreversible. By contrast, non-neuronal cells in the CNS, which clearly impact the course of ALS (e.g., astrocytes, microglia, oligodendrocytes [165]), are not post-mitotic and so do have the ability to repopulate. Arguably, the window for therapeutic intervention in these non-neuronal cells may extend to later in the disease course than that for motor neurons. Second, the causes of ALS are highly heterogenous, as underscored by the diversity of ALS-causing mutant genes. Diverse ALS genes differ in their proximal pathological mechanisms; this is probably also true for different mutations in the same gene. One indicator of heterogeneity of mechanism is the variability in ages of onset and rates of disease progression. For example, ALS-causing FUS mutations are often more clinically severe than mutations in other ALS genes. Although the peak age of onset of ALS overall is 58–63 years and for familial ALS patients 47–52 years [166], the mean of age of onset for FUS patient is decades earlier at 21 years [167]. Moreover, the cases of FUSP525L frequently show onset in childhood. The survival in SOD1-mediated ALS can vary dramatically with the specific mutation. At one extreme, the mean survival for SOD1A5V is rarely more than 1 year, while survival of ALS cases in northern Sweden with the SOD1D91A/D91A genotype can be more than a decade [168]. A perplexing unknown in the field is the age at which the preclinical biological pathology begins. Because the offending mutations are present at conception, preclinical biological dysfunction could theoretically precede clinical manifestations by decades, raising the possibility that the optimal time to treat asymptomic gene carriers is prior to, or just at, disease onset. Confounding the matter is that in both USA and western Europe, the typical time from symptom onset to diagnosis is 10–16 months [169], a delay that significantly reduces the therapeutic window. This is of high consequence for ALS clinical trial design. It is impossible to know if a treatment such as gene therapy fails because it does not rescue biological function or because it is administered too late in the disease course.
Dosing: Blood-Brain Barrier and Repeated Administration
Several ASOs targeting ALS genes are currently in clinical trials, and some preliminary findings are promising. However, there are challenges associated with the use of ASOs for ALS or other neurodegenerative disorders. ASOs do not typically cross the BBB and thus must be directly infused into the CNS. This is potentially a problem when one must also consider the role of peripheral cells and tissues in ALS, such as skeletal muscle [170]. The ASOs in human trials described in this review all employed intrathecal delivery. Intrathecal injections not uncommonly trigger adverse findings such as post-lumbar puncture headaches due to persistent CSF leakage from the puncture site. Another hurdle is that ASOs require repeated administration. This requirement for regular treatments, combined with the challenges of intrathecal delivery, adds additional strain on already physically vulnerable ALS patients. Clearly, optimization of delivery systems and routes of administration will strengthen the therapeutic potential of ASOs. Various promising carriers and modifications, such as lipid nanoparticles [171] and cell-penetrating peptides [172], could address this.
Immunoreactivity to AAV and ASOs
A potential advantage of AAV-mediated gene modulation (silencing, editing, augmentation) is that the year-long episomal persistence of AAV, and the corresponding continuous expression of therapeutic cargoes, should eliminate the need for recurrent dosing. For this reason, as noted above, AAV therapy has been piloted in ALS, using intrathecal delivery. A recently reported study of gene augmentation therapy in Tay-Sachs disease combined intrathecal and intraparenchymal delivery [173]. Three substantial benefits of intrathecal administration are by-passing the blood–brain barrier, avoiding exposure to the peripheral immune system and using markedly lower doses than are required in attempted peripheral AAV delivery. However, while AAV is generally considered safe, clinically significant immune responses to AAV therapies have been observed in animals and in human studies, as in the first AAVRh10.miR-SOD1-treated patient described above [127]. The main components that trigger immune responses are the capsid, vector genome, and transgene. Furthermore, pre-existing neutralizing antibodies may reduce AAV vector delivery efficiency, making some serotypes unsuitable for certain patients [174]. Additionally, AAV-infected cells can be targeted by cytotoxic T lymphocytes, leading to transgene loss and cell death [174]. In studies in pigs, neuroinflammatory toxicity targeting the dorsal root ganglia has initiated severe neuronal cell body loss and immune cell infiltrates after intrathecal AAV delivery [175]. Fortunately, these points notwithstanding, it is encouraging that pilot studies suggest immunoreactivity can be attenuated and avoided with appropriate immunotherapy [127, 173].
By contrast with early AAV experience, initial studies with ASOs are, in general, not associated with strongly adverse immunologic responses. The FDA-approved ASO therapies have not stimulated major immune toxicity. That said, a note of caution is that there are infrequent references to meningo-radiculitis after intrathecal ASO treatments (e.g., [76]).
The issue of immunoreactivity to AAV arises in the context of CRISPR/Cas9 therapy, which has been associated with immune responses triggered by long-term expression of a bacterial protein (Cas9) [176]. Other important cautionary issues in AAV-mediated CRISPR/Cas9 therapy include increased genomic instability, off-target editing due to the sustained activity of an active nuclease [177], and high rates of AAV integration at the double-stranded break site [115]. These concerns have underscored the view that other delivery strategies are more appropriate for DNA editing, such as the delivery of Cas9 and gRNA ribonucleoprotein or mRNA by lipid nanoparticles [178].
Conclusions and Future Approaches
Therapies that suppress gene expression are emerging as a powerful therapeutic approach to treat many diseases, including ALS. Currently, six gene modulation therapies for ALS are in human trials: one ASO and one AAV targeting SOD1, one ASO targeting FUS, 2 ASOs targeting C9ORF72, and one ASO targeting ATXN2. The pilot studies of AAV therapy for SOD1 and ASO therapies for C9orf72 and FUS offer compelling evidence that the offending, dominantly acting genes have been suppressed, as reviewed above. On the other hand, while some promising clinical responses have emerged, most of the treated cases have succumbed to the disease. One can speculate at length about this, but two points are salient. First, in every case, the disease was well underway at the time of treatment, raising the possibility that the above-noted therapeutic window had closed. And second, in each case, both the mutant and the wild-type non-mutant alleles were suppressed. To the extent that the normal allele may confer some neuroprotective benefit, it may be desirable in the future to target the mutant allele selectively. Ideally, gene suppression therapies that target the mutated genes specifically, and thereby blunt acquired, gain-of-function pathologies, will not exacerbate potential loss of function consequences of the mutation. Selective allele suppression has been extremely challenging in preclinical studies. This has suggested that another approach will be to address both GOF and LOF pathologies by using an AAV dual-function vector in which a microRNA that suppresses both copies of the gene (mutant and wild type) is administered simultaneously with a copy of the same gene modified to be resistant to the microRNA; the latter is intended to replace the function of the suppressed gene. In preclinical proof-of-concept studies, this approach looks promising in therapy of alpha-1 anti-trypsin disease [179].
Immense progress has been made in the field of gene therapy and modulation of gene function over the past decade. Virtually all of the major new technologies have potential application to the treatment of ALS. These technologies, and early pilot studies in ALS, instill hope that gene therapy, either alone or in combination with other more conventional therapies targeting various pathological mechanisms in ALS, will significantly slow ALS disease progression. There is little doubt that continued study of gene modulation methods, vehicles, and strategies, and improved understanding of the associated therapeutic challenges and limitations, will build the foundation for clinical success.
References
Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4(1):3.
Masrori P, Beckers J, Gossye H, Van Damme P. The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD. Mol Neurodegener. 2022;17(1):22.
Zaepfel BL, Rothstein JD. RNA is a double-edged sword in ALS pathogenesis. Front Cell Neurosci. 2021;19(15):708181.
Obrador E, Salvador-Palmer R, López-Blanch R, Jihad-Jebbar A, Vallés SL, Estrela JM. The link between oxidative stress, redox status, bioenergetics and mitochondria in the pathophysiology of ALS. Int J Mol Sci. 2021;22(12):6352.
De Vos KJ, Hafezparast M. Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol Dis. 2017;105:283–99.
Evans CS, Holzbaur ELF. Autophagy and mitophagy in ALS. Neurobiol Dis. 2019;122:35–40.
King AE, Woodhouse A, Kirkcaldie MTK, Vickers JC. Excitotoxicity in ALS: overstimulation, or overreaction? Exp Neurol. 2016;275(Pt 1):162–71.
Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci [Internet]. 2019 [cited 2022 Mar 15];13. Available from: https://www.frontiersin.org/article/10.3389/fnins.2019.01310.
French PW, Ludowyke R, Guillemin GJ. Fungal neurotoxins and sporadic amyotrophic lateral sclerosis. Neurotox Res. 2019;35(4):969–80.
Vasta R, Chia R, Traynor BJ, Chiò A. Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine. 2021;30(75): 103795.
Peters OM, Ghasemi M, Brown RH. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125(6):2548.
Brenner D, Weishaupt JH. Update on amyotrophic lateral sclerosis genetics. Curr Opin Neurol. 2019;32(5):735–9.
Deng Z, Lim J, Wang Q, Purtell K, Wu S, Palomo GM, et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2020;16(5):917–31.
Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623–7.
Wong C, Stavrou M, Elliott E, Gregory JM, Leigh N, Pinto AA, et al. Clinical trials in amyotrophic lateral sclerosis: a systematic review and perspective. Brain Commun. 2021;3(4):242.
Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17(1):4–31.
Breiner A, Zinman L, Bourque PR. Edaravone for amyotrophic lateral sclerosis: barriers to access and lifeboat ethics. CMAJ Can Med Assoc J J Assoc Medicale Can. 2020;192(12):E319–20.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62.
Bernard E, Pegat A, Svahn J, Bouhour F, Leblanc P, Millecamps S, et al. Clinical and molecular landscape of ALS patients with SOD1 mutations: novel pathogenic variants and novel phenotypes. A single ALS center study. Int J Mol Sci. 2020;21(18):6807.
Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. 1986;247(1):1–11.
ALSoD [Internet]. [cited 2022 Mar 21]. Available from: https://alsod.ac.uk/output/gene.php/SOD1.
Yamashita S, Ando Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener. 2015;24(4):13.
Cleveland DW, Laing N, Hurse PV, Brown RH. Toxic mutants in Charcot’s sclerosis. Nature. 1995;378(6555):342–3.
Hayashi Y, Homma K, Ichijo H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul. 2016;1(60):95–104.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5.
Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–38.
Saccon RA, Bunton-Stasyshyn RKA, Fisher EMC, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136(8):2342–58.
Andersen PM, Nordström U, Tsiakas K, Johannsen J, Volk AE, Bierhals T, et al. Phenotype in an infant with SOD1 homozygous truncating mutation. N Engl J Med. 2019;381(5):486–8.
Park JH, Elpers C, Reunert J, McCormick ML, Mohr J, Biskup S, et al. SOD1 deficiency: a novel syndrome distinct from amyotrophic lateral sclerosis. Brain. 2019;142(8):2230–7.
Hayward C, Brock DJ, Minns RA, Swingler RJ. Homozygosity for Asn86Ser mutation in the CuZn-superoxide dismutase gene produces a severe clinical phenotype in a juvenile onset case of familial amyotrophic lateral sclerosis. J Med Genet. 1998;35(2):174–174.
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–9.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature. 2022;603(7899):124–30.
Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD. ALS genetics: gains, losses, and implications for future therapies. Neuron. 2020;108(5):822–42.
Wood A, Gurfinkel Y, Polain N, Lamont W, Lyn RS. Molecular mechanisms underlying tdp-43 pathology in cellular and animal models of ALS and FTLD. Int J Mol Sci. 2021;22(9):4705.
Yamashita M, Nonaka T, Hirai S, Miwa A, Okado H, Arai T, et al. Distinct pathways leading to TDP-43-induced cellular dysfunctions. Hum Mol Genet. 2014;23(16):4345–56.
Janssens J, Wils H, Kleinberger G, Joris G, Cuijt I, Ceuterick-de Groote C, et al. Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol. 2013;48(1):22–35.
Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–8.
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–11.
Picher-Martel V, Brunet F, Dupré N, Chrestian N. The occurrence of FUS mutations in pediatric amyotrophic lateral sclerosis: a case report and review of the literature. J Child Neurol. 2020;35(8):556–62.
Buratti E. Trends in understanding the pathological roles of TDP-43 and FUS proteins. In: Ghetti B, Buratti E, Boeve B, Rademakers R, editors. Frontotemporal Dementias : Emerging Milestones of the 21st Century [Internet]. Cham: Springer International Publishing; 2021 [cited 2022 Mar 27]. p. 243–67. (Adv Exp Med Biol). Available from: https://doi.org/10.1007/978-3-030-51140-1_15.
ALSoD [Internet]. [cited 2022 Mar 26]. Available from: https://alsod.ac.uk/output/gene.php/FUS.
Tyzack GE, Luisier R, Taha DM, Neeves J, Modic M, Mitchell JS, et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain. 2019;142(9):2572–80.
Notaro A, Messina A, La Bella V. A deletion of the nuclear localization signal domain in the Fus protein induces stable post-stress cytoplasmic inclusions in SH-SY5Y cells. Front Neurosci [Internet]. 2021 [cited 2022 Mar 27];15. Available from: https://www.frontiersin.org/article/10.3389/fnins.2021.759659.
Scekic-Zahirovic J, Sendscheid O, El Oussini H, Jambeau M, Sun Y, Mersmann S, et al. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J. 2016;35(10):1077–97.
Ishigaki S, Sobue G. Importance of functional loss of FUS in FTLD/ALS. Front Mol Biosci. 2018;3(5):44.
DeJesus-Hernandez M, Mackenzie IRR, Boeve BFF, Boxer ALL, Baker M, Rutherford NJJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Majounie E, Renton AE, Mok K, Dopper EGPP, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11(4):323–30.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
Devenney E, Hornberger M, Irish M, Mioshi E, Burrell J, Tan R, et al. Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol. 2014;71(3):331–9.
Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C. Relationship between C9orf72 repeat size and clinical phenotype. Vol. 44, Current Opinion in Genetics and Development. Elsevier Ltd; 2017. p. 117–24.
Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol Commun. 2019;7(1):115.
Amick J, Ferguson SM. C9orf72: at the intersection of lysosome cell biology and neurodegenerative disease. Vol. 18, Traffic. Blackwell Munksgaard; 2017. p. 267–76.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–25.
Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–24.
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol (Berl). 2013;126(6):845–57.
Gendron TF, Bieniek KF, Zhang YJJ, Jansen-West K, Ash PEAA, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol (Berl). 2013;126(6):829–44.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–8.
Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci. 2013;110(51):E4968–77.
Ash PEA, Bieniek KF, Gendron TF, Caulfield T, Lin WL, DeJesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–46.
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol (Berl). 2014;128(4):485–503.
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–25.
Sun Y, Eshov A, Zhou J, Isiktas AU, Guo JU. C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat Commun. 2020;11(1):3354.
West RJH, Sharpe JL, Voelzmann A, Munro AL, Hahn I, Baines RA, et al. Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age- and combination-specific phenotypic profiles in Drosophila. Acta Neuropathol Commun. 2020;8(1):158.
Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019;363(6428).
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018;24(8):1136–42.
Belikova AM, Zarytova VF, Grineva NI. Synthesis of ribonucleosides and diribonucleoside phosphates containing 2-chloroethylamine and nitrogen mustard residues. Tetrahedron Lett. 1967;37:3557–62.
Migliorati JM, Liu S, Liu A, Gogate A, Nair S, Bahal R, et al. Absorption, distribution, metabolism, and excretion of FDA-approved antisense oligonucleotide drugs. Drug Metab Dispos [Internet]. 2022 Jan 1 [cited 2022 Mar 21]; Available from: https://dmd.aspetjournals.org/content/early/2022/02/27/dmd.121.000417.
Bennett CF. Therapeutic antisense oligonucleotides are coming of age. Annu Rev Med. 2019;27(70):307–21.
Rothstein JD, Bristol LA, Hosler B, Brown RH, Kuncl RW. Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc Natl Acad Sci U S A. 1994;91(10):4155–9.
Troy CM, Shelanski ML. Down-regulation of copper/zinc superoxide dismutase causes apoptotic death in PC12 neuronal cells. Proc Natl Acad Sci U S A. 1994;91(14):6384–7.
Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290–6.
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–42.
McCampbell A, Cole T, Wegener AJ, Tomassy GS, Setnicka A, Farley BJ, et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest. 2018;128(8):3558–67.
Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109–19.
Miller T, Cudkowicz M. Results from the Phase 3 VALOR study and its open-label extension: evaluating the clinical efficacy and safety of tofersen in adults with ALS and confirmed SOD1 mutation [Internet]. Published online October 19, 2021. Available from: https://biogen.gcs-web.com/static-files/b2154d4e-f69f-49d4-9a61-e834387293ea. Accessed 1 Apr 2022.
Biogen announces topline results from the tofersen phase 3 Study and its open-label extension in SOD1-ALS | Biogen [Internet]. Published online October 17, 2021. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its. Accessed 2 April 2022.
Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation [Internet]. clinicaltrials.gov; 2022 Mar [cited 2022 Mar 31]. Report No.: NCT04856982. Available from: https://clinicaltrials.gov/ct2/show/NCT04856982.
Sareen D, O’Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149-208ra149.
Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–28.
O’Rourke JG, Bogdanik L, Muhammad AKMG, Gendron TF, Kim KJ, Austin A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901.
Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47).
Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–50.
Cook CN, Wu Y, Odeh HM, Gendron TF, Jansen-West K, del Rosso G, et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med. 2020;12(559):eabb3774.
Liu Y, Dodart JC, Tran H, Berkovitch S, Braun M, Byrne M, et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat Commun. 2021;8(12):847.
Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140(4):887–97.
Tran H, Moazami MP, Yang H, McKenna-Yasek D, Douthwright CL, Pinto C, et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat Med. 2022;28(1):117–24.
Biogen and Ionis Announce Topline Phase 1 Study Results of Investigational Drug in C9orf72 Amyotrophic Lateral Sclerosis | Biogen [Internet]. Published online March 28, 2022. Available from: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results. Accessed 6 Apr 2022.
Liu Y, Andreucci A, Iwamoto N, Yin Y, Yang H, Liu F, et al. WVE-004, an investigational stereopure antisense oligonucleotide for the treatment of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (2302). Neurology [Internet]. 2021 Apr 13 [cited 2022 Apr 7];96(15 Supplement). Available from: https://n.neurology.org/content/96/15_Supplement/2302.
Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, Shneider NA. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022;28(1):104–16.
Promising ALS Therapy Moves Closer to Clinic [Internet]. Columbia University Irving Medical Center. Published online January 24, 2022. Available from: https://www.cuimc.columbia.edu/news/promising-als-therapy-moves-closer-clinic. Accessed 9 Apr 2022.
Ionis Pharmaceuticals, Inc. A phase 1-3 study to evaluate the efficacy, safety, pharmacokinetics and pharmacodynamics of intrathecally administered ION363 in amyotrophic lateral sclerosis patients with fused in sarcoma mutations (FUS-ALS) [Internet]. clinicaltrials.gov; 2022 Mar [cited 2022 Apr 7]. Report No.: NCT04768972. Available from: https://clinicaltrials.gov/ct2/show/NCT04768972.
Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–75.
Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, et al. Therapeutic reduction of ataxin 2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544(7650):367–71.
Biogen. A phase 1 multiple-ascending-dose study to assess the safety, tolerability, and pharmacokinetics of BIIB105 administered intrathecally to adults with amyotrophic lateral sclerosis with or without Poly-CAG expansion in the ataxin-2 gene [Internet]. clinicaltrials.gov; 2022 Jan [cited 2022 Apr 10]. Report No.: NCT04494256. Available from: https://clinicaltrials.gov/ct2/show/NCT04494256.
Turner BJ, Cheah IK, Macfarlane KJ, Lopes EC, Petratos S, Langford SJ, et al. Antisense peptide nucleic acid-mediated knockdown of the p75 neurotrophin receptor delays motor neuron disease in mutant SOD1 transgenic mice. J Neurochem. 2003;87(3):752–63.
Marc G, Leah R, Ofira E, Oded A, Zohar A, Hanna R. Presymptomatic treatment with acetylcholinesterase antisense oligonucleotides prolongs survival in ALS (G93A-SOD1) mice. BioMed Res Int. 2013;2013:845345.
Koval ED, Shaner C, Zhang P, du Maine X, Fischer K, Tay J, et al. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum Mol Genet. 2013;22(20):4127–35.
Shijo T, Warita H, Suzuki N, Ikeda K, Mitsuzawa S, Akiyama T, et al. Antagonizing bone morphogenetic protein 4 attenuates disease progression in a rat model of amyotrophic lateral sclerosis. Exp Neurol. 2018;1(307):164–79.
Zhang S, Cooper-Knock J, Weimer AK, Shi M, Moll T, Marshall JNG, et al. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron. 2022;110(6):992-1008.e11.
Kwok A, Raulf N, Habib N. Developing small activating RNA as a therapeutic: current challenges and promises. Ther Deliv. 2019;10(3):151–64.
Li C, Jiang W, Hu Q, Li LC, Dong L, Chen R, et al. Enhancing DPYSL3 gene expression via a promoter-targeted small activating RNA approach suppresses cancer cell motility and metastasis. Oncotarget. 2016;7(16):22893–910.
Schwartz JC, Younger ST, Nguyen NB, Hardy DB, Monia BP, Corey DR, et al. Antisense transcripts are targets for activating small RNAs. Nat Struct Mol Biol. 2008;15(8):842–8.
Sarker D, Plummer R, Meyer T, Sodergren MH, Basu B, Chee CE, et al. MTL-CEBPA, a small activating RNA therapeutic upregulating C/EBP-α, in patients with advanced liver cancer: a first-in-human, multicenter, open-label, phase i trial. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26(15):3936–46.
Fimiani C, Goina E, Su Q, Gao G, Mallamaci A. RNA activation of haploinsufficient Foxg1 gene in murine neocortex. Sci Rep. 2016;20(6):39311.
Monian P, Shivalila C, Lu G, Shimizu M, Boulay D, Bussow K, et al. Endogenous ADAR-mediated RNA editing in non-human primates using stereopure chemically modified oligonucleotides. Nat Biotechnol. 2022;7:1–10.
Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, et al. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis. 2012;45(3):1121–8.
ALSoD [Internet]. [cited 2022 Apr 25]. Available from: https://alsod.ac.uk/output/gene.php/TARDBP.
Hoggan MD, Blacklow NR, Rowe WP. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci U S A. 1966;55(6):1467–74.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18(5):358–78.
McIntosh NL, Berguig GY, Karim OA, Cortesio CL, De Angelis R, Khan AA, et al. Comprehensive characterization and quantification of adeno associated vectors by size exclusion chromatography and multi angle light scattering. Sci Rep. 2021;11(1):3012.
Dong JY, Fan PD, Frizzell RA. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum Gene Ther. 1996;7(17):2101–12.
Smith RH. Adeno-associated virus integration: virus versus vector. Gene Ther. 2008;15(11):817–22.
Hanlon KS, Kleinstiver BP, Garcia SP, Zaborowski MP, Volak A, Spirig SE, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun. 2019;10(1):4439.
Flotte TR, Afione SA, Conrad C, McGrath SA, Solow R, Oka H, et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci U S A. 1993;90(22):10613–7.
Flotte T, Carter B, Conrad C, Guggino W, Reynolds T, Rosenstein B, et al. A phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung disease. Johns Hopkins Children’s Center, Baltimore, Maryland. Hum Gene Ther. 1996 Jun 10;7(9):1145–59.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27(1):59–65.
Tanguy Y, Biferi MG, Besse A, Astord S, Cohen-Tannoudji M, Marais T, et al. Systemic AAVrh10 provides higher transgene expression than AAV9 in the brain and the spinal cord of neonatal mice. Front Mol Neurosci. 2015;28(8):36.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–22.
Foust KD, Salazar DL, Likhite S, Ferraiuolo L, Ditsworth D, Ilieva H, et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol Ther. 2013;21(12):2148–59.
Iannitti T, Scarrott JM, Likhite S, Coldicott IRP, Lewis KE, Heath PR, et al. Translating SOD1 gene silencing toward the clinic: a highly efficacious, off-target-free, and biomarker-supported strategy for fALS. Mol Ther Nucleic Acids. 2018;3(12):75–88.
Thomsen GM, Gowing G, Latter J, Chen M, Vit JP, Staggenborg K, et al. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J Neurosci Off J Soc Neurosci. 2014;34(47):15587–600.
Borel F, Gernoux G, Cardozo B, Metterville JP, Toro Cabreja GC, Song L, et al. Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1G93A mice and nonhuman primates. Hum Gene Ther. 2016;27(1):19–31.
Stoica L, Todeasa SH, Cabrera GT, Salameh JS, ElMallah MK, Mueller C, et al. Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann Neurol. 2016;79(4):687–700.
Borel F, Gernoux G, Sun H, Stock R, Blackwood M, Brown RH, et al. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci Transl Med. 2018;10(465).
Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 suppression with adeno-associated virus and microRNA in familial ALS. N Engl J Med. 2020;383(2):151–8.
Wang H, Yang B, Qiu L, Yang C, Kramer J, Su Q, et al. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23(3):668–81.
Keeler AM, Zieger M, Semple C, Pucci L, Veinbachs A, Brown RH, et al. Intralingual and intrapleural AAV gene therapy prolongs survival in a SOD1 ALS mouse model. Mol Ther Methods Clin Dev. 2019;24(17):246–57.
Biferi MG, Cohen-Tannoudji M, Cappelletto A, Giroux B, Roda M, Astord S, et al. A new AAV10-U7-mediated gene therapy prolongs survival and restores function in an ALS mouse model. Mol Ther. 2017;25(9):2038–52.
Duan W, Guo M, Yi L, Liu Y, Li Z, Ma Y, et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020;27(3–4):157–69.
Gaj T, Ojala DS, Ekman FK, Byrne LC, Limsirichai P, Schaffer DV. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci Adv. 2017;3(12):eaar3952.
Wang L, Yi F, Fu L, Yang J, Wang S, Wang Z, et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell. 2017;8(5):365–78.
Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, et al. Adeno-associated virus–mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther. 2014;22(3):498–510.
Ghadge GD, Pavlovic JD, Koduvayur SP, Kay BK, Roos RP. Single chain variable fragment antibodies block aggregation and toxicity induced by familial ALS-linked mutant forms of SOD1. Neurobiol Dis. 2013;1(56):74–8.
Ghadge GD, Kay BK, Drigotas C, Roos RP. Single chain variable fragment antibodies directed against SOD1 ameliorate disease in mutant SOD1 transgenic mice. Neurobiol Dis. 2019;1(121):131–7.
Statement from Novartis Gene Therapies: OAV301 program for familial ALS caused by SOD1 mutation [Internet]. Les Turner ALS Foundation. Published online November 30, 2020. Available from: https://lesturnerals.org/oav301-program-for-familial-als-caused-by-sod1-mutation-statement-from-novartis-gene-therapies/. Accessed 19 Apr 2022.
By. Apic Bio Receives FDA Fast Track Designation for APB-102 | Apic Bio [Internet]. Published online July 28, 2021. Available from: https://apic-bio.com/apic-bio-receives-fda-fast-track-designation-for-apb-102-for-the-treatment-of-patients-with-sod1-als/. Accessed 20 Apr 2022.
Lim CKW, Gapinske M, Brooks AK, Woods WS, Powell JE, Zeballos CMA, et al. Treatment of a mouse model of ALS by in vivo base editing. Mol Ther. 2020;28(4):1177–89.
Pozzi S, Thammisetty SS, Codron P, Rahimian R, Plourde KV, Soucy G, et al. Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J Clin Invest. 2019;129(4):1581–95.
Martier R, Liefhebber JM, Miniarikova J, van der Zon T, Snapper J, Kolder I, et al. Artificial microRNAs targeting C9orf72 can reduce accumulation of intra-nuclear transcripts in ALS and FTD patients. Mol Ther Nucleic Acids. 2019;1(14):593–608.
Martier R, Liefhebber JM, García-Osta A, Miniarikova J, Cuadrado-Tejedor M, Espelosin M, et al. Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol Ther Nucleic Acids. 2019;11(16):26–37.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–45.
Zu T, Guo S, Bardhi O, Ryskamp DA, Li J, Khoramian Tusi S, et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc Natl Acad Sci U S A. 2020;117(31):18591–9.
Piao X, Meng D, Zhang X, Song Q, Lv H, Jia Y. Dual-gRNA approach with limited off-target effect corrects C9ORF72 repeat expansion in vivo. Sci Rep. 2022;12(1):5672.
Gouel F, Rolland AS, Devedjian JC, Burnouf T, Devos D. Past and future of neurotrophic growth factors therapies in ALS: from single neurotrophic growth factor to stem cells and human platelet lysates. Front Neurol. 2019;2(10):835.
Henriques A, Pitzer C, Schneider A. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand? Front Neurosci. 2010;11(4):32.
Dodge JC, Haidet AM, Yang W, Passini MA, Hester M, Clarke J, et al. Delivery of AAV-IGF-1 to the CNS extends survival in ALS mice through modification of aberrant glial cell activity. Mol Ther J Am Soc Gene Ther. 2008;16(6):1056–64.
Dodge JC, Treleaven CM, Fidler JA, Hester M, Haidet A, Handy C, et al. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol Ther. 2010;18(12):2075–84.
Lin H, Hu H, Duan W, Liu Y, Tan G, Li Z, et al. Intramuscular delivery of scAAV9-hIGF1 prolongs survival in the hSOD1G93A ALS mouse model via upregulation of D-amino acid oxidase. Mol Neurobiol. 2018;55(1):682–95.
Wang W, Wen D, Duan W, Yin J, Cui C, Wang Y, et al. Systemic administration of scAAV9-IGF1 extends survival in SOD1G93A ALS mice via inhibiting p38 MAPK and the JNK-mediated apoptosis pathway. Brain Res Bull. 2018;1(139):203–10.
Wang LJ, Lu YY, Muramatsu SI, Ikeguchi K, Fujimoto KI, Okada T, et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J Neurosci Off J Soc Neurosci. 2002;22(16):6920–8.
Thomsen GM, Alkaslasi M, Vit JP, Lawless G, Godoy M, Gowing G, et al. Systemic injection of AAV9-GDNF provides modest functional improvements in the SOD1G93A ALS rat but has adverse side effects. Gene Ther. 2017;24(4):245–52.
Mòdol-Caballero G, García-Lareu B, Herrando-Grabulosa M, Verdés S, López-Vales R, Pagès G, et al. Specific expression of glial-derived neurotrophic factor in muscles as gene therapy strategy for amyotrophic lateral sclerosis. Neurotherapeutics. 2021;18(2):1113–26.
Henriques A, Pitzer C, Dittgen T, Klugmann M, Dupuis L, Schneider A. CNS-targeted viral delivery of G-CSF in an animal model for ALS: improved efficacy and preservation of the neuromuscular unit. Mol Ther. 2011;19(2):284–92.
Lee SH, Kim S, Lee N, Lee J, Yu SS, Kim JH, et al. Intrathecal delivery of recombinant AAV1 encoding hepatocyte growth factor improves motor functions and protects neuromuscular system in the nerve crush and SOD1-G93A transgenic mouse models. Acta Neuropathol Commun. 2019;12(7):14.
Lee SH, Lee N, Kim S, Lee J, Choi W, Yu SS, et al. Intramuscular delivery of HGF-expressing recombinant AAV improves muscle integrity and alleviates neurological symptoms in the nerve crush and SOD1-G93A transgenic mouse models. Biochem Biophys Res Commun. 2019;517(3):452–7.
Leyton-Jaimes MF, Kahn J, Israelson A. AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc Natl Acad Sci U S A. 2019;116(29):14755–60.
Tung YT, Peng KC, Chen YC, Yen YP, Chang M, Thams S, et al. Mir-17∼92 confers motor neuron subtype differential resistance to ALS-associated degeneration. Cell Stem Cell. 2019;25(2):193-209.e7.
Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29(8):1037–47.
Porter JJ, Heil CS, Lueck JD. Therapeutic promise of engineered nonsense suppressor tRNAs. Wiley Interdiscip Rev RNA. 2021;12(4):e1641.
Wang J, Zhang Y, Mendonca CA, Yukselen O, Muneeruddin K, Ren L, et al. AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice. Nature. 2022;604(7905):343–8.
Torkzaban B, Kawalerski R, Coller J. Development of a tethered mRNA amplifier to increase protein expression. Biotechnol J. 2022:2200214.
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–6.
Vahsen BF, Gray E, Thompson AG, Ansorge O, Anthony DC, Cowley SA, et al. Non-neuronal cells in amyotrophic lateral sclerosis — from pathogenesis to biomarkers. Nat Rev Neurol. 2021;17(6):333–48.
Logroscino G, Traynor BJ, Hardiman O, Chiò A, Mitchell D, Swingler RJ, et al. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry. 2010;81(4):385–90.
Lehky T, Grunseich C. Juvenile amyotrophic lateral sclerosis: a review. Genes. 2021;12(12):1935.
Andersen PM, Nilsson P, Ala-Hurula V, Keränen ML, Tarvainen I, Haltia T, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet. 1995;10(1):61–6.
Richards D, Morren JA, Pioro EP. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J Neurol Sci. 2020;15(417):117054.
Loeffler J, Picchiarelli G, Dupuis L, Gonzalez De Aguilar J. The role of skeletal muscle in amyotrophic lateral sclerosis. Brain Pathol. 2016;26(2):227–36.
Yan Y, Liu XY, Lu A, Wang XY, Jiang LX, Wang JC. Non-viral vectors for RNA delivery. J Control Release Off J Control Release Soc. 2022;342:241–79.
Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci. 2016;113(39):10962–7.
Flotte TR, Cataltepe O, Puri A, Batista AR, Moser R, McKenna-Yasek D, et al. AAV gene therapy for Tay-Sachs disease. Nat Med. 2022;28(2):251–9.
Shirley JL, de Jong YP, Terhorst C, Herzog RW. Immune responses to viral gene therapy vectors. Mol Ther. 2020;28(3):709–22.
Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther. 2018;29(3):285–98.
Nelson CE, Wu Y, Gemberling MP, Oliver ML, Waller MA, Bohning JD, et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat Med. 2019;25(3):427–32.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Efficient delivery of genome-editing proteins in vitro and in vivo. Nat Biotechnol. 2015;33(1):73–80.
Duan L, Ouyang K, Xu X, Xu L, Wen C, Zhou X, et al. Nanoparticle delivery of CRISPR/Cas9 for genome editing. Front Genet [Internet]. 2021 [cited 2022 Apr 26];12. Available from: https://www.frontiersin.org/article/10.3389/fgene.2021.673286.
Mueller C, Tang Q, Gruntman A, Blomenkamp K, Teckman J, Song L, et al. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol Ther J Am Soc Gene Ther. 2012;20(3):590–600.
Wave Life Sciences Ltd. A multicenter, randomized, double-blind, placebo-controlled, phase 1b/2a study of WVE-004 administered intrathecally to patients with C9orf72-associated amyotrophic lateral sclerosis (ALS) or frontotemporal dementia (FTD) [Internet]. clinicaltrials.gov; 2022 Mar [cited 2022 May 2]. Report No.: NCT04931862. Available from: https://clinicaltrials.gov/ct2/show/NCT04931862.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This work was funded by the NIH (R01 NS111990 to RHB), the Angel Fund for ALS Research, ALSOne, ALS Finding a Cure, the Cellucci Fund for ALS Research, the Pierre de Bourgknecht ALS Research Fund, and the Max Rosenfeld Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meijboom, K.E., Brown, R.H. Approaches to Gene Modulation Therapy for ALS. Neurotherapeutics 19, 1159–1179 (2022). https://doi.org/10.1007/s13311-022-01285-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01285-w