
Abstract
Bacillus spp. have been widely reported with the ability to control plant diseases. In this work, we analyzed the whole genome of LJBS06, which was isolated from grapevine rhizosphere soil. In view of physiological and biochemical characteristics, genome data, and phylogenetic analysis of 16S rRNA, LJBS06 was affiliated with Bacillus stercoris. LJBS06 showed antagonistic activities against a variety of plant pathogens. The inhibition rate of Magnaporthe oryzae was up to 75.05% and the inhibition rates of Colletotrichum gloeosporioides, Coniothyrium diplodiella, and Botrytis cinerea were all above 50% in the plate assays. The genome of LJBS06 had a 4,154,362-bp circular chromosome, with an average GC content of 43.96%, containing an 82,935-bp plasmid with a GC content of 35.18%. The circular chromosome of LJBS06 contained 4231 protein-coding genes, 30 rRNA genes, and 87 tRNA genes, including genes related to the synthesis of plant defense-related enzymes and the promotion of plant growth. Meanwhile, 11 gene clusters involved in biosynthesis of secondary metabolites were present in the genome of LJBS06. In conclusion, our findings indicated that LJBS06 strain had the necessary genetic machinery to control plant pathogens and provided insights for future studies of the biocontrol mechanisms of B. stercoris LJBS06.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacillus species are spherical or rod-shaped bacteria, which can survive under harsh environmental for long period of time due to the formation of endospores (Nicholson et al. 2000). Many Bacillus spp. were proved to be effective against broad-spectrum phytopathogens. B. amyloliquefaciens, B. megaterium, B. licheniformis, and B. subtilis have been commercially produced as biocontrol agent (Chen et al. 2018). Some among them, specifically B. subtilis, were widely used to control plant pathogens and play important role in different agro-ecosystems (Ahemad and Kibret 2014; Blake et al. 2021). Recently, B. subtilis subsp. stercoris was promoted to species status as B. stercoris (Dunlap et al. 2020). Many studies had investigated the potential of B. stercoris for biological control. For example, B. stercoris JS inhibited tobacco fungal and oomycete diseases through inducing plant resistance (Kim et al. 2015). B. stercoris Fmb60 isolated from compost, had significant broad-spectrum antimicrobial activities through producing aurantinins B–D to destroy pathogen cell membranes (Yang et al. 2016). Furthermore, B. stercoris A053, B. stercoris JNUCC and B. stercoris X2 showed strong antifungal activity against plant pathogens (Byun et al. 2020; Dhruw et al. 2020; Guo et al. 2015).
The occurrence of the diseases lead to crop yield and quality reduction, and chemical pesticides were the main way to control diseases. However, environmental pollution, pesticide residues, and fungicide resistance caused by pesticides were becoming more and more serious (Aboutorabi 2018). The use of microorganisms as biocontrol agent was a powerful alternative to prevent and suppress of plant diseases. Besides, beneficial microorganisms could promote plant growth to enhance disease resistance. For example, the consecutive application of B. amyloliquefaciens NJN-6 as biofertilizer controlled banana wilt by changing soil bacterial and fungal community composition (Fu et al. 2017). B. subtilis 9407 suppressed apple ring rot disease by producing lipopeptides (Fan et al. 2017). B. velezensis ZSY-1, B. subtilis PTS-394, and B. subtilis CF-3 released volatile organic compounds (VOCs) to promote plant growth and suppress plant pathogens (Gao et al. 2017, 2018; Qiao et al. 2017).
Bacillus species as benefit microorganisms were environmentally friendly alternative to pesticides. It was able to inhabit a wide range of plant pathogens through diverse mechanisms, including the synthesis of defense-related enzymes, secondary metabolites, and systemically induced disease resistance (Blake et al. 2021; Valenzuela-Ruiz et al. 2019). Meanwhile, Bacillus spp. also had the potential of promoting growth, helping plant to defend against pathogens indirectly. Defense-related enzymes like chitinases, glucanases, cellulases and proteases inhibited the growth and reproduction of pathogens by hydrolyzing the major components of the fungal and bacterial cell walls (Miljakovic et al. 2020). A range of secondary metabolites produced by Bacillus species, such as ribosomally and non-ribosomally synthesized peptides, terpenes, siderophores as well as polyketides are important weapons against plant pathogens (Kaspar et al. 2019). Ribosomally synthesized peptides, such as Bacteriocins, showed bactericidal activity against pathogens by interfering with the synthesis of the cell wall and forming pores in the cell membrane (Shafi et al. 2017). Cyclic lipopeptides including surfactin, iturin and fengycin families, were non-ribosomally synthesized peptides and acted against a wide range of plant pathogens by interacting with the cell membrane to cause changes in structure and permeability of target cell (Djordje et al. 2018). Other synthesized antibiotics, such as siderophores and polyketides showed diverse antifungal and antibacterial activities via diverse pathway. Bacillus spp. can also trigger resistance to protect the plant from attack against various pathogenic organisms by modifications of plant biochemical reaction and structures, activation of plant-resistance factors, and production of phenolic compounds and defensive enzymes (Shafi et al. 2017).
In this study, we isolated LJBS06 strain with biocontrol effects from grapevine rhizosphere soil and performed genome sequencing. Through morphological, physiological, biochemical, phylogenetic and genome analysis, the LJBS06 strain was identified as B. stercoris. Various genes related to the biosynthesis of defense-related enzymes, secondary metabolites and the promotion of plant growth were found in the LJBS06 genome by genome annotation. These results provided a better understanding of the mechanisms of Bacillus spp. as biocontrol agents and the scientific basis for application and optimization.
Materials and methods
Bacterial isolation
Bacillus stercoris LJBS06 was isolated from the grapevine rhizosphere soil at Shanghai, China, in October 2019. Soil samples were collected from the rhizosphere about 20 cm from the ground and were dissolved in sterile NaCl solution (0.85% w/v) by vibrating violently for 2 min. The soil suspension was incubated at 80 °C for 30 min, then the supernatant was serially diluted (10−1–10−3) with sterile distilled water, and 100 μL aliquot from each dilution was spread on Luria–Bertani (LB) agar plate (5.0 g yeast extract, 10.0 g peptone, 10.0 g NaCl and 15.0 g agar per liter, pH 7.0) (Santana et al. 2008). After incubating for 24 h at 37 °C, milky white colonies with biofilm were sub-cultured on the LB agar plates using a streak method following Sari et al. (2019). The screened bacteria were co-cultured with Colletotrichum gloeosporioides, Coniothyrium diplodiella and Botrytis cinerea on PDA medium (200.0 g potato, 20.0 g glucose, 20.0 g agar per liter, pH 7.0). The strain LJBS06, which can inhibit the growth of the hyphae of above pathogens, was selected and stored at − 20 °C with glycerol.
Morphological, physiological and biochemical analysis
The morphological characteristics of bacterial colonies were observed after growing on LB plates for 24 h. Gram staining and spore staining were performed according to Khurshed and Shakoori (2005) method. The physiological characteristics of LJBS06 were identified according to the procedures outlined in the Bergey Manual of Deterministic Bacteriology (Guerrero 2001). Plate analysis was performed to evaluate the activities of cellulase (Cel) and protease (Prt), according to reported methods with modifications (Paudel and Qin 2015; Rajput and Tiwari 2012). Test carboxymethyl cellulose (CMC) plates (10.0 g CMC, 3.0 g yeast extract, 3.0 g K2HPO4, 1 g KH2PO4, 0.5 g MgSO4·7H2O, and 20.0 g agar per liter) and Prt plates (10.0 g casein, 3.0 g yeast extract, 5.0 g Na2HPO4·12H2O, 5.0 g NaCl, and 20 g agar per liter) with LJBS06 were at 37 °C for 24 h to evaluate Cel and Prt activity. The Cel plate was stained with 0.1% (w/v) congo red solution for 30 min, and then washed with 1 M NaCl solution for 15 min. Experiments were repeated three times.
16S rRNA sequencing and phylogenetic analysis
According to the manufacturer's instructions, a commercially available DNA extraction kit (Shanghai Sangon Biotech Co., Ltd.) was used to extract and purify the DNA of LJBS06. Forward primer 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and reverse primer 1492R (5′-ACGGCTACCTTGTTACGACTT-3′) were used for PCR amplification of the extracted DNA. The PCR product was sequenced by Shanghai Sunny Biotechnology Co., Ltd. Homologous sequences were searched and downloaded from EZBiocloud (https://www.ezbiocloud.net/) database, then aligned by Clustal X. MEGA 7.0 software was used to analyze and calculate the genetic distance between the sequences, and constructed the 16S rRNA phylogenetic tree according to neighbor-joining method.
Genome sequencing and bioinformatic analysis
Whole-genome sequencing was performed by the third generation single molecular sequencing technique based on the Oxford Nanopore Technologies (ONT) platform. The reads of the third generation single molecular sequencing were assembled into contigs using HGAP V4 and CANU v1.7.1 (Chin et al. 2016; Koren et al. 2017). The reads of the Next-Generation Sequencing were used to correct the contigs by software Pilon v1.18 (Walker et al. 2014). Based on the OrthoANI algorithm, the average nucleotide identity of LJBS06 with closely related strains was compared through Jspecies software (http://jspecies.ribohost.com/jspeciesws/) (Richter and Rossello-Mora 2009). Genome-to-Genome Distance Calculator (GGDC) v2.1 (https://ggdc.dsmz.de/) was used to judge the species according to the whole genome information (Meier-Kolthoff et al. 2013). GeneMarkS (http://topaz.gatech.edu/GeneMark/) was used to predict the open reading frame (ORF) of the bacterial genome (Besemer et al. 2001). The tRNAscan-SE, Barrnap, Rfam (http://rfam.xfam.org/) were used for predicting tRNA genes, rRNA genes, and non-coding RNAs (Kalvari et al. 2018a, b; Lowe and Eddy 1997), respectively. Multiple databases were used to annotate protein-coding gene functions, including nonredundant (NR) database (http://ftp.ncbi.nih.gov/blast/db/) (Buchfink et al. 2015), evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) database (http://eggnogdb.embl.de/#/app/home/) (Jensen et al. 2008), Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) (Ogata et al. 1999), Swiss-Prot database (http://www.uniprot.org/) (Boeckmann et al. 2003) and Gene Ontology (GO) database (http://www.geneontology.org/) (Ashburner et al. 2000). The graphical views of genome alignments was completed by CGView (http://stothard.afns.ualberta.ca/cgview_server/) (Chin et al. 2016). In addition, signal peptides and transmembrane helices were predicted by SignalP v4.1 (Petersen et al. 2011) and TMHMM v2.0 (Chen et al. 2003), respectively. IslandViewer v4 (Bertelli et al. 2017), hmmscan v3.2.1 (Krogh et al. 1994), and CRISPR finder (Grissa et al. 2007) were used to predict genomics islands (GIs), carbohydrate active enzyme (CAZy), and clustered regularly interspaced short palindromic repeats (CRISPRs), respectively. AntiSMASH v6.0 (https://antismash.secondarymetabolites.org/) was used to identify the putative secondary metabolite genes in the LJBS06 genome (Blin et al. 2021).
Enzymatic assays
LJBS06 was cultured in LB liquid medium using an orbital shaker at 180 rpm, 37 °C for 48 h. The cell-free supernatant of LJBS06 was obtained by centrifuging fermentation solution at 12,000×g at 4 °C for 10 min and sequentially filtered through a 0.22 μm organic filter membrane. The supernatant was used for determining enzymatic activities (Mohammad and Alireza 2007).
A modified quantitative estimation assay using 3,5-dinitro salicylic acid (DNS) method was used to measure the activities of chitinase (Liang et al. 2014), chitosanase (Liang et al. 2014), and cellalase (Miller 1959). Briefly, 1% (w/v) colloidal chitin, 5% (w/v) colloidal chitosan, and 1% (w/v) CMC in 50 mM phosphate buffer (pH 6.0) were used as the substrate for the measurement of chitinase, chitosanase, and cellalase activities, respectively. One unit (U) of these enzymes was defined as 1 μmol of N-acetyl-d-glucosamine, d-glucosamine, and d-glucose formed, respectively, per min. Protease activity was assayed using casein as a substrate with the method according to Yang et al. (2012). One unit of protease activity caused the production of 1 μg of tyrosine per min.
Polyphenol oxidase (PPO) activity assay was assayed with the method by Mayende et al. (2006). One unit of PPO activity was defined as a change in absorbance at 420 nm of 0.01 per min. For superoxide dismutase (SOD) activity determination, each sample and substrate (10 μM cytochrome c, 50 μM xanthine, and sufficient xanthine oxidase) were reacted in 50 mM potassium phosphate buffer (pH 7.8) at 30 °C (Hakamada et al. 1997). One unit of SOD activity was defined as the amount of protein required to inhibit the reduction rate of cytochrome c by 50%. The activity of peroxidase (POD) was measured according to Mohammad and Alireza (2007) with modification. One unit of POD activity caused a change in absorbance at 470 nm of 0.01 per min. Catalase activity (CAT) assay was measured with the method by Xu et al. (2020a). One unit of CAT resulted in releases 1 μmol H2O2 per min.
Antifungal activity
Magnaporthe oryzae, Colletotrichum gloeosporioides, Coniothyrium diplodiella, Phytophthora capsica, Fusarium graminearum, Fusarium solani, Exserohilum rostratum are were cultured in a dark incubator at 28 ± 1 °C. Botrytis cinerea, Fusarium equiseti, Verticiltium dahliae, Sclerotinia selerotiorum, Fusarium oxysporum were cultured in a dark incubator at 22 ± 1 °C. The above pathogens were cultured on PDA medium and stored at 4 °C. The antagonistic activity of LJBS06 was evaluated using dual culture on PDA plates (Zhang et al. 2017). After activating LJBS06 on the LB medium, the activated strain was streaked horizontally in the middle of a plate (9 cm diameter) with PDA medium. Two discs (6 mm diameter) of the same pathogen were placed at 2.2 cm from the center of the plate, one on the right and one on the left of the LJBS06. Pathogen discs placed on the PDA plates without LJBS06 strain served as the control (Zhang et al. 2016). The inhibitory area of the treatment was observed and measured when the hyphae of the control were overgrown. The antagonistic activity was expressed as the rate of inhibition of mycelial growth compared to the control, following the equation: ((R1−R2)/R1) × 100%. R1 and R2 were the radii of the pathogen mycelium in the control and treatment, respectively (Zhang et al. 2016). Each treatment was repeated three times, and each replicate consisted of three samples.
Statistical analysis
All data were analyzed by one-way analysis of variance (ANOVA), using SPSS 22.0 (SPSS Institute, Chicago, IL). Data were expressed as the means of replicates with standard errors. Significance analysis between means was compared by the least significant difference (LSD) test (p = 0.05).
Results and discussion
Identification and characterization of LJBS06
Bacillus stercoris LJBS06 was a gram-positive, spore-producing, rod-shaped and aerobic bacterium, the colonies of which on LB plates were rough, creamy white, with uneven wavy edges and a dry wrinkle on the surface (Fig. S1). According to physiological and biochemical characteristics (Table 1), LJBS06 belonged to Bacillus genus. To further understand the genetic relationships between LJBS06 with other Bacillus strains, the 16S rRNA sequence of LJBS06 was compared by the EZBioCloud database. LJBS06 had 100% 16S rRNA similarity with type strain B. stercoris JCM 30051 (MN536904). Strains showing 98.7% or higher 16S rRNA similarity with the sequence of LJBS06 were selected. These selected strains were compared with LJBS06 at the genome level by average nucleic identity based on Blast + (ANIb), MUMmer (ANIm) and genome-to-genome distance calculator (GGDC) via BLAST. The estimate values of ANIb, ANIm, and GGDC between strains LJBS06 with B. stercoris JS were 98.93%, 99.06% and 92.3%, respectively (Table 2). Based on the cut-off values for species delimitation for ANI > 95–96% and GGDC > 70% (Chun et al. 2018), LJBS06 strain was affiliated with B. stercoris. The phylogenetic tree using the 16S rRNA gene (MW940707) was constructed with the neighbor-joining method confirming the taxonomic position of LJBS06 (Fig. 1). LJBS06 was identified as B. stercoris by morphological, biochemical, and molecular analysis. It has been confirmed that B. stercoris had strong antifungal activity against plant pathogens (Byun et al. 2020; Dhruw et al. 2020; Guo et al. 2015). Strain LJBS06 as B. stercoris may be effective against a broad range of pathogenic microorganisms.

Phylogenetic tree based on 16S rRNA gene sequences showing the relationship between B. stercoris LJBS06 and closely related Bacillus species. The phylogenetic tree was constructed using the Neighbor-Joining method. Bootstrap values (%) based on 1000 replications were given at nodes. The scale bar indicates 0.005 nucleotide substitution per site
Genomic features of B. stercoris LJBS06
Minimum information about the genome sequence (MIGS) of B. stercoris LJBS06 is presented in Table S1. The complete genome of B. stercoris LJBS06 contained a circular chromosome and a plasmid. The chromosome sequence was 4,154,362 bp with an average GC content of 43.96%. The full length of the plasmid sequence was 82,935 bp and the GC content was 35.18%. There was a total of 4231 ORFs predicted in the genome with 30 rRNA genes and 87 tRNA genes in the chromosome (Table 3). A graphical circular map of the chromosome of the LJBS06 was shown in Fig. 2A. The plasmid contained 95 ORFs, in which the function of most genes was unclear. A circular map of the plasmid was presented in Fig. 2B. 228 (5.39%) and 1115 (26.35%) of the ORFs on the chromosome were classified into different groups using the SignalP and TMHMM databases, respectively (Table S2). Moreover, the LJBS06 encoded 111 secreted proteins, 269 genomic islands, 158 CAZys and 2 CRISPR loci (Table S2). The genome size, GC content and number of genes of LJBS06 were similar to those of known Bacillus spp. genomes. To further analyze genetic information, ORFs in the LJBS06 chromosome were classified into different COG categories using eggNOG database. There were 3540 ORFs in the LJBS06 chromosome, among which 6.71% were associated with amino acid transport and metabolism, 6.31% with carbohydrate transport and metabolism, 4.30% with energy production and conversion, 1.70% with secondary metabolites biosynthesis, transport and catabolism, and 0.85% with cell motility (Table S3). Amino acid transport and metabolism, carbohydrate transport and metabolism, and energy production and conversion may endow LJBS06 with a wide variety of antimicrobial compounds, such as lytic enzymes. The abundance of secondary metabolites biosynthesized by LJBS06 played key roles for antagonism. Overall, these biological functions were important for the biocontrol function of LJBS06.

The genome map of the chromosome (A) and the plasmid (B) of B. stercoris LJBS06 using the CGview server. From inner to outer, ring 1 represents the scale marks of the genome. Ring 2 represents the GC skew (green, positive skew; purple, negative skew). Ring 3 represents the G + C content. Ring 5 represents reverse CDSs and ring 4 represents reverse COG Annotated coding sequences. Ring 6 represents forward CDSs. Ring 7 represents forward COG Annotated coding sequences. Very short features were enlarged to enhance visibility. Clustered genes, such as several rRNA genes, may appear as one line due to space limitation
Antifungal activity of LJBS06
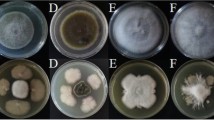
In the antagonistic plate assays, LJBS06 had a broad inhibitory spectrum against plant pathogens. The inhibition rate of M. oryzae (rice blast) was up to 75.05% and the inhibition rates of C. gloeosporioides, C. diplodiella, and B. cinerea were all above 50% (Table S4). Prevention and cure of crop diseases was the primary condition for the development of agriculture. LJBS06 had the potential to inhibit the growth of pathogens and replace pesticides as environmentally friendly biocontrol agent. Moreover, the mycelia of C. gloeosporioides on the dual culture plate were severely enlarged, twisted and broken, with the leakage of intracellular protoplasm under microscopic observations (Fig. S2), whereas no similar changes were noted in the control mycelia. Enzyme assays showed B. stercoris LJBS06 synthesized Prt and Cel (Fig. S3). Lytic enzymes, such as Prt and Cel, can hydrolyze the major components of cell walls, resulting in mycelial expansion of pathogens (Shafi et al. 2017). In general, B. stercoris LJBS06 produced lytic enzymes and showed a broad inhibitory spectrum against plant pathogens (Fig. 3).

Antagonistic assay of B.stercoris LJBS06 against 12 pathogens. Uppercase letter indicated the treatment and lowercase letter indicated the control. A Magnaporthe oryzae. B Colletotrichum gloeosporioides. C Coniothyrium diplodiella. D Botrytis cinerea. E Phytophthora capsica. F Verticiltium dahliae. G Fusarium graminearum. H Exserohilum rostratum. I Fusarium solani. J Sclerotinia selerotiorum. K Fusarium oxysporum. L Rhizoctonia solani
Genes involved in defense-related enzymes and plant growth promotion
Defense-related enzymes produced by Bacillus spp. are active in response to disease defense and considered to be important features in biocontrol activities (Leal et al. 2021). Bacillus species producing mycolytic enzymes like chitinase and cellulase are effective against a variety of plant pathogenic diseases by degrading the cell wall of pathogens (Shafi et al. 2017). For example, Bacillus sp. X-b secreting a complex of hydrolytic enzymes had broad-spectrum activity against pathogenic fungi (Helisto et al. 2001). The genes encoding chitinase (chi01), chitosanase (csn), cellulase (bglC, bglS, and bglH) and 35 genes related to proteases were present in the genome of LJBS06 (Table S5).
It was reported that defense-related oxidative enzymes such as PPO and PO produced by Bacillus species involve in plant defense by inducing lignin and phenol oxidation. The production of PPO and POD in plant–pathogen interactions reduced severity of disease symptoms, showing oxidative enzymes played an important role against plant pathogens (Jiang et al. 2018; Thipyapong and Steffens 1997). Besides, SOD and CAT produced by Bacillus spp. reduced the level of reactive oxygen species in plant, alleviating pathogens from plant tissue damage (Leal et al. 2021). There were genes involved in the synthesis of oxidoreductases, including PPO (ylmD), SOD (sodA), POD (osmC and oxyR), and CAT (katE) in Bacillus stercoris LJBS06 (Table S5). The function of annotated genes related to the synthesis of defense-related enzymes was validated at a metabolic level, observing that LJBS06 was able to produce chitinase (0.10 ± 0.00 U mL−1), chitosanase (0.10 ± 0.00 U mL−1), cellulase (0.50 ± 0.02 U mL−1), protease (1.26 ± 0.03 U mL−1), PPO (2.10 ± 0.08 U mL−1), SOD (3.28 ± 0.22 U mL−1), POD (145.65 ± 5.34 U mL−1), and CAT (3.01 ± 0.13 U mL−1). It was widely reported that Bacillus spp. promoted growth indirectly enhancing plant resistance (Shafi et al. 2017). The genome of B. stercoris LJBS06 contained a series of genes associated with plant growth promotion, including phosphate metabolism (pho and pst), nitrogen metabolism (nif and glt), IAA production (trpABCDEFS), volatile organic compounds (aco, acu, als, and isp), and arginine (spe) (Table S6).
Genes related to antifungal secondary metabolites
The bioactive secondary metabolites from Bacillus species have been applied as plant pathogen control agents (Kaspar et al. 2019). Eleven secondary metabolite biosynthetic gene clusters were found, covering about 13.4% of the genome of LJBS06. There were two non-ribosomal peptide synthetase (NRPS) gene clusters, two terpene gene clusters, one type III polyketides (T3PKS) gene cluster, one sactipeptide gene cluster, one NRPS-T1PKS gene cluster, one NRPS-T3PKS-transAT-PKS gene cluster, one NRPS-betalactone gene cluster, one LAP-thiopeptide gene cluster, and one ‘other’ type of gene cluster in the genome of B. stercoris LJBS06 (Table 4). Gene cluster 1 had 82% similarity with surfactin biosynthetic clustered genes, suggesting that LJBS06 could produce a new kind of surfactin. Gene clusters 3, 4, 8, 10, and 11 had 100% similarity with bacillaene, fengycin, bacillibactin, subtilosin A, and bacilysin biosynthetic clustered genes, respectively. The above highly similar gene clusters and their core genes are shown in Fig. 4. In addition, gene cluster 2 had 18% similarity with zwittermicin A and 4 of the 11 clusters did not show any similarity clusters in the database.

The known secondary metabolite core gene clusters identified in B. stercoris LJBS06. AntiSMASH 6.0 was used to predict potential secondary metabolite biosynthetic gene clusters. A minimum of 80% cut-off was set for visualization of gene clusters
Cyclic lipopeptides, synthesized by NRPSs, were well known for their powerful antagonistic action against a wide range of plant pathogens (Fira et al. 2018). Surfactins exhibited a wide range of antimicrobial and antiviral activities relying on its membrane permeabilization properties (Kaspar et al. 2019). For example, the mycelia of phytopathogens were significantly enlarged, twisted and broken after coculturing with surfactin-producing B.velezensis ZF2 (Xu et al. 2020a, b). Fengycins showed the spectrum of antifungal activity through a membrane disruption mechanism to cause the death of pathogens. Importantly, surfactins as biosurfactants working together with fengycin, exhibited a synergistic inhibitory effect on phytopathogens. Cyclic lipopeptides (surfactin and fengycin) were secreted from B. amyloliquefaciens FJAT-2349 as potential biocontrol agents against tomato bacterial wilt (Chen et al. 2019). Surfactin and fengycin from B. subtilis contributed to the highly active against grape downy mildew (Li et al. 2019). The lipopeptides surfactin and fengycin as signals induce plant defense responses by increasing the expression of the biocontrol genes PR-1, PR-4, SOD-2, PIN-1, and PIN-3 (Ongena et al. 2007; Yanez-Mendizabal and Falconi 2021). Besides, surfactins were essential for biofilm formation and root colonization, which facilitated the ability of promoting growth to increase plant resistance. Primary gene clusters responsible for surfactin (srfAA to srfAD) and fengycin (fenA to fenE) biosynthesis were present in the genome, indicating that B. stercoris LJBS06 has the potential to produce surfactin and fengycin to control diseases.
In addition, gene clusters encoding other antimicrobial compounds were also present in the genome of LJBS06, including zwittermicin A (zmaA to zmaQ), bacilysin (bacA to bacE), siderophore bacillibactin (dhbABCDEF), bacillaene (bae) and subtilosin A (sbo and ywiA). Zwittermicin A was a highly polar, water-soluble, broad-spectrum linear aminopolyol antibiotic with significant activity against bacteria, fungi, and protists. For example, B. cereus UW85 had the ability to suppress damping-off of alfalfa by producing Zwittermicin A (Milner et al. 1996). Bacilysin as a dipeptide antibiotic exhibited activity against a wide range of pathogens by destroying the structure of the microbial cell wall. Bacillus spp. with the production of bacilysin were able to control plant diseases, such as bacterial diseases of rice, and potato ring rot (Wu et al. 2015a, b). Siderophores as metal-chelating can deprive iron to alter pathogen fitness and aggressiveness, especially under iron starvation conditions (Khan et al. 2018). Furthermore, microbial siderophores provided iron for plant growth to enhance plant disease resistance indirectly. Bacillus species able to produce siderophores as an important mechanism for inhibiting the growth of phytopathogens, has been proven to be a potential biocontrol agent (Hu and Xu 2011). Bacillaene, a polyene antibiotic, selectively inhibits protein biosynthesis in prokaryotes (Olishevska et al. 2019). Subtilosin A as a cyclical peptide, against phytopathogenic fungi and bacterial, were detected in the genome. The existence of bacillaene and subtilosin A, which worked with other bioactive compounds, enhanced the antibacterial activity of LJBS06. In summary, there were genes responsible for surfactin, fengycin, zwittermicin A, bacilysin, siderophore bacillibactin, bacillaene, and subtilosin A in the genome of LJBS06, showing that LJBS06 had the potential as natural antagonists of phytopathogens through multiple secondary metabolites.
Conclusion
Bacillus stercoris LJBS06 isolated from grape rhizosphere soil, had a broad inhibitory spectrum against fungal pathogens. Whole-genome sequencing and genomic analysis suggested that it encoded numerous potential genes responsible for plant biocontrol mechanisms. B. stercoris LJBS06 harbored genes involved in the production of plant defense-related enzymes to prevent further infection of pathogens and the promotion of plant growth to enhance resistance indirectly. Meanwhile, there were 11 gene clusters related to the synthesis of secondary metabolites as important antimicrobial compounds. In conclusion, our data indicated that LJBS06 strain had the necessary genetic machinery to control plant diseases through potential direct and indirect mechanisms, including production of defense-related enzymes, secondary metabolites, and promotion of plant growth. These features made it a promising candidate for field application.
Genome sequence accession number
The complete sequence of the chromosome of the strain B. stercoris LJBS06 and the sequence of the plasmid have been deposited in GenBank database under the accession number CP070483 and CP070484, respectively. The strain B. stercoris LJBS06 was publicly available from China General Microbiological Culture Collection Center (CGMCC) under the accession number CGMCC No. 21263.
Availability of data and material
The GenBank accession number for the 16S rRNA gene of Bacillus stercoris LJBS06 is MW940707. The complete sequence of the chromosome of the strain B. stercoris LJBS06 and the sequence of the plasmid have been deposited in GenBank database under the accession number CP070483 and CP070484, respectively. The strain B. stercoris LJBS06 was publicly available from China General Microbiological Culture Collection Center (CGMCC) under the accession number CGMCC No. 21263. General characteristics of B. stercoris LJBS06, effect of LJBS06 on the mycelia of C. gloeosporioides, plate assays for the activities of extracellular enzymes, genome statistics, COG categories of the chromosome and the plasmid of B. stercoris LJBS06, inhibition diameter of B. stercoris LJBS06 against 12 pathogens, genes related to defense-related enzymes in the LJBS06 genome, and genes attributed to plant growth promoting traits in the LJBS06 genome are available as Supplementary Materials.
Code availability
Not applicable.
References
Aboutorabi M (2018) A review on the biological control of plant diseases using various microorganisms. J Res Med Dent Sci 6(4):30–35
Ahemad M, Kibret M (2014) Mechanisms and applications of plant growth promoting rhizobacteria: current perspective. J King Saud Univ Sci 26(1):1–20. https://doi.org/10.1016/j.jksus.2013.05.001
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene Ontology C (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25–29. https://doi.org/10.1038/75556
Bertelli C, Laird MR, Williams KP, Lau BY, Hoad G, Winsor GL, Brinkman FSL, Simon Fraser Univ Res Comp G (2017) IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res 45(W1):W30–W35. https://doi.org/10.1093/nar/gkx343
Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29(12):2607–2618. https://doi.org/10.1093/nar/29.12.2607
Blake C, Christensen MN, Kovacs AT (2021) Molecular aspects of plant growth promotion and protection by Bacillus subtilis. Mol Plant Microbe Interact 34(1):15–25. https://doi.org/10.1094/MPMI-08-20-0225-CR
Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, Weber T (2021) antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. https://doi.org/10.1093/nar/gkab335
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31(1):365–370. https://doi.org/10.1093/nar/gkg095
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12(1):59–60. https://doi.org/10.1038/nmeth.3176
Byun H-D, Bin HS, Bae S, Han J-M, Hyun C-G (2020) Bacillus subtilis JNUCC Isolated from Galchisokjeot: draft genome sequence and alpha-glucosidase and Tyrosinase inhibitory activities. J Pure Appl Microbiol 14(1):189–193. https://doi.org/10.22207/jpam.14.1.20
Chen YJ, Yu P, Luo JC, Jiang Y (2003) Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm Genome 14(12):859–865. https://doi.org/10.1007/s00335-003-2296-6
Chen L, Gu W, Xu HY, Yang GL, Shan XF, Chen G, Wang CF, Qian AD (2018) Complete genome sequence of Bacillus velezensis 157 isolated from Eucommia ulmoides with pathogenic bacteria inhibiting and lignocellulolytic enzymes production by SSF. 3 Biotech 8(2):114. https://doi.org/10.1007/s13205-018-1125-2
Chen MC, Wang JP, Zhu YJ, Liu B, Yang WJ, Ruan CQ (2019) Antibacterial activity against Ralstonia solanacearum of the lipopeptides secreted from the Bacillus amyloliquefaciens strain FJAT-2349. J Appl Microbiol 126(5):1519–1529. https://doi.org/10.1111/jam.14213
Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O’Malley R, Figueroa-Balderas R, Morales-Cruz A, Cramer GR, Delledonne M, Luo C, Ecker JR, Cantu D, Rank DR, Schatz MC (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13(12):1050. https://doi.org/10.1038/nmeth.4035
Chun J, Oren A, Ventosa A, Christensen H, Arahal DR, da Costa MS, Rooney AP, Yi H, Xu XW, De Meyer S, Trujillo ME (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 68(1):461–466. https://doi.org/10.1099/ijsem.0.002516
Dhruw C, Husain K, Kumar V, Sonawane VC (2020) Novel xylanase producing Bacillus strain X2: molecular phylogenetic analysis and its application for production of xylooligosaccharides. 3 Biotech. https://doi.org/10.1007/s13205-020-02322-1
Djordje F, Ivica D, Tanja B, Jelena L, Slaviša S (2018) Biological control of plant pathogens by Bacillus species. J Biotechnol 285:44–55
Dunlap CA, Bowman MJ, Zeigler DR (2020) Promotion of Bacillus subtilis subsp. inaquosorum, Bacillus subtilis subsp. spizizenii and Bacillus subtilis subsp. stercoris to species status. Antonie Van Leeuwenhoek 113(1):1–12. https://doi.org/10.1007/s10482-019-01354-9
Fan H, Ru J, Zhang Y, Wang Q, Li Y (2017) Fengycin produced by Bacillus subtilis 9407 plays a major role in the biocontrol of apple ring rot disease. Microbiol Res 199:89–97. https://doi.org/10.1016/j.micres.2017.03.004
Fira D, Dimkić I, Berić T, Lozo J, Stanković S (2018) Biological control of plant pathogens by Bacillus species. J Biotechnol 285:44–55. https://doi.org/10.1016/j.jbiotec.2018.07.044
Fu L, Penton CR, Ruan Y, Shen Z, Xue C, Li R, Shen Q (2017) Inducing the rhizosphere microbiome by biofertilizer application to suppress banana Fusarium wilt disease. Soil Biol Biochem 104:39–48. https://doi.org/10.1016/j.soilbio.2016.10.008
Gao Z, Zhang B, Liu H, Han J, Zhang Y (2017) Identification of endophytic Bacillus velezensis ZSY-1 strain and antifungal activity of its volatile compounds against Alternaria solani and Botrytis cinerea. Biol Control 105:27–39. https://doi.org/10.1016/j.biocontrol.2016.11.007
Gao H, Li P, Xu X, Zeng Q, Guan W (2018) Research on volatile organic compounds from Bacillus subtilis CF-3: biocontrol effects on fruit fungal pathogens and dynamic changes during fermentation. Front Microbiol. https://doi.org/10.3389/fmicb.2018.00456
Grissa I, Vergnaud G, Pourcel C (2007) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. https://doi.org/10.1093/nar/gkm360
Guerrero R (2001) Bergey’s manuals and the classification of prokaryotes. Int Microbiol 4(2):103–109
Guo W, Chen Z, Cui P, Chen X (2015) Draft genome of Bacillus sp A053 isolated from the Arctic seawater with antimicrobial activity. Mar Genom 22:19–21. https://doi.org/10.1016/j.margen.2015.03.008
Hakamada Y, Koike K, Kobayashi T, Ito S (1997) Purification and properties of mangano-superoxide dismutase from a strain of alkaliphilic Bacillus. Extremophiles 1(2):74–78. https://doi.org/10.1007/s007920050017
Helisto P, Aktuganov G, Galimzianova N, Melentjev A, Korpela T (2001) Lytic enzyme complex of an antagonistic Bacillus sp., X-b: isolation and purification of components. J Chromatogr B 758(2):197–205. https://doi.org/10.1016/s0378-4347(01)00181-5
Hu Q-P, Xu J-G (2011) A simple double-layered chrome azurol S agar (SD-CASA) plate assay to optimize the production of siderophores by a potential biocontrol agent Bacillus. Afr J Microbiol Res 5(25):4321–4327. https://doi.org/10.5897/ajmr11.238
Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, Bork P (2008) eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 36:D250–D254. https://doi.org/10.1093/nar/gkm796
Jiang C-H, Liao M-J, Wang H-K, Zheng M-Z, Xu J-J, Guo J-H (2018) Bacillus velezensis, a potential and efficient biocontrol agent in control of pepper gray mold caused by Botrytis cinerea. Biol Control 126:147–157. https://doi.org/10.1016/j.biocontrol.2018.07.017
Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, Bateman A, Finn RD, Petrov AI (2018a) Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 46(D1):D335–D342. https://doi.org/10.1093/nar/gkx1038
Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, Petrov AI (2018b) Non-Coding RNA analysis using the Rfam database. Curr Protoc Bioinform 62(1):e51–e51. https://doi.org/10.1002/cpbi.51
Kaspar F, Neubauer P, Gimpel M (2019) Bioactive secondary metabolites from Bacillus subtilis: a comprehensive review. J Nat Prod 82(7):2038–2053. https://doi.org/10.1021/acs.jnatprod.9b00110
Khan A, Singh P, Srivastava A (2018) Synthesis, nature and utility of universal iron chelator—siderophore: a review. Microbiol Res 212:103–111. https://doi.org/10.1016/j.micres.2017.10.012
Khurshed N, Shakoori AR (2005) Characterization of locally isolated strains of Bacillus and their evaluation as potential biocides against house fly, Musca domestica. Pak J Zool 37(4):249–255
Kim JS, Lee J, Lee CH, Woo SY, Kang H, Seo SG, Kim SH (2015) Activation of pathogenesis-related genes by the rhizobacterium, Bacillus sp. JS, which induces systemic resistance in Tobacco plants. Plant Pathol J 31(2):195–201. https://doi.org/10.5423/PPJ.NT.11.2014.0122
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27(5):722–736. https://doi.org/10.1101/gr.215087.116
Krogh A, Brown M, Mian IS, Sjolander K, Haussler D (1994) Hidden Markov models in computational biology: applications to protein modeling. J Mol Biol 235(5):1501–1531. https://doi.org/10.1006/jmbi.1994.1104
Leal C, Fontaine F, Aziz A, Egas C, Clément C, Trotel-Aziz P (2021) Genome sequence analysis of the beneficial Bacillus subtilis PTA-271 isolated from a Vitis vinifera (cv. Chardonnay) rhizospheric soil: assets for sustainable biocontrol. Environ Microbiome. https://doi.org/10.1186/s40793-021-00372-3
Li Y, Heloir M-C, Zhang X, Geissler M, Trouvelot S, Jacquens L, Henkel M, Su X, Fang X, Wang Q, Adrian M (2019) Surfactin and fengycin contribute to the protection of a Bacillus subtilis strain against grape downy mildew by both direct effect and defense stimulation. Mol Plant Pathol 20(8):1037–1050. https://doi.org/10.1111/mpp.12809
Liang TW, Chen YY, Pan PS, Wang SL (2014) Purification of chitinase/chitosanase from Bacillus cereus and discovery of an enzyme inhibitor. Int J Biol Macromol 63:8–14. https://doi.org/10.1016/j.ijbiomac.2013.10.027
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964. https://doi.org/10.1093/nar/25.5.955
Mayende L, Wilhelmi B, Pletschke B (2006) Cellulases (CMCases) and polyphenol oxidases from thermophilic Bacillus spp. isolated from compost. Soil Biol Biochem 38(9):2963–2966. https://doi.org/10.1016/j.soilbio.2006.03.019
Meier-Kolthoff JP, Auch AF, Klenk H-P, Goeker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. https://doi.org/10.1186/1471-2105-14-60
Miljakovic D, Marinkovic J, Balesevic-Tubic S (2020) The significance of Bacillus spp. in disease suppression and growth promotion of field and vegetable crops. Microorganisms. https://doi.org/10.3390/microorganisms8071037
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428. https://doi.org/10.1021/ac6014a030
Milner JL, Stohl EA, Handelsman J (1996) Zwittermicin a resistance gene from Bacillus cereus. J Bacteriol 178(14):4266–4272. https://doi.org/10.1128/jb.178.14.4266-4272.1996
Mohammad FG, Alireza T (2007) Isolation and characterization of polyphenol oxidase- and peroxidase-producing Bacillus strains from fully fermented tea (Camellia sinensis). World J Microbiol Biotechnol 23(9):1327–1332. https://doi.org/10.1007/s11274-007-9363-1
Nicholson WL, Munakata N, Horneck G, Melosh HJ, Setlow P (2000) Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol Mol Biol Rev 64(3):548. https://doi.org/10.1128/mmbr.64.3.548-572.2000
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M (1999) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 27(1):29–34. https://doi.org/10.1093/nar/27.1.29
Olishevska S, Nickzad A, Deziel E (2019) Bacillus and Paenibacillus secreted polyketides and peptides involved in controlling human and plant pathogens. Appl Microbiol Biotechnol 103(3):1189–1215. https://doi.org/10.1007/s00253-018-9541-0
Ongena M, Jourdan E, Adam A, Paquot M, Brans A, Joris B, Arpigny J-L, Thonart P (2007) Surfactin and fengycin lipopeptides of Bacillus subtilis as elicitors of induced systemic resistance in plants. Environ Microbiol 9(4):1084–1090. https://doi.org/10.1111/j.1462-2920.2006.01202.x
Paudel YP, Qin W (2015) Characterization of novel cellulase-producing bacteria isolated from rotting wood samples. Appl Biochem Biotechnol 177(5):1186–1198. https://doi.org/10.1007/s12010-015-1806-9
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10):785–786. https://doi.org/10.1038/nmeth.1701
Qiao J, Yu X, Liang X, Liu Y, Borriss R, Liu Y (2017) Addition of plant-growth-promoting Bacillus subtilis PTS-394 on tomato rhizosphere has no durable impact on composition of root microbiome. BMC Microbiol 17:1–12. https://doi.org/10.1186/s12866-017-1039-x
Rajput S, Tiwari E (2012) Study the effect of various pH on the protease producing capacity of Bacillus. J Pure Appl Microbiol 6(1):501–503
Richter M, Rossello-Mora R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106(45):19126–19131. https://doi.org/10.1073/pnas.0906412106
Santana MA, Moccia-V CC, Gillis AE (2008) Bacillus thuringiensis improved isolation methodology from soil samples. J Microbiol Methods 75(2):357–358. https://doi.org/10.1016/j.mimet.2008.06.008
Sari G, Trihadiningrum Y, Ni’matuzahroh N (2019) Isolation and Identification of native bacteria from total petroleum hydrocarbon polluted soil in wonocolo public oilfields, Indonesia. J Ecol Eng 20(8):60–64
Shafi J, Tian H, Ji M (2017) Bacillus species as versatile weapons for plant pathogens: a review. Biotechnol Biotechnol Equip 31(3):446–459. https://doi.org/10.1080/13102818.2017.1286950
Thipyapong P, Steffens JC (1997) Tomato polyphenol oxidase—differential response of the polyphenol oxidase F promoter to injuries and wound signals. Plant Physiol 115(2):409–418. https://doi.org/10.1104/pp.115.2.409
Valenzuela-Ruiz V, Robles-Montoya RI, Parra-Cota FI, Santoyo G, Del Carmen O-MM, Rodriguez-Ramirez R, de Los S-VS (2019) Draft genome sequence of Bacillus paralicheniformis TRQ65, a biological control agent and plant growth-promoting bacterium isolated from wheat (Triticum turgidum subsp. durum) rhizosphere in the Yaqui Valley, Mexico. 3 Biotech 9(11):436. https://doi.org/10.1007/s13205-019-1972-5
Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, Earl AM (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. https://doi.org/10.1371/journal.pone.0112963
Wu L, Wu H, Chen L, Lin L, Borriss R, Gao X (2015a) Bacilysin overproduction in Bacillus amyloliquefaciens FZB42 markerless derivative strains FZBREP and FZBSPA enhances antibacterial activity. Appl Microbiol Biotechnol 99(10):4255–4263. https://doi.org/10.1007/s00253-014-6251-0
Wu L, Wu H, Chen L, Yu X, Borriss R, Gao X (2015b) Difficidin and bacilysin from Bacillus amyloliquefaciens FZB42 have antibacterial activity against Xanthomonas oryzae rice pathogens. Sci Rep. https://doi.org/10.1038/srep12975
Xu L, Ning X, Chai H, Fei G (2020a) Preliminary evaluation of irradiated medium and the optimization of conditions for a catalase produced by Bacillus firmus GL3. J Food Meas Charact 14(2):1073–1084. https://doi.org/10.1007/s11694-019-00357-4
Xu S, Xie X, Zhao Y, Shi Y, Chai A, Li L, Li B (2020b) Whole-genome analysis of Bacillus velezensis ZF2, a biocontrol agent that protects cucumis sativus against corynespora leaf spot diseases. 3 Biotech 10(4):186. https://doi.org/10.1007/s13205-020-2165-y
Yanez-Mendizabal V, Falconi CE (2021) Bacillus subtilis CtpxS2-1 induces systemic resistance against anthracnose in Andean lupin by lipopeptide production. Biotech Lett 43(3):719–728. https://doi.org/10.1007/s10529-020-03066-x
Yang J-H, Kim I-S, Oh C-H, Oh N-S (2012) Protease and γ-glutamyltranspeptidase activities of Bacillus spp. isolated from rice straw. Food Sci Biotechnol 21(1):291–294. https://doi.org/10.1007/s10068-012-0039-8
Yang J, Zhu X, Cao M, Wang C, Zhang C, Lu Z, Lu F (2016) Genomics-inspired discovery of three antibacterial active metabolites, aurantinins B, C, and D from compost-associated Bacillus subtilis fmb60. J Agric Food Chem 64(46):8811–8820. https://doi.org/10.1021/acs.jafc.6b04455
Zhang M, Li J, Shen A, Tan S, Yan Z, Yu Y, Xue Z, Tan T, Zeng L (2016) Isolation and Identification of Bacillus amyloliquefaciens IBFCBF-1 with potential for biological control of phytophthora blight and growth promotion of pepper. J Phytopathol 164(11–12):1012–1021. https://doi.org/10.1111/jph.12522
Zhang X, Zhou Y, Li Y, Fu X, Wang Q (2017) Screening and characterization of endophytic Bacillus for biocontrol of grapevine downy mildew. Crop Prot 96:173–179. https://doi.org/10.1016/j.cropro.2017.02.018
Funding
This work was supported by Shanghai Municipal Commission for Science and Technology (18391900400), National Key R & D Program of China (2019YFD1002501), and China Postdoctoral Science Foundation (18Z102060107 and 20Z102060031).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by BW, HP, WW, BY, YC, FX, YP, PF. The first draft of the manuscript was written by BW and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
All authors approved the manuscript.
Consent for publication
Written informed consent for publication was obtained from all participants.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, B., Peng, H., Wu, W. et al. Genomic insights into biocontrol potential of Bacillus stercoris LJBS06. 3 Biotech 11, 458 (2021). https://doi.org/10.1007/s13205-021-03000-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-021-03000-6