
Abstract
A Bacillus strain X2 that produced extracellular endo-xylanase (GH 11) (EC: 3.2.1.8) was isolated from the soil of the Northeast India region. This aerobic culture was Gram positive and endospore forming. Chemotaxonomic characterization showed variance with the fatty acid profile of related species in the Bacillus subtilis group. In Bacillus strain X2, distinct occurrence of iso-C14:0 lipids is absent in other related species. The 16S rRNA gene sequence homology showed 99% similarity with Bacillus subtilis subsp. inaquosorum. The phylogenetic analysis by the multilocus sequence analysis (MLSA) of the nucleotide sequence of six concatenated genes (16S rRNA, groEL, gyrA, polC, purH and rpoB) resolved the taxonomic position of the Bacillus strain X2 in the Bacillus subtilis subsp. group. The MLSA showed that it is a member of a clade that includes Bacillus subtilis subsp. stercoris. In in silico DNA–DNA hybridization (DDH), the highest matching score was obtained with Bacillus subtilis subsp. stercoris (87%). The in silico DDH of the genome (G + C 43.7 mol %) shared 48.5%, with Bacillus subtilis subsp. inaquosorum. The MLSA phylogenetic tree and the highest degree of DNA hybridization, indicating that it belongs to the Bacillus subtilis subspecies stercoris.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Bacillus genus comprises a rod-shaped aerobic, endospore-forming group of bacteria that have been classified under the phylum Firmicutes (Logan and De Vos 2009). They are ubiquitous in nature and are found in diverse ecological habitats due to their ability to metabolize a wide range substrates and to form endospores in adverse conditions (Zwick et al. 2012). Recently, many new Bacillus species which were classified as Bacillus subtilis have been reclassified as B. atrophaeus, B. velezensis, B. tequilensis and more (Nakamura 1989; Roberts et al. 1994, 1996; Palmisano et al. 2001; Gatson et al. 2006; Wang et al. 2007, 2008).
A xylanolytic Bacillus strain X2 was isolated from decaying wood samples collected from the northeast state of India, Assam. This aerobic culture was Gram positive and endospore forming. The 16S rRNA gene sequence homology showed 99% similarity with Bacillus subtilis subsp. inaquosorum; however, the in silico DDH study revealed that the homology shared was only 48.5%. Due to this anomaly in these two taxonomic properties, exact identification was not done and it was necessary to resolve the taxonomic position of the isolate X2 for further characterization (Tindall et al. 2010). It is known that due to the complexity in the classification of Bacillus subtilis species, the phylogenetic association between different subspecies under this group cannot be determined only on the basis of phenotypic and biochemical characteristics (polyphasic) or 16S rRNA gene systematics. Genomics based approach offers an opportunity for the resolution of the taxonomic description of a new strain putatively identified as Bacillus subtilis on the basis of 16S rRNA gene homology. The taxonomic position of many species in the genus Bacillus has been ascertained and reclassified by using many parameters such as MLSA and the whole genome sequence identity method (Stackebrandt et al. 2002; Gevers et al. 2005). In 1995, two subgroups of Bacillus subtilis were separated based on the multilocus sequence alignment (MLSA) method (Roberts and Cohan 1995). These groups had 58–69% homology on the basis of DNA–-DNA hybridization and sexual isolation on the basis of DNA donor/recipient ability. On this basis, Nakamura et al. (Nakamura et al. 1999) proposed two groups, i.e., B. subtilis subsp. subtilis and B. subtilis subsp. spizizenii. Further resolution of the subspecies spizizenii was done by Rooney et al. (Rooney et al. 2009) using MLSA combined with MALDI-ToF analysis to determine the existence of two different taxonomic subspecies as B. subtilis subsp. spizizenii and B. subtilis subsp. inaquosorum. The uses of high-throughput data (Genome sequence) in taxonomy have enabled circumscribing the classification of the Bacillus group. Based on molecular phylogeny and in silico DDH, Dunlap and his coworkers rationalized the taxonomical classification among Bacillus amyloliquefaciens subsp. plantarum, Bacillus velezensis, Bacillus axarquiensis, and Bacillus malacitensis (Dunlap et al. 2016a, b).
In this study, we investigated the taxonomical position of Bacillus strain X2 using the multilocus sequence phylogenetics as well as phylogenomics analysis for comparing it with a novel taxa, Bacillus subtilis subspecies stercoris (Adelskov and Patel 2017) within the Bacillus subtilis complex, which has become a model microorganism for the study of bacterial divergence (Cohan and Kane 2001; Cohan 2002, 2006; Perry et al. 2006).
Most of the species of genus Bacillus are non-pathogenic and non-toxic, hence they have been given the generally recognized as safe (GRAS) status and are important candidates for industrial applications. The metabolic capacity of Bacillus has been harnessed by the industry for the production of riboflavin, streptavidin, and beta-lactamase (Maughan and Van der Auwera 2011; Oraei et al. 2018). Various Bacillus subtilis strains are the major source of lignocellulose degrading enzymes, particularly xylanase, which are used for the production of prebiotics such as xylooligosaccharides using the xylan from various plant sources (Rhee et al. 2014; Reddy and Krishnan 2016a). Xylan is the major hemicellulose of plant origin, which can be used as the substrate for the production of xylooligosaccharides (XOS) (Vázquez et al. 2001; Huang et al. 2017). This novel strain of Bacillus has the ability to produce extracellular endo-xylanase which can convert xylan of agricultural residue origin for the production of prebiotics.
Materials and methods
Chemicals
Oat spelt xylan was obtained from Sigma Chemicals (St. Louis, MO, US), beechwood xylan from CDH, India, birchwood xylan was from Sigma Chemicals (St. Louis, MO, USA), and corncob xylan was obtained from CDH, India. Congo red was procured from Hi-Media, India. Reference standard xylooligosaccharides xylobiose and xylotriose were purchased from Megazyme (Bray, Ireland). All other chemicals used were of analytical grade.
Microorganisms
Type strains of Bacillus subtilis subsp. subtilis MCC 2049T (ATCC 6051T) and Bacillus subtilis subsp. spizizenii MCC 2480T (JCM 12233T) were procured from microbial culture collection NCCS Pune, Maharashtra, India.
Isolation and preservation of the Bacillus strain X2
Decaying wood from the guest house garden of CSIR-NEIST Jorhat, Assam, India (26.74° N, 94.16° E) was serially tenfold diluted with sterile phosphate buffer saline. Aliquot of 100 μl of every dilution was spread over the nutrient agar (NA) plate and incubated at 37 °C for 24 h. Based on the morphology of the individual isolated colony, different isolates were picked up and spot inoculated on two nutrient agar plates containing 0.5% birchwood xylan and further incubated for 24 h. After 24 h, one of the nutrient birchwood agar plates was flooded with 5 ml 1% Congo red dye in distilled water. After 5 min in the dye solution, the excess dye was discarded and the plate was serially rinsed with 1 M NaCl three to four times. The colonies producing unstained zone around them were selected as xylanases-producing strain. The most promising strain producing a 15 mm zone of discoloration was selected for further investigation. After confirming the purity of the strain by streaking on the NA plate as well as Gram staining, the strain was maintained in the laboratory as Bacillus sp. strain X2. All cultures were maintained on NA slant at 4˚C in the refrigerator and long-term storage was done in 15% glycerol in a deep freezer at – 80 °C.
Phenotypic and physiological characterization
Cell morphology and cell surface structure of pure culture bacterial cells were inspected under the scanning electron microscope (EVO 40, ZEISS), Bacteria were grown in LB broth for 24 h, followed by washing with phosphate-buffered saline (PBS). The washed bacterial pellet was resuspended in sterile PBS and a smear was made on a sterile clean glass slide. The slide was kept in modified Karnovsky’s fixative for 3 h following washing and dehydrated gradually. After dehydration of cells adhering to the slide, the slide was treated with tert-butyl alcohol for 45 min. The treated slide was freeze dried. The specimen was metal coated by gold palladium sputtering at 12–15 mA. The scanning electron micrograph was obtained at 20.0 kV operating voltage and magnification obtained was 10 kX. The image was recorded by an in-built Nikon digital camera. Spore staining using Malachite green was done for confirming the endospore formation and was observed under the phase contrast microscope after growing the strain X2 on the sporulating agar medium for a week (Logan et al. 2009). Gram reaction of the strains was carried out with KOH lysis (Moaledj 1986).
Different biochemical tests such as nitrate reduction, aesculin hydrolysis, catalase, indole production, oxidase activities, H2S production, urea, and gelatin hydrolysis of species were carried out as described by Lanyí (1988) and Smibert and Krieg (1994). Many extracellular enzymatic activities such as protease, lipase, and amylase were tested using the activity measurement described by Srinivas et al. (2009). Antibiotic susceptibility, acid production, and carbon substrate utilization of the strains were executed using the earlier described methods by Anil et al. (2012). Following the manufacturer’s protocol, biochemical and enzyme characterization was also performed using the Vitek 2 GP system (bioM'erieux). Standard phenotypic tests were performed to characterize the strain X2. Growth at different NaCl concentrations at 2–12% (w/v) and different temperatures range 5–70 °C were examined. To determine the pH range for growth, TSA was buffered with citric acid/NaOH (for pH 5.0 and 6.0), NaHPO4/Na2HPO4 (for pH 7.0–8.0), glycine/NaOH (for pH 9.0–10.0), Na2HPO4/NaOH (for pH 11) and KCl/NaOH (for pH 12) (Xu et al. 2005).
Chemotaxonomy characterization
For cellular fatty acid analysis of strain X2, Bacillus subtilis subsp. subtilis MCC 2049T and Bacillus subtilis subsp. spizizenii MCC 2480T were grown on TSA plates at 37 °C for 20 h, and at the logarithmic phase, the growth was harvested. MIDI system (MIS operating manual version 6.1) protocol was performed for cellular fatty acid methyl esters (FAMEs) sample preparation by saponification and methylation, followed by extraction. FAMEs were analyzed by gas chromatography (Agilent model 7890A). Samples were heated by increasing the temperature at a rate of 1 °C min−1 till 300 °C. Fatty acid profiles were compared with those in the MIS library of midi (TSBA 6.0 library).
MALDI-ToF MS was used to determine the lipopeptide biomarker profiles followed the method of Price et al. (2007). Mass spectra were acquired on a Bruker-Daltoniks Microflex instrument operating in reflectron mode. Ion source 1 and 2 was set at 20.0 kV and 18.25 kV, respectively, with lens voltage 9.9 kV and reflector voltages of 20.00 kV. 2,5-Dihydrobenzoic acid matrix (10 mg/ml in acetonitrile) with aqueous samples were applied on Bruker-Daltoniks MSP 96 polished steel target. Bruker bacterial test standard was used as the reference. The instrument was run by flex control software and data were analyzed by flex analysis software in conjunction with MALDI BIOTYPER real time (3.0).
Genotyping of Bacillus strain X2 and MLSA
For genomic DNA isolation, Bacillus strain X2 was grown overnight at 37 °C in the NA medium. The bacterial culture was treated as the process described by Marmur (1961). 16S rRNA gene sequencing was done as described earlier (Lane 1991). The 16S rRNA gene sequence of the strain Bacillus strain X2 (1526 bp) was queried in the EzTaxon server (Chun et al. 2007). Nucleotide BLAST sequence similarity search (Altschul et al. 1990) was used to find the neighboring taxa. All the 16S rRNA gene sequences of closely related species of the genus Bacillus were extracted from the NCBI database (www.ncbi.nlm.nih.gov). For MLSA, partial sequence of the groEL, gyrA, polC, purH, and rpoB genes from 36 Bacillus species strain’s genome were taken. Sequences of each gene from all strains were aligned using MUSCLE. Genes sequence lengths were: 16S rRNA—1270 bp, groEL—1605 bp, gyrA—2077 bp, polC—2407 bp, purH—1542 bp and rpoB—3444 bp. These sequences were concatenated into a single sequence of 12,345 bp long in a series of 16S-groEL-gyrA-polC-purH-rpoB. MEGA software version 7.0 was used for phylogenetic analysis (Kumar et al. 2016), Neighbor-joining method (Saitou and Nei 1987) and the Tamura–Nei model (Tamura and Nei 1993). Tamura–Nei model is suitable for high G + C content gene sequences and the above resultant sequences had 47% GC content. The resulting sequence was 12,058 bp after the elimination of all positions containing gaps or missing in the final construct. Bootstrap of 1000 replicates was performed to evaluate the confidence of the phylogenetic tree topology.
Genome sequencing and annotation
The phenol–chloroform method was used for isolating the total genomic DNA from Bacillus strain X2 (Sambrook and Russell 2006). The quality and integrity of the genomic DNA were confirmed by agarose gel electrophoresis. The purity and concentration of the genomic DNA preparation were determined by Nanodrop instrument (Thermo Scientific, MA, USA). DNA was used for library preparation with Illumina Nextseq 500 Kit protocol. DNA libraries were paired-end sequenced using Illumina Nextseq 500 adapters on an Illumina Nextseq 500 platform. Raw reads were analyzed using the FastQC tool, CLC bio Genomics Workbench version 7.50 (CGWB). 16S rRNA gene sequences from the genomes were extracted using RNAmmer 1.2 server (Lagesen et al. 2007) and the EzTaxon server was used to characterize the sequences (Chun et al. 2007). tRNA encoded in the genome was identified in the genome by tRNAScan-SE (Lowe and Eddy 1996). The genome annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline (https://www.ncbi.nlm.nih.gov/genome/annotation_prok/) and Rapid Annotation System Technology (RAST) pipeline (Aziz et al. 2008). SignalP 4.1 database was used to screen the genes with signal peptides (Petersen et al. 2011).
Construction of phylogenomic tree based on housekeeping protein sequences
Based on 16S rRNA gene sequence similarity according to EzTaxon, we retrieved the genomes of all the neighboring Bacillus species from the GenBank. The names of Bacillus species and their type species have been listed in Table 1S. The genomes of all the listed Bacillus species were loaded onto Amphoranet 2 server (Kerepesi et al. 2014) and amino acid sequences of housekeeping proteins were extracted from the respective genomes. The lists of 31 housekeeping proteins selected for the estimation of amino acid sequence similarity are given in Table 2S. The amino acid sequence of 31 housekeeping proteins was manually concatenated as a sequence from each strain and alignment of all concatemers was done using MUSCLE and the phylogenetic tree was constructed in MEGA 7 software (Kumar et al. 2016). The evolutionary history was calculated through the neighbor-joining method (Saitou and Nei 1987).
Sequence analysis of a Xyl_1 gene
Genomic DNA of B. subtilis X2 was used as a template for polymerase chain reaction (PCR) amplification. Specific primers Xyl_1F (5′ GATGGATCCGCAACTACAATCACCTCAAATCAA 3′) and Xyl_1R (5′ GATTCTAGATTATTGACTTTTCCCCCCAAC 3′) were designed for amplification of Xyl_1 gene. PCR conditions were as follows: step 1—hot start at 94 °C for 5 min, step 2—35 cycles of 94 °C for 30 s, step 3—55 °C for 30 s, step 4—72 °C for 3 min, and step 5—10 min of extension at 72 °C. The PCR product was purified and verified by sequencing. Structure-based sequence alignment was performed by using CLUSTAL W and exported by ESPript (Robert and Gouet 2014). All protein sequences were retrieved from GenBank.
Oligosaccharides production and quantification
Xylanase from the Bacillus strain X2 was isolated and purified by the conventional method of purification comprising ultrafiltration and fractionation by chromatography on Q-Sepharose and phenyl Sepharose (Manuscript under preparation). Purified xylanase was assayed by following Bailey’s method (Bailey et al. 1992). The definition of 1 IU of xylanase activity is the amount of enzyme that releases 1 µmol of reducing sugar as xylose per minute from birchwood xylan in 50 mM Tris–HCl, pH 8.0. Conversion of birchwood xylan into XOS was done by incubating 500 μl of 1% birchwood xylan in Tris–HCl buffer, pH8, with 1 IU of xylanase at 50 °C for 1 h. After incubation, the reaction products were filtered through a 0.2-µm filter. For the analysis of the reaction products, 20 µl content was injected onto the sugar pack I column (300 × 6.5 mm Waters, USA) connected to the HPLC (La’Chrome, Merck, Hitachi,) fitted with an RI detector (Waters 2414). The mobile phase was distilled water containing 50 mg/L calcium EDTA. The flow rate was 0.5 ml/min and the temperature of the column and detector was 70 °C and 30 °C, respectively. HPLC-grade xylose, glucose, xylobiose (Megazyme), and xylotriose (Megazyme) were used as standard.
Results and discussion
Phenotypic and biochemical characterization
Bacillus strain X2 formed irregular colonies with lobate margin, rough surface, and opaque in density (Fig. 1S). Based on electron microscopy, the cell morphology was confirmed as rod shaped (Fig. 1S). It was a terminal spore-forming, Gram-positive bacteria. The strain was alkali tolerant as it can grow in a pH range of 4.0–10.0. It is also a halophile, as it can tolerate and grow in 10% (w/v) NaCl concentration. The rest of the biochemical characteristics were similar to the reference type strains, viz. Bacillus subtilis subsp. subtilis and Bacillus subtilis subsp. spizizenii. The biochemical properties of strain X2 were almost similar and indistinguishable from that of Bacillus subtilis. Since the phenotypic characterization could not resolve any differences with respect to the reference strains, lipopeptide and fatty acid content analysis of bacterial cells through MALDI-ToF and FAME, respectively, were further conducted.
MALDI-ToF MS biomarker profiles over the m/z 300–1200 (Table 3S) and m/z 2000–20,000 (data not given) mass unit range were obtained. In the m/z 2000–20,000 range, any significant differential peak was not found. The abundance of lipopeptides ions was observed in the range m/z 300–1200 and subdivided into three mass ranges: 850–950 m/z, which includes kurstakins, 1000–1100 m/z, which includes surfactins and iturins, and 1100–1200 m/z, which includes polymyxins (Price et al. 2007). MALDI-ToF mass spectrometry analysis showed that Bacillus subtilis subsp. stercoris and strain X2 produce a novel Kurstakins-like lipopeptide with mass m/z 894.8, which was not observed in the Bacillus subtilis subsp. subtilis and Bacillus subtilis subsp. spizizenii. The mass spectra also revealed that the Bacillus strain X2 did not produce any polymyxins, whereas subsp. subtilis, subsp. spizizenii and subsp. stercoris produced it. Similarly, Rooney et.al. have also classified the Bacillus subtilis subsp. inaquosorum based on the production of a novel lipopeptides having m/z 1120.8 and 1106.8. A small ion (m/z 1106.8) was also found in B. subtilis subsp. spizizenii (Rooney et al. 2009). This small ion m/z 1106.5 was also found in subsp. stercoris and subsp. spizizenii in this study. It has been reported that the production pattern of different lipopeptides by different strains does not change significantly by using different media (Calderaro et al. 2014). Hence, the differential expression of lipopeptides in the strains belonging to the same subspecies could be the effect of environment pressure.
The FAME analysis
The total cellular fatty acid profile of the Bacillus strain X2 and other type strains were obtained by FAME analysis (Table 1). The FAME data indicated that three fatty acids iso-C15:0, anteiso-C15:0, and anteiso-C17:0 dominated the total fatty acid contents in the Bacillus strain X2. The abundance of iso-C15:0, in Bacillus strain X2 was 28.86%, nearly equal to that in the Bacillus subtilis subsp. subtilis (MCC 2049T), but nearly double that in Bacillus subtilis subsp. spizizenii (MCC 2480T). The occurrence of iso-C17:1 ω10c and iso-C18:1 H could be used as a differentiating feature, as both were detected only in Bacillus subtilis subsp. spizizenii (MCC 2480T). Summed feature 4 and summed feature 5 were absent in Bacillus subtilis subsp. subtilis (MCC 2049T); however, they were detected in Bacillus strain X2 in relatively double quantity concerning Bacillus subtilis subsp. spizizenii (MCC 2480T). The occurrence of Iso-C14:0 was uniquely associated with the Bacillus strain X2, being absent in both reference strains. Thus, fatty acid profiling of Bacillus strain X2 and its comparison with two type subsp. provided some data useful for differentiating among the three subsp.
Genotyping and MLSA
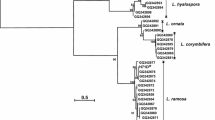
16S rRNA gene sequence 1526 bp was sequenced from the strain X2. Similarity search for the 16S rRNA gene sequence of the strain X2 was performed in the EzBioCloud server (Chun et al. 2007). Similarity search indicated that, amongst 11 very similar Bacillus species, Bacillus subtilis subsp. inaquosorum shared the highest similarity with the Bacillus strain X2 (99.86%). 16S rRNA sequence-based phylogenetic analysis (Fig. 2S) showed that the Bacillus strain X2 was closely linked with different subspecies of Bacillus subtilis that contained Bacillus subtilis subsp. natto, Bacillus subtilis subsp. inaquosorum, Bacillus subtilis subsp. subtilis and Bacillus subtilis subsp. stercoris. All share > 99.5% similarity confirming that the Bacillus strain X2 was a member of B. subtilis complex and therefore further mention this strain as Bacillus subtilis X2. However, due to the sharing of more than 99.5% similarity with multiple Bacillus species, the exact taxonomic identification of the Bacillus subtilis X2 was not possible (Saitou and Nei 1987; Chun and Bae 2000; Palmisano et al. 2001; Ruiz-García et al. 2005; Gatson et al. 2006). Since the Bacillus subtilis species complex comprises a closely associated group of related species, and could not be differentiated taxonomically based on 16S rRNA gene alone, the MLSA tool was used (Rooney et al. 2009). Apart from the 16S rRNA gene, groEL, gyrA, polC, purH, and rpoB genes were selected for MLSA, as they had been analyzed for assigning the taxonomical position to a Natto fermenting Bacillus strain in the Bacillus subtilis subgroup (Kubo et al. 2011). MLSA phylogenetic analysis provided some resolution of the taxonomic position of the Bacillus subtilis X2 in the Bacillus subtilis complex. The nucleotide sequence percent similarity of concatenated 16S rRNA, groEL, gyrA, polC, purH, and rpoB genes of the Bacillus subtilis X2 was found 99% with Bacillus subtilis subsp. stercoris, 97% with Bacillus subtilis subsp. subtilis 168 and 96% with Bacillus subtilis subsp. subtilis 6051 HGW, Bacillus subtilis subsp. spizizenii and Bacillus subtilis subsp. inaquosorum. The MLSA result Fig. 1 indicated that the Bacillus subtilis X2 belongs to Bacillus subtilis subsp. stercoris. Using MLSA, we could ascertain the taxonomic position of the Bacillus subtilis X2.

MLSA (16S rRNA, groEL, gyrA, polC, purH and rpoB)-based neighbor-joining phylogenetic tree. Tamura–Nei method was used to calculate the evolutionary distances (Tamura and Nei 1993). The bootstrap percentage after 1000 replication is shown (> 50%) (Felsenstein 1985). The bar represents 0.05 substitutions per site. B. cereus ATCC 14579T is an outgroup taxon. The final dataset was 12,058 bp after the removal of all gaps and missing data
Genome sequencing and annotation
The whole genome was de novo assembled into 38 contigs with a predictable size of 4,125,163 bp and GC content of 43.7%. The final N50 and N75 values were 245,478 and 163,885 bp, respectively, of the genome, with a genome coverage of 150 ×. Genome statistics are presented in Table 2 and the genome was compared with Bacillus subtilis subsp. subtilis and Bacillus subtilis subsp. spizizenii is shown in Fig. 2 indicating the presence of additional sequences. The overall genomic features of strain X2 were similar to the genome of B. subtilis (Yi et al. 2014). The strain X2 draft genome was composed of 4041 coding sequences, and 82 tRNAs, 4 rRNAs, and 5 ncRNA were identified. BActeriocin GEnome mining tooL 3 (BAGEL3) was used to identify one putative bacteriocin (sactipeptides) (van Heel et al. 2013), and PHAge Search Tool Enhanced Release (PHASTER) Web server was unable to find any prophage (Arndt et al. 2016). PlasmidFinder did not give any information about any type of plasmid (Carattoli et al. 2014). Additionally, genes for potential resistance to tetracycline resistance and aminoglycoside resistance were identified when the genome was matched against the ResFinder database (Zankari et al. 2012).

Genomic representation of the Bacillus subtilis subsp. stercoris X2 genome. Ring from inside: 1—GC content, ring 2—GC skew, ring 3—tRNA, ring 4—rRNA, ring 5–7—genomic sequence identity at minimum 80% similarity with the genome of reference type strains of B. subtilis, B. spizizenii, B. inaquosorum. The figure was prepared using BRIG software (Alikhan et al. 2011)
Genes of strains used in the study were assigned to COG (Cluster of Orthologous Group) functional categories. Bacillus subtilis X2 genome in COG comparison with B. subtilis subsp. subtilis, B. subtilis subsp. spizizenii and B. subtilis subsp. inaquosorum has shown differences in the secondary metabolite production and defense mechanism sector, indicating the presence of strong defense strategies. Those results are presented in Table 3.
Genome homology
Biochemical and phenotypic characterization and 16S rRNA sequence homology grossly indicated that Bacillus strain X2 belongs to the Bacillus subtilis complex. MLSA gave us the real clue about Bacillus strain X2 being the nearest to Bacillus subtilis subsp. stercoris. The gold standard for the species delineation is the DDH. However, it is rigorous and yet sometimes may not produce consistent results (Colston et al. 2014). Richer and Rossellό-Mόra (2009) suggested that information obtained from the genome sequence such as ANIb could be used for delineation of species in place of genomic DNA–DNA hybridization. ANIb similarity threshold of 95–96% could replace the 70% DDH criterion in the current prokaryotic systematics for circumscribing a species. Due to the advent of new generation sequencing (NSG), it has become easy to obtain ANIb data that can be processed to conclude the taxonomical identity of the species. The genome sequence data of Bacillus subtilis X2 was subjected to different bioinformatics tools available to ascertain the identity of the Bacillus subtilis X2.
The ANIb values or the tetranucleotide signature frequency correlation coefficient (TETRA) was calculated using JspeciesWS (Richter et al. 2015). The DSMZ GGDC platform was used for the digital DDH analysis (Meier-Kolthoff et al. 2013). Among the four Bacillus subtilis subspecies type strain including the strain X2, the highest DDH and ANIb values obtained were 87.3% and 98.33% for the subspecies stercoris; the other three type subspecies, i.e., Bacillus subtilis subsp. subtilis, Bacillus subtilis subsp. spizizenii and Bacillus subtilis subsp. inaquosorum are distant relative with ANIb and DDH values 95.23%, 92.33%, 92.36% and 62.9, 48.5, 48.9, respectively (Table 4). This is in contrast with the highest similarity in the 16S rRNA gene sequence of Bacillus inaquosorum. Differences in both the ANIb and dDDH values for the three type strains Bacillus subtilis subsp. subtilis, Bacillus subtilis subsp. spizizenii and Bacillus subtilis subsp. inaquosorum placed them below the identity threshold mark and the strain X2 as the as one more distinct strain in the Bacillus subtilis subsp. stercoris. Similar results for the identification of strains from three subsp. of B. subtilis, i.e., subspecies subtilis, spizizenii, and inaquosorum have been reported by Yi et al. (2014), Adelskov and Patel (2017) and helped us to draw this conclusion.
Construction of phylogenomic trees based on housekeeping protein sequences
The amino acid sequence of 31 homologous housekeeping proteins encoded by highly conserved and single copy genes in genomes from various Bacillus species was extracted using Amphoranet 2 (Kerepesi et al. 2014) server (Table 2S).
Phylogenomics of the concatenated protein (Fig. 3) analysis supported the differentiation of four subspecies in the groups of Bacillus subtilis. Different subspecies created distinct phylogenomic lines within the coverage of B. subtilis, suggesting that subspecies inaquosorum, spizizenii, and stercoris are least distinguished from subtilis. These results are in accordance with those obtained from ANIb, dDDH and MLSA phylogenetic tree analysis.

Protein marker-based neighbor-joining phylogenomics. The Tamura–-Nei method was used to calculate evolutionary distances (Tamura and Nei 1993). The number at branch points indicate bootstrap percentage after 1000 replication is shown (> 50%) (Felsenstein 1985). B. cereus ATCC 14579T is an outgroup taxon. The final dataset after the deletion of all gaps and missing data was 7026 bp
Sequence analysis of a Xyl_1 gene and xylooligosaccharides production
A xylanase gene (Xyl_1) from B. subtilis strain X2 was amplified. The gene encoding 205 amino acids protein with a predicted molecular mass of 22.7 kDa and a theoretical pI of 9.0. The gene sequence has been submitted to the NCBI database under accession number MT505314. Xyl_1 gene encoding the product displayed relatively high identities with the reported GH family 11 xylanase, sharing the highest identity of 95.5% with the GH 11 xylanase from Bacillus subtillis B230, in which 3–196 amino acid sequence is GH 11 domain (1IGO) (Oakley et al. 2003), followed by the GH 11 xylanase from Bacillus sp. strain 41M-1 (6KKA, 75.8%) (Takita et al. 2019), Bacillus agaradhaerens (1H4G, 71.1%) (Sabini et al. 1999), Streptomyces lividans (5EJ3, 53.2%) (Gagné et al. 2016) and Thermopolyspora flexuosas (1M4W, 57.5%) (Hakulinen et al. 2003) (Fig. 4). The triad Pro132−Ser133−Ile134 is highly conserved (> 90%) in the GH11 domain; Ile134 is sometimes substituted by Val or Leu, which have roughly the same spatial occupancy and hydrophobicity. Finally, Pro106 in the middle of the cord is present in 80% of the sequences (Paës et al. 2012). The xylan hydrolysis profile of birchwood xylan by Xyl_1 indicated that xylotriose, xylotetraose and xylopentaose were major product components. This pattern of product formation is the unique characteristic of the GH11 xylanase (Yagi et al. 2019). The high homology similarity of amino acid sequence and product profile suggest that Xyl_1 should be a member of the GH family 11 xylanase.

Multiple sequence alignment of Xyl_1 with its closely characterized homologs GH11 xylanase (α-helices are displayed as squiggles, β-strands are rendered as arrows and strict β-turns are represented by the letters TT). Identical residues are shown in white on a red background. Some of the identical residues are conserved domains of the GH 11 family (Paës et al. 2012). Catalytic residues (Glu94, Glu183) are marked by red dots. Abbreviations of the GH family 11 enzymes in the alignment are as follows: B. subtilis strain X2 (Xyl_1), Bacillus subtillis B230 (1IGO), Bacillus sp. strain 41M-1 (6KKA), Bacillus agaradhaerens (1H4G), Streptomyces lividans (5EJ3) and Thermopolyspora flexuosas (1M4W)
As Bacillus subtilis subsp. stercoris X2 was found to produce endo-xylanase GH11 (EC: 3.2.1.8) that could convert birchwood xylan into xylooligosaccharides. HPLC analysis of reaction products of xylanase with birchwood xylan did not produce xylose as a reaction product. Dimer, trimer, tetramer, and pentamers of xylose were detected by HPLC (Fig. 5). This indicates that the xylanase produced by B. subtilis subsp. stercoris strain X2 is an endo-xylanase. One unit of xylanase produced 0.8 mg/ml xylotriose and 0.13 mg/ml xylobiose in 1 h.

HPLC chromatogram of XOS. a Xylotriose (RT = 7.5 min), b xylobiose (RT = 8.7 min), c glucose (RT = 9.8 min), and d xylose (RT = 10.7 min), e Hydrolysis profile of birchwood xylan substrate control (no enzyme). f Hydrolysis products formed after 60 min incubation contains xylopentaose (RT = 6.24 min), xylotetraose (RT = 6.74 min), xylotriose (RT = 7.5 min) and xylobiose (RT = 8.7 min)
Many Bacillus subtilis species are known for the production of xylanolytic enzymes, but due to the co-production of β-xylosidase conversion of valuable XOS to xylose takes place which is subsequently metabolized by microbes (Bhalla et al. 2014; Reddy and Krishnan 2016b). Hence, screening such microbes that can produce β-xylosidase-free xylanase is a prerequisite for the production of low cost XOS. Bacillus subtilis subsp. stercoris X2 was found to synthesize β-xylosidase-free endo-xylanase. Bacillus subtilis KCX006 has also been reported to produce xylanase in solid state fermentation without β-xylosidase (Reddy and Krishnan 2016a). Xylanase from different species of Bacillus such as Bacillus mojavensis A21 (Haddar et al. 2012) and Bacillus aerophilus KGJ2 (Gowdhaman and Ponnusami 2015) have been described for the production of xylooligosaccharides by hydrolysis of xylan. Endo-xylanase is more suitable for the production of XOS as a prebiotic, as they produce longer xylooligosaccharides. XOS has wide applications in the food and feed industry. It is used as a prebiotic that helps to maintain the gut microflora of the gastrointestinal tract in humans and animals and augments the probiotic efficacy. (Gullón et al. 2010; Teng et al. 2010). XOS has been reported to have antimicrobial activity against Gram-positive and Gram-negative bacteria (Christakopoulos et al. 2003). Xylobiose is used as a low calorie sweetener.
Conclusion
We have isolated potent xylanase (GH 11) (EC:3.2.1.8) producer which can convert xylan into XOS which are important in the food and feed industry. We identified the isolate X2 as Bacillus subtilis subsp. stercoris. Phylogenetic investigation using MLSA in conjunction with the chemotaxonomic characterization and genome-based analysis of ANIb as well as digital DDH was applied to ascertain the taxonomic position of the Bacillus strain X2. Based on the phylogenetic, phenotypic, and genomic analysis, we propose a new member in subsp. stercoris. The novel strain X2 has shown distinctive features such as being unable to produce polymyxin in contrast to our study, in which we found that the type strain of subsp. stercoris produced three different types of polymyxins, even though, the gene responsible for polymyxin production was available in both strains. This contrasting behavior might be necessary for the survival of bacteria in adverse habitats. This is the first report of endo-xylanase production and its application in the production of XOS from this Bacillus subtilis subspecies stercoris. Bacillus strain X2 is the first strain of the Bacillus subtilis subsp. stercoris group that has been studied by both approaches, polyphasic as well as genomic analysis for assigning its taxonomical position namely, Bacillus subtilis subsp. stercoris.
Change history
09 September 2020
Unfortunately, the funding information of the article has been missed in the publication of the article.
Abbreviations
- MALDI-ToF:
-
Matrix-assisted laser desorption ionization-time of flight
- XOS:
-
Xylooligosaccharides
- MLSA:
-
Multilocus sequence analysis
- FAMEs:
-
Fatty acid methyl esters
References
Adelskov J, Patel BKC (2017) Erratum to: A molecular phylogenetic framework for Bacillus subtilis using genome sequences and its application to Bacillus subtilis subspecies stecoris strain D7XPN1, an isolate from a commercial food-waste degrading bioreactor. 3 Biotech 6:1
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. https://doi.org/10.1186/1471-2164-12-402
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Anil Kumar P, Aravind R, Francis K et al (2012) Shivajiella indica gen. nov., sp. nov., a marine bacterium of the family “ Cyclobacteriaceae” with nitrate reducing activity. Syst Appl Microbiol 35:320–325. https://doi.org/10.1016/j.syapm.2012.04.004
Arndt D, Grant JR, Marcu A et al (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw387
Aziz RK, Bartels D, Best A et al (2008) The RAST Server: Rapid annotations using subsystems technology. BMC Genomics. https://doi.org/10.1186/1471-2164-9-75
Bailey MJ, Biely P, Poutanen K (1992) Interlaboratory testing of methods for assay of xylanase activity. J Biotechnol 23:257–270. https://doi.org/10.1016/0168-1656(92)90074-J
Bhalla A, Bischoff KM, Sani RK (2014) Highly thermostable GH39 β-xylosidase from a Geobacillus sp. strain WSUCF1. BMC Biotechnol. https://doi.org/10.1186/s12896-014-0106-8
Calderaro A, Arcangeletti M-C, Rodighiero I et al (2014) Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry applied to virus identification. Sci Rep. https://doi.org/10.1038/srep06803
Carattoli A, Zankari E, Garciá-Fernández A et al (2014) In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.02412-14
Christakopoulos P, Katapodis P, Kalogeris E et al (2003) Antimicrobial activity of acidic xylo-oligosaccharides produced by family 10 and 11 endoxylanases. Int J Biol Macromol. https://doi.org/10.1016/S0141-8130(02)00079-X
Chun J, Bae KS (2000) Phylogenetic analysis of Bacillus subtilis and related taxa based on partial gyrA gene sequences. Antonie Van Leeuwenhoek. https://doi.org/10.1023/A:1026555830014
Chun J, Lee J-H, Jung Y et al (2007) EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57:2259–2261. https://doi.org/10.1099/ijs.0.64915-0
Cohan FM (2002) What are bacterial species? Annu Rev Microbiol 56:457–487. https://doi.org/10.1146/annurev.micro.56.012302.160634
Cohan FM (2006) Towards a conceptual and operational union of bacterial systematics, ecology, and evolution. Philos Trans R Soc B Biol Sci 361:1985–1996. https://doi.org/10.1098/rstb.2006.1918
Cohan FM, Kane M (2001) Bacterial species and speciation. Syst Biol 50:513–524. https://doi.org/10.1080/10635150118398
Colston SM, Fullmer MS, Beka L et al (2014) Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using aeromonas as a test case. MBio. https://doi.org/10.1128/mBio.02136-14
Dunlap CA, Bowman MJ, Schisler DA, Rooney AP (2016a) Genome analysis shows Bacillus axarquiensis is not a later heterotypic synonym of Bacillus mojavensis; reclassification of Bacillus malacitensis and Brevibacterium halotolerans as heterotypic synonyms of Bacillus axarquiensis. Int J Syst Evol Microbiol 66:2438–2443. https://doi.org/10.1099/ijsem.0.001048
Dunlap CA, Kim SJ, Kwon SW, Rooney AP (2016b) Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens; Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. Plantarum and ‘Bacillus oryzicola’ are later heterotypic synonyms of Bacillus velezensis based on phylogenom. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijsem.0.000858
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution. https://doi.org/10.2307/2408678
Gagné D, Narayanan C, Nguyen-Thi N et al (2016) Ligand binding enhances millisecond conformational exchange in xylanase B2 from Streptomyces lividans. Biochemistry. https://doi.org/10.1021/acs.biochem.6b00130
Gatson JW, Benz BF, Chandrasekaran C et al (2006) Bacillus tequilensis sp. nov., isolated from a 2000-year-old Mexican shaft-tomb, is closely related to Bacillus subtilis. Int J Syst Evol Microbiol 56:1475–1484. https://doi.org/10.1099/ijs.0.63946-0
Gevers D, Cohan FM, Lawrence JG et al (2005) Re-evaluating prokaryotic species. Nat Rev Microbiol 3:733–739
Gowdhaman D, Ponnusami V (2015) Production and optimization of xylooligosaccharides from corncob by Bacillus aerophilus KGJ2 xylanase and its antioxidant potential. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2015.05.046
Gullón B, Yáñez R, Alonso JL, Parajó JC (2010) Production of oligosaccharides and sugars from rye straw: a kinetic approach. Bioresour Technol. https://doi.org/10.1016/j.biortech.2010.03.080
Haddar A, Driss D, Frikha F et al (2012) Alkaline xylanases from Bacillus mojavensis A21: production and generation of xylooligosaccharides. Int J Biol Macromol. https://doi.org/10.1016/j.ijbiomac.2012.06.036
Hakulinen N, Turunen O, Jänis J et al (2003) Three-dimensional structures of thermophilic β-1,4-xylanases from Chaetomium thermophilum and Nonomuraea flexuosa: Comparison of twelve xylanases in relation to their thermal stability. Eur J Biochem. https://doi.org/10.1046/j.1432-1033.2003.03496.x
Huang D, Liu J, Qi Y et al (2017) Synergistic hydrolysis of xylan using novel xylanases, β-xylosidases, and an α-l-arabinofuranosidase from Geobacillus thermodenitrificans NG80-2. Appl Microbiol Biotechnol 101:6023–6037. https://doi.org/10.1007/s00253-017-8341-2
Huerta-Cepas J, Forslund K, Coelho LP et al (2017) Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. https://doi.org/10.1093/molbev/msx148
Kerepesi C, Bánky D, Grolmusz V (2014) AmphoraNet: the webserver implementation of the AMPHORA2 metagenomic workflow suite. Gene 533:538–540. https://doi.org/10.1016/j.gene.2013.10.015
Kubo Y, Rooney AP, Tsukakoshi Y et al (2011) Phylogenetic analysis of Bacillus subtilis strains applicable to natto (fermented soybean) production. Appl Environ Microbiol. https://doi.org/10.1128/AEM.00448-11
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Lagesen K, Hallin P, Rødland EA et al (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108. https://doi.org/10.1093/nar/gkm160
Lane DJ (1991) 16S/23S rRNA sequencing. Nucleic Acid Tech Bact Syst. https://doi.org/10.1007/s00227-012-2133-0
Lányi B (1988) Classical and rapid identification methods for medically important bacteria. Methods Microbiol 19:1–67. https://doi.org/10.1016/S0580-9517(08)70407-0
Logan NA, De Vos P (2009) Genus I. Bacillus Cohn 1872, 174AL. In: Bergey’s Manual of Systematic Bacteriology Volume Three The Firmicutes. pp 21–130
Logan NA, Berge O, Bishop AH et al (2009) Proposed minimal standards for describing new taxa of aerobic, endospore-forming bacteria. Int J Syst Evol Microbiol 59:2114–2121. https://doi.org/10.1099/ijs.0.013649-0
Lowe TM, Eddy SR (1996) TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. https://doi.org/10.1093/nar/25.5.0955
Marmur J (1961) A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J Mol Biol 3:IN1–218. https://doi.org/10.1016/S0022-2836(61)80047-8
Maughan H, Van der Auwera G (2011) Bacillus taxonomy in the genomic era finds phenotypes to be essential though often misleading. Infect Genet Evol 11:789–797. https://doi.org/10.1016/j.meegid.2011.02.001
Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M (2013) Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. https://doi.org/10.1186/1471-2105-14-60
Moaledj K (1986) Comparison of Gram-staining and alternate methods, KOH test and aminopeptidase activity in aquatic bacteria: their application to numerical taxonomy. J Microbiol Methods 5:303–310. https://doi.org/10.1016/0167-7012(86)90056-4
Nakamura LK (1989) Taxonomic relationship of black-pigmented Bacillus subtilis strains and a proposal for Bacillus atrophaeus sp. nov. Int J Syst Bacteriol 39:295–300. https://doi.org/10.1099/00207713-39-3-295
Nakamura LK, Roberts MS, Cohan FM (1999) Relationship of Bacillus subtilis clades associated with strains 168 and W23: a proposal for Bacillus subtilis subsp. subtilis subsp. nov. and Bacillus subtilis subsp. spizizenii subsp. nov. Int J Syst Bacteriol. https://doi.org/10.1099/00207713-49-3-1211
Oakley AJ, Heinrich T, Thompson CA, Wilce MCJ (2003) Characterization of a family 11 xylanase from Bacillus subtillis B230 used for paper bleaching. Acta Crystallogr Sect D Biol Crystallogr. https://doi.org/10.1107/S0907444903001227
Oraei M, Razavi SH, Khodaiyan F (2018) Optimization of effective minerals on riboflavin production by Bacillus subtilis subsp. subtilis ATCC 6051 using statistical designs. Avicenna J Med Biotechnol 10(1):49–55
Paës G, Berrin JG, Beaugrand J (2012) GH11 xylanases: Structure/function/properties relationships and applications. Biotechnol Adv 30(3):564–592. https://doi.org/10.1016/j.biotechadv.2011.10.003
Palmisano MM, Nakamura LK, Duncan KE et al (2001) Bacillus sonorensis sp. nov., a close relative of Bacillus licheniformis, isolated from soil in the Sonoran Desert Arizona. Int J Syst Evol Microbiol 51:1671–1679. https://doi.org/10.1099/00207713-51-5-1671
Perry EB, Koeppel A, Krizanc D et al (2006) Identifying the fundamental units of diversity among Bacillus isolates from "Evolution Canyon" III. Isr J Ecol Evol 52:543–552. https://doi.org/10.1560/IJEE_52_3-4_543
Petersen TN, Brunak S, Von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Price NPJ, Rooney AP, Swezey JL et al (2007) Mass spectrometric analysis of lipopeptides from Bacillus strains isolated from diverse geographical locations. FEMS Microbiol Lett 271:83–89. https://doi.org/10.1111/j.1574-6968.2007.00702.x
Reddy SS, Krishnan C (2016a) Production of xylooligosaccharides in SSF by Bacillus subtilis KCX006 producing β-xylosidase-free endo-xylanase and multiple xylan debranching enzymes. Prep Biochem Biotechnol. https://doi.org/10.1080/10826068.2014.970694
Reddy SS, Krishnan C (2016b) Production of high-pure xylooligosaccharides from sugarcane bagasse using crude β-xylosidase-free xylanase of Bacillus subtilis KCX006 and their bifidogenic function. LWT Food Sci Technol. https://doi.org/10.1016/j.lwt.2015.08.013
Rhee MS, Wei L, Sawhney N et al (2014) Engineering the xylan utilization system in Bacillus subtilis for production of acidic xylooligosaccharides. Appl Environ Microbiol. https://doi.org/10.1128/AEM.03246-13
Richter M, Rossello-Mora R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.0906412106
Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J (2015) JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv681
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. https://doi.org/10.1093/nar/gku316
Roberts MS, Cohan FM (1995) Recombination and migration rates in natural populations of Bacillus subtilis and Bacillus mojavensis. Evolution. https://doi.org/10.2307/2410433
Roberts MS, Nakamura LK, Cohan FM (1994) Bacillus mojavensis sp. nov., distinguishable from Bacillus subtilis by sexual isolation, divergence in DNA sequence, and differences in fatty acid composition. Int J Syst Bacteriol 44:256–264. https://doi.org/10.1099/00207713-44-2-256
Roberts MS, Nakamura LK, Cohan FM (1996) Bacillus vallismortis sp. nov., a close relative of Bacillus subtilis, isolated from soil in Death Valley. California Int J Syst Bacteriol 46:470–475. https://doi.org/10.1099/00207713-46-2-470
Rooney AP, Price NPJ, Ehrhardt C et al (2009) Phylogeny and molecular taxonomy of the Bacillus subtilis species complex and description of Bacillus subtilis subsp. inaquosorum subsp. nov. Int J Syst Evol Microbiol 59:2429–2436. https://doi.org/10.1099/ijs.0.009126-0
Ruiz-García C, Béjar V, Martínez-Checa F et al (2005) Bacillus velezensis sp. nov, a surfactant-producing bacterium isolated from the river Vélez in Málaga, southern Spain. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijs.0.63310-0
Sabini E, Sulzenbacher G, Dauter M et al (1999) Catalysis and specificity in enzymatic glycoside hydrolysis: a 2,5 B conformation for the glycosyl-enzyme intermediate revealed by the structure of the Bacillus agaradhaerens family 11 xylanase. Chem Biol. https://doi.org/10.1016/S1074-5521(99)80066-0
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. https://doi.org/10.1093/oxfordjournals.molbev.a040454
Sambrook J, Russell DW (2006) Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harb Protoc 2006:pdb.prot4455. https://doi.org/10.1101/pdb.prot4455
Smibert RM, Krieg NR (1994) Phenotypic characterization. In: Gerhardt P, Murray RGE, Wood WA, Krieg NR (eds) Methods for general and molecular bacteriology. American Society for Microbiology, Washington DC, pp 607–654
Srinivas TNR, Nageswara Rao SSS, Vishnu Vardhan Reddy P et al (2009) Bacterial diversity and bioprospecting for cold-active lipases, amylases and proteases, from culturable bacteria of Kongsfjorden and Ny-Ålesund, Svalbard, Arctic. Curr Microbiol 59:537–547. https://doi.org/10.1007/s00284-009-9473-0
Stackebrandt E, Frederiksen W, Garrity GM et al (2002) Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52:1043–1047. https://doi.org/10.1099/ijs.0.02360-0
Takita T, Nakatani K, Katano Y et al (2019) Increase in the thermostability of GH11 xylanase XynJ from Bacillus sp. strain 41M–1 using site saturation mutagenesis. Enzyme Microb Technol. https://doi.org/10.1016/j.enzmictec.2019.109363
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526. https://doi.org/10.1093/molbev/msl149
Teng C, Yan Q, Jiang Z et al (2010) Production of xylooligosaccharides from the steam explosion liquor of corncobs coupled with enzymatic hydrolysis using a thermostable xylanase. Bioresour Technol. https://doi.org/10.1016/j.biortech.2010.05.004
Tindall BJ, Rosselló-Móra R, Busse HJ et al (2010) Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijs.0.016949-0
van Heel AJ, de Jong A, Montalbán-López M et al (2013) BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt391
Vázquez MJ, Alonso JL, Domínguez H, Parajó JC (2001) Xylooligosaccharides: manufacture and applications. Trends Food Sci Technol 11:387–393
Wang LT, Lee FL, Tai CJ et al (2007) Reclassification of Bacillus axarquiensis Ruiz-García et al. 2005 and Bacillus malacitensis Ruiz-García et al. 2005 as later heterotypic synonyms of Bacillus mojavensis Roberts et al. 1994. Int J Syst Evol Microbiol 57:1663–1667. https://doi.org/10.1099/ijs.0.64808-0
Wang LT, Lee FL, Tai CJ, Kuo HP (2008) Bacillus velezensis is a later heterotypic synonym of Bacillus amyloliquefaciens. Int J Syst Evol Microbiol 58:671–675. https://doi.org/10.1099/ijs.0.65191-0
Xu P, Li WJ, Tang SK et al (2005) Naxibacter alkalitolerans gen. nov., sp. Nov., a novel member of the family “Oxalobacteraceae” isolated from China. Int J Syst Evol Microbiol. https://doi.org/10.1099/ijs.0.63407-0
Yagi H, Takehara R, Tamaki A et al (2019) Functional characterization of the GH10 and GH11 xylanases from Streptomyces olivaceoviridis E-86 provide insights into the advantage of GH11 xylanase in catalyzing biomass degradation. J Appl Glycosci. https://doi.org/10.5458/jag.jag.jag-2018_0008
Yi H, Chun J, Cha CJ (2014) Genomic insights into the taxonomic status of the three subspecies of Bacillus subtilis. Syst Appl Microbiol 37:95–99. https://doi.org/10.1016/j.syapm.2013.09.006
Zankari E, Hasman H, Cosentino S et al (2012) Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. https://doi.org/10.1093/jac/dks261
Zwick ME, Joseph SJ, Didelot X et al (2012) Genomic characterization of the Bacillus cereus sensu lato species: backdrop to the evolution of Bacillus anthracis. Genome Res 22:1512–1524. https://doi.org/10.1101/gr.134437.111
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dhruw, C., Husain, K., Kumar, V. et al. Novel xylanase producing Bacillus strain X2: molecular phylogenetic analysis and its application for production of xylooligosaccharides. 3 Biotech 10, 328 (2020). https://doi.org/10.1007/s13205-020-02322-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-020-02322-1